
Abstract
We present two-feldspar thermometry and diffusion chronometry from sanidine, orthopyroxene and quartz from multiple samples of the Bishop Tuff, California, to constrain the temperature stratification within the pre-eruptive magma body and the timescales of magma mixing prior to its evacuation. Two-feldspar thermometry yields estimates that agree well with previous Fe–Ti oxide thermometry and gives a ~80 °C temperature difference between the earlier- and later-erupted regions of the magma chamber. Using the thermometry results, we model diffusion of Ti in quartz, and Ba and Sr in sanidine as well as Fe–Mg interdiffusion in orthopyroxene to yield timescales for the formation of overgrowth rims on these crystal phases. Diffusion profiles of Ti in quartz and Fe–Mg in orthopyroxene both yield timescales of <150 years for the formation of overgrowth rims. In contrast, both Ba and Sr diffusion in sanidine yield nominal timescales 1–2 orders of magnitude longer than these two methods. The main cause for this discrepancy is inferred to be an incorrect assumption for the initial profile shape for Ba and Sr diffusion modelling (i.e. growth zoning exists). Utilising the divergent diffusion behaviour of Ba and Sr, we place constraints on the initial width of the interface and can refine our initial conditions considerably, bringing Ba and Sr data into alignment, and yielding timescales closer to 500 years, the majority of which are then within uncertainty of timescales modelled from Ti diffusion in quartz. Care must be thus taken when using Ba in sanidine geospeedometry in evolved magmatic systems where no other phases or elements are available for comparative diffusion profiling. Our diffusion modelling reveals piecemeal rejuvenation of the lower parts of the Bishop Tuff magma chamber at least 500 years prior to eruption. Timescales from our mineral profiling imply either that diffusion coefficients currently used are uncertain by 1–2 orders of magnitude, or that the minerals concerned did not experience a common history, despite being extracted from the same single pumice clasts. Introduction of the magma initiating crystallisation of the contrasting rims on sanidine, quartz, orthopyroxene and zircon was prolonged, and may be a marker of other processes that initiated the Bishop Tuff eruption rather than the trigger itself.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Understanding the role of recharge in the assembly, stratification and evacuation of magma chambers has been a long-standing question in the study of large-scale silicic magma systems, particularly those generating supereruptions. These eruptions, of >1015 kg (~450 km3) magma (Self 2006; Miller and Wark 2008), have the potential to cause massive destruction, as well as having catastrophic effects on global climate (Self and Blake 2008). In terms of mitigating potential hazards, the more that can be determined regarding the timescales of the processes occurring in the parental magma systems (regardless of size), the more information that can be fed into monitoring programs at currently active silicic volcanoes.
Recharge of volcanic systems has often been cited as a trigger for eruptions (e.g. Sparks et al. 1977; Jellinek and DePaolo 2003; Wark et al. 2007; Arienzo et al. 2011; Matthews et al. 2012a; Saunders et al. 2012) whether due to direct increase in volume causing failure of the magma chamber wall rocks (e.g. Jellinek and DePaolo 2003), the buoyancy-driven effects of accumulating magma (e.g. Caricchi et al. 2014; Malfait et al. 2014), or by indirectly causing changes in volume of saturated gases and crystal cargo (e.g. Snyder 2000; Wark et al. 2007). Evidence of magmatic recharge or mixing is commonly preserved as zonations within crystal phases already present in the magma chamber (e.g. Morgan et al. 2004; Ginibre et al. 2007; Wark et al. 2007; Martin et al. 2008; Humphreys et al. 2009; Allan et al. 2013). However, two factors make interpretation complex. The first is whether the crystal phases are recording consistently the same event, resolution of which is hampered (in general) by the use of single mineral phases for study (e.g. Wark et al. 2007; Gualda et al. 2012a). The second is whether evidence for recharge or magma mixing is automatically evidence for an eruption trigger (e.g. Wark et al. 2007), when other, external factors may be important in causing both the recharge and the eruption (e.g. Allan et al. 2012, 2013).
Diffusional geochronometry aims to model the evolution of compositional profiles within zoned crystals to determine the time elapsed since compositional variations were introduced. Typically, an initial starting profile shape is assumed—usually a sharp, step change—and this is modelled forwards in time until it matches the observed profile. The timescale determined via this method can be regarded as a maximum, as the exact initial condition is not known. We will return to the validity of this initial assumption later in this paper. Although diffusion modelling of single or multiple elements has been frequently utilised to explore timescales of crystal residence in magma chambers (e.g. Zellmer et al. 1999; Costa et al. 2003; Morgan et al. 2004, 2006; Costa and Dungan 2005; Wark et al. 2007; Gualda et al. 2012a; Saunders et al. 2012; Till et al. 2012; Allan et al. 2013), multiple phases from the same sample have not been routinely investigated.
The Bishop Tuff (eruption age of 767 ka: Rivera et al. 2011) is an iconic example of a supereruption, which displays evidence of magma mixing preserved as zonation in quartz, sanidine and orthopyroxene crystals (Hildreth 1977, 1979; Anderson et al. 2000; Peppard et al. 2001; Morgan and Blake 2006; Wark et al. 2007; Gualda et al. 2012a). Whilst the Bishop Tuff has been the subject of many volcanological and geochemical investigations (see Hildreth and Wilson 2007 for review), the timescales associated with magma chamber assembly, residence and pre-eruptive mobilisation remain controversial. For example, radiogenic isotopic studies on crystal and glass separates from the Bishop Tuff pointed to a long residence time for the magma (300–1,300 kyr: Christensen and Halliday 1996; Davies and Halliday 1998), apparently supported by theoretical models of felsic magma genesis (Fowler and Spera 2010). However, more recent studies of zircon U–Pb ages and crystal size distribution (CSD) modelling indicate that this is unrealistic and infer timescales of between ~160 kyr to a few thousand years (e.g. Gualda et al. 2012a; Pamukcu et al. 2012; Reid and Schmitt 2012; Chamberlain et al. 2014) for crystals residing in the melt-dominant magma body. Previous workers have investigated the timescales for magma mobilisation within the Bishop Tuff magma chamber, primarily by modelling Ti diffusion with quartz (Wark et al. 2007; Gualda et al. 2012a), which yields timescales of <100 years [for the ‘bright rim’ (in CL imagery) overgrowths] to ~2,700 years (for core-interior zonation). However, no comprehensive combined study of the diffusion records of multiple crystal phases within the Bishop Tuff has been undertaken. Here, we compare and contrast the timescales inferred from Fe–Mg interdiffusion in orthopyroxene, Ti diffusion in quartz (cf. Wark et al. 2007; Gualda et al. 2012a), Sr diffusion in sanidine and Ba diffusion in sanidine (cf. Morgan and Blake 2006) from multiple samples in the Bishop Tuff. We examine how comparable each of these timescales are, whether they are tracing the same magmatic events, and constrain the relatively short-lived injection of magma preceding the eruption. We also compare our data with alternative information on timescales of the processes involved from high-precision U–Pb chronometry and textural studies in co-erupted zircon (Chamberlain et al. 2014).
Geological setting
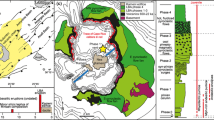
The Bishop Tuff eruption was the culmination of ~3.8 Myr of magmatic activity in the Long Valley region and is the product of one of the largest eruptions on Earth in the last 1 Myr. Over 600 km3 of magma was erupted to generate fall deposits and ignimbrite, along with voluminous infill from the formation of Long Valley caldera (Fig. 1; Bailey et al. 1976; Hildreth 1979, 2004; Wilson and Hildreth 1997; Hildreth and Wilson 2007). The volume of rhyolite erupted as the Bishop Tuff is greater than all other felsic eruptions combined in the Long Valley area before or since (Hildreth 2004), and so questions arise as to how, and over what time period, such a large body of magma was accumulated and what the triggering processes were for the eruption.

a Map of the Long Valley area, eastern California, USA, and b summary of the Bishop Tuff stratigraphy, after Hildreth and Wilson (2007). a The topographic outline of Long Valley caldera is shown as a dashed line; regions of Bishop ignimbrite are filled in grey. The envelope enclosing mapped vent locations for the precursory Glass Mountain (GM) eruptions is highlighted in light grey. The line marked ‘Fall deposition envelope’ marks the westerly limits of where Bishop fall deposits are found in this area. The lines marked i and ii relate to the respective labels in panel b. Line i shows a hypothetical cross section from NW to NE across the northern ignimbrite lobes as viewed from the centre of Long Valley caldera. Line ii is a schematic proximal to distal cross section, approximately along the line of the Owens River gorge. b Schematic stratigraphy of the Bishop Tuff in its proximal area based on sections along lines i and ii. Ignimbrite units are coloured; fall units are grey. Samples collected for this paper cover most of the stratigraphic units and are detailed in Electronic Appendix 1
Proximal deposits of the eruption consist of nine fall units (F1–F9) and eleven ignimbrite packages (Wilson and Hildreth 1997; Fig. 1). A systematic change from ‘early’, crystal-poor (<6 wt% crystals), more-evolved (78.2–74.8 wt% SiO2 whole rock) to ‘late’, crystal-rich (12–25 wt% crystals), less-evolved (77.8–73.4 wt% SiO2 whole rock) compositions is observed within the stratigraphy of the Bishop Tuff (see Hildreth and Wilson 2007 for details). In the context of this paper, ‘early’ refers to fall units F1–F8 and their coeval Ig1 ignimbrite packages, ‘late’ refers to Ig2N and Ig2NW, and ‘intermediate’ refers to the Ig2E ignimbrite packages and fall unit F9. Changes in pumice composition accompany an overall change in volatile contents and species in quartz hosted melt inclusions from water-richer, CO2-poorer to water-poorer, CO2-richer (Dunbar and Hervig 1992; Wallace et al. 1999) magma. For this study, single pumice samples were collected from every ignimbrite unit of the Bishop Tuff, and pumices covering the range of crystallinities were sampled (Fig. 1; see Electronic Appendix 1 for sample descriptions and localities). Pumice clasts are categorised within two spectra: the ‘normal’ crystal-poor (xp) to crystal-rich (xr) scale, and the ‘variant’ types, following Hildreth and Wilson (2007). The latter are of particular interest because they often show evidence of physical mixing on millimetre-to-centimetre scales, and so eight samples of the ‘swirly’ and ‘chocolate’ variant pumice types of Hildreth and Wilson (2007) were inspected from packages Ig1Eb through to Ig2Nb.
Methods
Sample preparation
Single pumices were selected, washed to remove adhering matrix, and then crushed to chips using a Boyd crusher at Victoria University of Wellington. Crushed material was sieved, and the sample was water-panned to separate glass from crystals. Quartz and sanidine crystals were then hand-picked under optical microscope from the 1- to 2-mm-size fraction. Orthopyroxene crystals were hand-picked from the 250- to 500-µm-size fraction (the sieved fraction in which orthopyroxene crystals were most common). Sanidine and orthopyroxene crystals were orientated with the crystal b- and c-axes in the plane of the section, so that these axes were exposed once the epoxy mount was ground and polished for imaging and analysis.
BSE imaging
Zoning patterns in sanidine and orthopyroxene were imaged by back-scattered electron (BSE) methods on a JEOL JXA 8230 Electron Microprobe (EPMA) at Victoria University of Wellington. Due to the dependence of BSE brightness on the effective atomic number of the sample per unit volume, changes in BSE brightness correspond to compositional changes within the crystal (Reed 2005). In sanidine, the major element affecting BSE brightness is Ba, and therefore, brighter BSE tones relate, in these samples, to areas of higher Ba concentration. Sanidine compositions vary by less 0.5 mol% anorthite, <3 mol% albite and <3 mol% orthoclase, though typically this does not exceed ~0.2, ~0.8 and ~1 mol%, respectively. In orthopyroxene, BSE brightness is controlled largely by changing Fe–Mg content. Ca can have an effect, but this can be neglected in this case (see Electronic Appendix 2 and Allan et al. 2013). All crystals were imaged completely and any zoning identified, before high-resolution images of the boundaries in the crystal were acquired to give improved spatial resolution across boundaries of interest. Image acquisition time was ~2 min, with a single integration.
CL imaging
Quartz zoning patterns were gathered by cathodoluminescence (CL) imaging, where the brightness of the image directly corresponds to Ti concentration (Wark et al. 2007; Matthews et al. 2012a, b). Initial lower-quality whole-crystal images were acquired at Stanford University using a JEOL LV 5600 Scanning Electron Microscope (SEM) so that a large number of crystals could be evaluated for their zoning patterns prior to detailed analysis. High-resolution CL images of Ti boundaries in quartz crystals were obtained on a FEI Quanta 650 FEG-SEM (field emission gun—scanning electron microscope) with a KE Centaurus panchromatic CL detector at the University of Leeds, with a dwell time of 100 µs per pixel.
Quantitative analyses
Quantitative major, minor and selected trace element analyses were undertaken on a JEOL JXA 8230 SuperProbe at Victoria University of Wellington. For all elements apart from Sr and Ba, an accelerating voltage of 15 kV, a beam current of 12 nA and a spot size of ~1 µm were used. Elements were analysed with a 30-s count on-peak. For Sr and Ba in sanidines, a 20-kV accelerating voltage and 40-nA beam current were used (following the method of Ginibre et al. 2002) with a defocused beam diameter of 5 µm. Spots were placed at a 7-µm spacing (between spot centres) so as to avoid significant convolution issues. Sr was analysed with a 240-s peak count, and background was measured within 2 mm of the peak location (unlike 5 mm for other elements) to remove the effect of Si. Ba was analysed with a 120-s count time on-peak.
Trace element analyses from the EPMA were compared with trace element concentrations measured using laser ablation inductively coupled plasma mass spectrometry (LA-ICPMS) at Victoria University of Wellington. In situ measurements were taken using a New Wave deep UV laser (193 nm solid state) coupled to an Agilent 7500cs ICPMS.
Modelling parameters
Greyscale profiles of Ba in sanidine, Fe–Mg in orthopyroxene and Ti in quartz were extracted from BSE or CL images using ImageJ® software. Sr profiles in sanidine were taken directly from EPMA measurements at 7-µm spacing. For all profiles, an initial condition of a step function was assumed. Sanidine and quartz modelling was carried out using a simple 1D model (e.g. Morgan et al. 2006). This assumption can hold due to the relatively small extent of diffusion relative to grain size, and follows the method of Morgan et al. (2006) and Morgan and Blake (2006). Diffusion anisotropy was considered small for Ti in quartz, and Ba and Sr in sanidine, consistent with results from previous studies (Cherniak and Watson 1992; Cherniak 2002, 2010; Cherniak et al. 2007). The modelling parameters used for each species are presented in Table 1. Given the high temperature dependence of elemental diffusion, the uncertainty on every timescale was calculated using ±30 °C in the temperature input, based on common uncertainties in Fe–Ti oxide thermometry (e.g. Hildreth and Wilson 2007), and that associated with the two-feldspar thermometry (see ‘Results’ section).
In order to model diffusion for Ti in quartz, Sr in sanidine and Ba in sanidine, the diffusivity of the element (D i ) being considered must be calculated. In many systems (including those listed above) where there is no compositional or oxygen fugacity dependence, these diffusivities are expressed in the form (see, e.g. Crank 1975; Zhang 2010):
where D i,0 is the D 0 for species i given in Table 1, E is the activation energy given in Table 1, R is the gas constant and T is temperature in Kelvins. Having calculated the appropriate values of D for each elemental system at the appropriate temperature, the following equation was solved for time (Crank 1975; Morgan et al. 2004):
where C is the normalised concentration of Ti, Sr or Ba, C 0 and C 1 refer to the initial amounts of the element on each side of an initial interface, D i is the calculated diffusivity in m2 s−1, t is the diffusion time and x is the position measured in metres along the profile and centred on the interface (the mid-point of the profile). The best-fit profiles were then determined by using a macro to minimise the difference between modelled profile shape and the input profile, yielding a timescale for the best-fit profile.
For modelling in orthopyroxene, Fe–Mg profiles were obtained along the crystallographic a- or b-axis, to be internally consistent and to avoid anisotropy effects. This is comparable with other Fe–Mg orthopyroxene interdiffusion chronometry where growth effects along the c-axis could be demonstrated to have occurred (e.g. Allan et al. 2013). Due to an expected fO2 dependence of D Fe–Mg in orthopyroxene (Ganguly and Tazzoli 1994), the formula of Ganguly and Tazzoli (1994) modified as in Allan et al. (2013) is used to calculate D Fe–Mg:
where X Fe is the molar proportion of the Fe end member (ferrosilite), T is temperature in Kelvins and fO2 is oxygen fugacity. In order to then model a timescale, finite difference methods were used, as detailed in Allan et al. (2013). We revisit the potential accuracy of this expression in the discussion.
In order to test that all profiles extracted were statistically significant, and to what degree variation on either end of the profile is significant, statistical analyses were conducted across every profile extracted from images. The uncertainty on the greyscale profile, being essentially random thermal noise in the BSE or CL detector, scales as \(\frac{1}{\sqrt n }\) where n is the number of pixels being averaged together, yielding a standard error (SE). If diffusion is the cause of the sigmoidal concentration profile, the plateaux at either end of the profile should be flat. Profiles which had variation in the plateaux that exceeded the calculated 2 SE were rejected.
Results
Two-feldspar thermometry
Where present, major element analyses of sanidine inclusions in plagioclase crystals (and vice versa) were used in combination with the Elkins and Grove (1990) calibration of the two-feldspar thermometer to iteratively calculate the temperatures at which these feldspars crystallised. We present 26 new temperature calculations from the early Bishop Tuff (Ig1Eb, Fig. 1) through to the latest Bishop Tuff (Ig2Nc, Fig. 1). The full results are presented in Table 2. All inclusions measured are within BSE-‘dark’ cores of crystals, showing that the temperature stratification of ~80 °C calculated by two-feldspar thermometry (Fig. 2) was present prior to any mixing with the contrasting magma composition that gave rise to the BSE-bright crystal rims. The ‘bright rim’ forming magma is inferred to have been of slightly higher temperature than the ‘normal’ Bishop Tuff magma due to its less-evolved melt composition (from increased Ba, Sr and Ti in overgrowth rims) and as indicated by increases in Fe–Ti oxide and oxygen-isotope model temperatures with inferred depth in the system (Hildreth 1979; Wallace et al. 1999; Bindeman and Valley 2002; Hildreth and Wilson 2007).

Temperature variations with stratigraphic unit of the Bishop Tuff from early (Ig1Eb) to late (Ig2N/Ig2NW). Results presented (diamonds) are from two-feldspar thermometry with an associated ±30 °C uncertainty. The grey fields represent the range in FeTi-oxide temperatures calculated by Hildreth (1977, 1979) and Hildreth and Wilson (2007). Stratigraphic units used are those of Fig. 1 and follow the nomenclature of Wilson and Hildreth (1997)
We then use these temperature results, in combination with previously published Fe–Ti oxide thermometry (Hildreth and Wilson 2007), to estimate temperatures at which diffusion occurred (Table 3). Samples from units Ig2Na yield cooler temperatures than those of the overlying Ig2Nb and Ig2Nc packages (Fig. 1; Table 2), and are thus modelled at 770 °C, representing a rounded average of the calculated model temperatures. Samples from Ig2Nb and Ig2Nc appear indistinguishable based on the feldspar thermometry and thus are modelled at 790 °C, representing again a rounded average of the results. Samples from Ig2NWa and Ig2NWb are modelled at 815 °C due to their higher model temperatures than Ig2N packages. Where zoning is present, crystals from the F9 sample are modelled using an average of 780 °C represented by the results from Ig2N. Some quartz crystals from Ig2SW preserved Ti zonation, and this diffusion was modelled at 753 °C, that is, an average of Ig2E and Ig2Na modelled temperatures. In all cases, a ±30 °C (1σ) uncertainty on thermometry is assumed, which is then propagated through to uncertainties on the diffusive timescales. The actual temperature controlling diffusion is, however, likely to have been slightly hotter than those modelled here from the cores of crystals given that the brighter rims grew from a less-evolved melt (Hildreth 1979; Hildreth and Wilson 2007; Chamberlain et al. 2014: see Table 3 for temperatures used).
Ti in quartz
Quartz from 16 samples from F1 (earliest erupted) to Ig2Nc (latest erupted: Fig. 1) were imaged, of which 11 samples showed bright-rim overgrowths on quartz crystals suitable for modelling (Fig. 3; Electronic Appendix 1). The 11 samples came from units Ig2Na, Ig2Nb, Ig2Nc, Ig2NWa, Ig2NWb, F9 as well as three boundaries from a sample of Ig2SW (Fig. 1). As previously shown, only quartz crystals from ‘late’ in the Bishop Tuff eruption sequence (and therefore from deeper in the magma chamber: Wallace et al. 1999) have high-Ti rims. ‘Early’ samples (which constitute approximately two-thirds of the eruptive volume, Hildreth and Wilson 2007) still retain some complex oscillatory zoning (Peppard et al. 2001; Wark et al. 2007; Gualda et al. 2012a), but this is not investigated further here.

CL images of representative quartz crystals from ignimbrite packages a Ig2NWb and b Ig2Nb. Areas across which greyscale profiles were extracted are highlighted as red boxes, and their corresponding profiles shown below the images. The modelled profile (age given next to the associated profiles) is shown as a black line, overlying the data (circles, connected by grey dashed line). The y-axis for both graphs is relative greyscale values, with 2σ uncertainty on greyscale value shown as the error bars. It is apparent that no matter which axis the greyscale profile is extracted from, timescales are comparable. White scale bars are 100 µm
One hundred and seventy-four timescales were modelled, with temperatures constrained using the results of the two-feldspar thermometry detailed above. Of these timescales, fifteen were rejected for not having a statistically significant contrast along the profile (see ‘Methods’ section). The boundaries between higher and lower Ti-bearing quartz range in width from ~5 to ~30 µm, yielding maximum timescales that range from as little as 15 days (+37/−8 days uncertainty) to 226 years (+556/−97 years uncertainty) with a mean value of 24 years, which is skewed by the dominance of shorter timescales. The best timescale resolution achievable from CL images was calculated by modelling the fictive apparent timescale across a known sharp contact, in this case, a crack within a quartz crystal. This yielded a ‘timescale’ of 0.56 years (at 815 °C), and therefore, any timescales shorter than this are effectively unresolvable via CL imaging and are discounted. The shortest resolvable timescale from our data is thus 1.0 years (+2.4/−0.45 years) (Fig. 4). All timescales are referred to as ‘maximum’ due to the assumption that the initial boundary between the horizontal concentration profiles was both sharp and oriented perpendicular to the sample surface (see Fig. 3 dotted lines), as no angular correction has been applied (Costa and Morgan 2010; Matthews et al. 2012a). Changing these assumptions would serve only to shorten the modelled timescales further.

Compiled cumulative frequency plot of modelled timescales from Ti diffusion in quartz (grey diamonds), Fe–Mg interdiffusion in orthopyroxene (yellow circles), Sr in sanidine (green crosses), and Ba in sanidine (blue triangles). The 1 SD uncertainty on each timescale modelled is shown as corresponding fields for each technique: yellow for Fe–Mg in orthopyroxene, grey for Ti in quartz, green for Sr in sanidine and blue for Ba in sanidine
There is no systematic difference in timescales obtained from individual samples or packages, although note that of the eighteen quartz crystals imaged from Ig2SW, only two showed a bright overgrowth. In all other late-erupted packages, the bright-rim development was far more common (see Electronic Appendix 3 for compiled, low-resolution images of representative samples; Electronic Appendix 4 for all modelled boundaries in quartz). This pattern is matched in Bishop zircons (Chamberlain et al. 2014) where samples from Ig2SW show only sparsely developed bright-rim overgrowths when compared with other late-erupted material from the northern and north-western vents.
Fe–Mg in orthopyroxene
There were twelve samples from which orthopyroxene could be extracted (from packages Ig2Nb, Ig2Nc, Ig2NWa and Ig2NWb; note that there was a lack of significant orthopyroxene in Ig2Na). Of these, nine produced zoned crystals suitable for modelling for diffusive timescales. The zoning textures in orthopyroxene are not as systematic as those in quartz, but can be split into three groups: unzoned (Fig. 5a), darker (i.e. Mg-richer) rims (Fig. 5b) and brighter (i.e. Fe-richer) rims (Fig. 5c). A direct correlation exists between BSE brightness and Fe–Mg content (e.g. Allan et al. 2013) and is demonstrated for the Bishop samples in Electronic Appendix 2. The variation in Mg content across the zones is relatively small, only up to 5.4 mol% enstatite, with the minimum resolvable change for diffusive modelling in this way being 0.5 mol% enstatite

BSE images of orthopyroxene showing the three observed zoning categories, with Mg# labelled in white. a Unzoned, b those with a dark rim (more Mg-rich rim), c those with a light (more Fe-rich rim). Areas across which greyscale profiles were extracted are shown by red boxes, with the corresponding profiles labelled with i, ii or iii. The profiles have been corrected to X Mg, and the modelled timescale is given next to the associated profile. Extracted profiles are shown as circles (connected with a grey line), and the modelled profiles are the black lines. 2σ uncertainty on greyscale value (corrected to X Mg) is shown as the error bars. White scale bar on all images is 100 µm
Thirty-six timescales were obtained from diffusive modelling within orthopyroxene crystals following the method of Allan et al. (2013), where different oxygen fugacities (+0.48 log units to +1.11 log units, relative to NNO) were used in combination with the range in temperatures (790 or 815 °C). Oxygen fugacity data were taken from Hildreth and Wilson (2007) and calculated relative to NNO using the formulation from Huebner and Sato (1970). The maximum resolution for orthopyroxene boundaries was calculated in the same way as for quartz crystals using a crystal edge as the known sharp boundary. The shortest time resolvable by this technique is equivalent to ~0.04 years, shorter than that from CL images of Ti in quartz due to the better spatial resolution of BSE imaging and the different values of D calculated for different diffusion regimes. Boundaries between lower Fe and higher Fe vary in full width from ~4 to ~30 µm, giving ranges in maximum timescales of 0.1 year (+0.1/−0.05 years) to 14 years (+13/−6.7 years: Fig. 4). The mean value of Fe–Mg interdiffusion timescales is 2.8 years, an order of magnitude shorter than that of Ti in quartz, and again shows a dominance of very short timescales (<4 years). Although different textural groups are observed, the timescales do not vary systematically with absolute rim compositions, relative changes in Mg content (i.e. lighter versus darker rims), or eruptive package. This observation implies that although the resulting rims have slightly different compositions, the process(es) causing the overgrowths occurred over similar timescales (Electronic Appendix 2).
Ba in sanidine
A total of 52 samples of sanidine crystals were imaged, spanning the entire eruptive sequence from F1 to Ig2Nc (Fig. 1). Of these, 15 samples showed sanidine crystals with a bright (higher Ba, Sr) overgrowth (Fig. 6). These crystals were found dominantly in samples from the Ig2N and Ig2NW packages (plus single samples from F9 and Ig2SW). As with quartz CL images (see above), the Ig2SW sanidines generally lack bright rims, with only two of twelve crystals having a bright overgrowth. All other early-erupted samples show no zonation (Fig. 6a). We cannot replicate the observations by Gualda (2007) and Pamukcu et al. (2012) that some feldspars from Ig2E have bright rims in our ten-sample suite from Ig2E pumices (representing a range in crystallinities from vxp to xr; Hildreth and Wilson 2007). The minimum resolvable timescale for Ba in sanidine was calculated using the same method as for orthopyroxene, yielding a value of ~30 years.

BSE images of sanidine crystals showing representative examples of a those that are unzoned and b those that have bright, Ba- and Sr-rich rims. Sr and Ba contents are labelled in white, from EPMA analyses. Areas over which greyscale profiles were extracted are shown as red boxes, which are labelled to correspond to the profiles shown below. All profiles are plotted as circles, connected with a grey dashed line; the modelled profiles are plotted as black curves. Ba profiles from b are shown in c with the y-axis being relative greyscale values; 2σ uncertainty on greyscale value plotted as error bars. The associated Sr profiles from EPMA are plotted in d, with 2 SD EPMA analytical uncertainties shown. Note the lack of a stable Sr plateau in (d, i), which means that this timescale is not included in our results (see text for discussion of profile classes and Fig. 8, below). White scale bars in images a and b are 100 µm
Fifty-four timescales were modelled from the statistically significant profiles (see ‘Methods’ section) between higher and lower Ba zones in sanidine crystals. The width of these boundaries varied from ~4 to ~40 µm, yielding maximum timescales that range from 44 years (+138/−33 years) to 150,000 years (+540,000/−114,000 years), although most are commonly on the order of 100 to 10,000 s of years (Fig. 4; Electronic Appendix 5 for all modelled timescales). The mean value of timescales for Ba diffusion in sanidine is 6,770 years, but if the two longest values are removed, this is reduced to 600 years. The median value of timescales for Ba diffusion in sanidine is 350 years. Even though these timescales are considered maxima, there remains substantial differences from the modelled timescales of Ti diffusion in quartz and Fe–Mg interdiffusion in orthopyroxene. Ba diffusion timescales in sanidine appear to be between one and three orders of magnitude longer than those from other two phases (Fig. 4).
Sr in sanidine
In order to assess whether the contrast in Ba in sanidine model ages is due to sanidine having a different crystallisation history to quartz and orthopyroxene, or reflects an issue related to the modelling of Ba behaviour, we undertook comparative modelling of Sr diffusion in sanidine. Strontium distribution in these sanidines is known to mimic that of Ba (Hildreth 1977, 1979; Lu 1991; Lu et al. 1992; Anderson et al. 2000; Hildreth and Wilson 2007). With the more rapid diffusion rates of Sr in alkali feldspar (Cherniak and Watson 1992), it would be expected that Sr profiles would be over a length scale that could be measured via EPMA or LA-ICPMS. Ten of the 15 examples of Ba-zoned sanidine were selected for investigation. Initial LA-ICPMS measurements were taken to confirm that the correlations of Sr and Ba in zoned sanidines noted by earlier authors (Lu 1991; Anderson et al. 2000) were widespread throughout the Bishop Tuff (Fig. 7: R 2 values of 0.87 for LA-ICPMS and 0.74 for EPMA). Given these positive correlations, a range of diffusion boundaries previously analysed for Ba were selected for Sr analysis. Due to the relatively narrow width of the diffusion boundaries (<50 µm), LA-ICPMS lacked the spatial resolution to definitively measure Sr changes across the boundary without serious convolution problems. Although the use of NanoSIMS (Saunders et al. 2014) would have been preferable in terms of spatial precision, the short time required to measure a Sr profile by EPMA (~1.5 h) meant that modelling of maximum timescales (reflecting the spatial resolution) across multiple boundaries was relatively straightforward. By using a 40-nA current and long (240 s) count time, our measurement uncertainties were improved to ±75 ppm (2 SD), with a detection limit of 150 ppm (4 SD), sufficient to define profiles in 19 grains.

Plot of Ba versus Sr from LA-ICPMS (dark circles) and EPMA (light diamonds) analyses of sanidine crystals from samples investigated in this study. Error crosses show 2 SD uncertainties: the dark grey cross corresponds to LA-ICPMS analyses, the larger light grey cross to EPMA analyses
Boundaries for Sr were located using the BSE images, as Sr and Ba were directly correlated (Figs. 6, 7). Although many boundaries were analysed, only seventeen yielded Sr profiles, which were deemed appropriate for diffusion modelling with acceptable definition, and lower degrees of analytical noise (e.g. Fig. 6d [i]). Only Sr profiles that were matched by changes in Ba concentration (from EPMA analyses) were used for modelling, to ensure that the change in Sr concentration was not an artefact of sub-optimal analytical procedures. The maximum width of these profiles varied between ~5 and ~50 µm, which yielded maximum timescales of 19 years (+65/−14 years) to 1,710 years (+5,790/−1,290 years: Fig. 4), with a mean timescale of 430 years. With analysis by EPMA and a defocused beam, these results will necessarily include some convolution, and so these timescales are effectively overestimates but at least provide upper limits for comparative purposes. However, given that the shortest resolvable time is 20 years (across a 2-point jump from plateau to plateau), the maximum convolution introduced will not be greater than 20 years for all modelled timescales.
Discussion
Temperature stratification within the Bishop magma body
Many estimates have been presented to magmatic temperatures of the Bishop Tuff. These thermometry calculations have utilised Fe–Ti oxides (Hildreth 1977, 1979; Hildreth and Wilson 2007; Evans and Bachmann 2013), Ti concentrations in quartz (Wark et al. 2007; Thomas et al. 2010; Thomas and Watson 2012; Wilson et al. 2012), Ti concentrations in zircon (Reid et al. 2011; Chamberlain et al. 2014), Δ18O fractionation between crystal phases (Bindeman and Valley 2002) and 2-pyroxene compositions (Hildreth 1979; Frost and Lindsley 1992). All of these approaches yield ~100 °C temperature difference between upper and lower regions in the magma chamber. In contrast, thermodynamic modelling of the Bishop Tuff system has been used to propose a very uniform temperature range (Gualda et al. 2012b). Calculations for thermometry can be somewhat limited as they often involve assumptions about the uniformity of pre-eruptive melt conditions (e.g. aTiO2 for Ti in quartz and Ti in zircon), or reveal only the most recent thermal stratification within the magma chamber (e.g. Fe–Ti oxides re-equilibrate rapidly and so cannot preserve evidence of any longer-term history: Buddington and Lindsley 1964; Hammond and Taylor 1982; Ghiorso and Sack 1991). By utilising two-feldspar thermometry, we avoid entering into ongoing debates over the use of Fe–Ti oxide and Ti in quartz thermometry (cf. Thomas et al. 2010; Wilson et al. 2012; Evans and Bachmann 2013; Ghiorso and Gualda 2013), although our conclusions are similar to those previously proposed regarding thermal gradients within the Bishop magma body (Bindeman and Valley 2002; Hildreth and Wilson 2007). Our two-feldspar thermometry does not make any assumptions other than that the two feldspars were in equilibrium with each other at the time of inclusion. The feldspar thermometer is not reset quickly due to the relatively slow diffusion timescales for Ca, Na and K within feldspar crystals and the need for coupled substitution of Al and Si for charge balance (Cherniak 2010). We therefore infer that our temperatures accurately record the magmatic temperatures at the point of crystallisation. All inclusion pairs analysed were within the cores of crystals and have therefore been unaffected by the late-stage mixing with the ‘bright-rim’ magma (Wark et al. 2007; Chamberlain et al. 2014). As such, we can thus investigate any temperature stratification within the Bishop Tuff magma body prior to the formation of the bright-rim overgrowths seen on quartz, sanidine and zircon.
Our results show that the ‘early’ units from Ig1Eb through to Ig2Eb (Fig. 1) have relatively uniform model temperatures of 740–760 °C (except for two analyses from Ig2Eb, Fig. 2; Table 2). This is up to 30 °C hotter than the reported Fe–Ti oxide and Δ18O (qtz-mt) temperatures of 714 °C (Bindeman and Valley 2002; Hildreth and Wilson 2007), but is more comparable (when the ±30 °C uncertainty is considered) with Ti-in-quartz analyses, which yield temperatures of 720–750 °C (Wark et al. 2007; Kularatne and Audétat 2014). Temperatures then start to increase in samples from Ig2Na, an ignimbrite package that appears to play a transitional role between the cooler, upper and hotter, lower regions of the magma chamber. We observe a general progression within the northern and north-western units to the hottest model temperature of 821 °C (Fig. 2; Table 2). The temperatures calculated for cores of late-erupted northern and north-western units are broadly similar to temperatures calculated from FeTi oxide, Δ18O (qtz-mt) and Ti-in-quartz thermometry (Hildreth 1977, 1979; Bindeman and Valley 2002; Hildreth and Wilson 2007; Wark et al. 2007), all of which are subject to late-stage re-equilibration due to mixing with the less-evolved ‘bright-rim’ magma (for example, in the case of Ti in quartz, the analyses are from within the bright, Ti rich rims). These similarities in model temperatures imply that the ‘bright-rim’ magma was not in fact significantly hotter than the pre-existing ‘normal’ Bishop magma. Our data thus support the notion that a general temperature and compositional stratification (Hildreth and Wilson 2007, and references therein) existed within the unitary magma chamber prior to the introduction of the ‘bright-rim’ magma.
In contrast, recent thermodynamic modelling (Gualda et al. 2012b) has led to the assertion that there was no significant thermal gradient through the Bishop Tuff magma chamber (and that in fact there existed two separate magma chambers: Gualda and Ghiorso 2013). This modelling suggests that the ‘late’ Bishop Tuff magma was held at ~760 °C, and that there was a <30 °C gradient across the ‘late’ north and north-western units. We do not find evidence of crystallisation at 760 °C for any of our ‘late’ ternary feldspar pairs, apart from those in Ig2Na (which fills a transitional role with respect to model temperatures), and in turn see evidence for ~80 °C variation across the erupted Bishop compositions.
Uncertainties in diffusion chronometry
The single most important factor in calculating the uncertainties associated with diffusion chronometry is temperature, which has an exponential effect (see Eqs. 1, 2) on the timescales modelled via the Arrhenius relationship in Eq. 1 (e.g. Morgan et al. 2006; Allan et al. 2013). Given uncertainties of ±30 °C in our thermometry estimates, this leads to an asymmetric uncertainty which, for the longer (+) timescale uncertainty, can be numerically more than double the calculated timescale (from modelling at the low temperature from uncertainties, see Electronic Appendix 7 for all timescales and relative uncertainties). Uncertainties in D 0 and fO2 are second order, but still play a considerable role. Uncertainty in fO2 is only considered in relation to Fe–Mg interdiffusion modelling, and a value of ±0.3 log units is assumed (following Allan et al. 2013). In the case of Ti in quartz, Ba in sanidine and Sr in sanidine, we calculate uncertainties of ±0.06, 0.12 and 0.03 log units (for Ti, Ba and Sr, respectively) for D 0 and E (Electronic Appendix 7). For further discussion on the effects of T and fO2 variation on orthopyroxene Fe–Mg interdiffusion modelling, see Allan et al. (2013).
Timescales in this paper are referred to as maximum timescales due to the spatial limitations of constraining the maximum profile width of the zone across which diffusion has occurred. Due to either pixel size (a more limited effect) or spot size and spacing (for Sr), the maximum distance across which diffusion has occurred is always likely to be slightly overestimated. Similarly, by assuming an initial step-change concentration profile, the full width of the profile is assumed to be caused by diffusion, yielding the maximum timescale (see Eq. 2). These assumptions will be discussed later in relation to sanidine diffusion modelling.
Trends in diffusion profiles
Three distinct categories of profile shape are evident on visual inspection of all modelled profiles from all four modelled systems (Fig. 8). They can be defined as class 1: ideal diffusion shape, good sigmoid and close fit to modelled profile shape; class 2: profiles with a spike on the side of one boundary, which sets an initially high/low condition on half of the profile; class 3: slightly asymmetric profiles, where one side of the measured profile deviates away from the modelled profile.

The three classes of diffusion profiles demonstrated in Ba profiles in sanidine (blue), Ti profiles in quartz (grey), Mg# number profiles in orthopyroxene (yellow) and Sr profiles in sanidine (green) (see text for details). All x-axes are distance (in µm), y-axes are concentrations (absolute or inferred) of the element in question. Class 1 is the ideal diffusion profile, being symmetrical and fitting the model (black line) well. Class 2 profiles reveal a false high/low on one side of the profile, where the ‘plateau’ is not horizontal as assumed in the modelling (black line). Class 3 shows the asymmetric boundaries that deviate away from the model (black line) within the concentration gradient. This relict of growth zoning would cause timescales to be overestimated
We interpret that the class 2 and class 3 profiles generally represent some relict growth zoning, which has been overprinted by diffusion and are not ideal for modelling. By their shape, they indicate that our initial assumption of the starting profile shape was not correct, and the profile must have started with a width somewhat greater than zero, i.e. less sharp than the modelled step change. Whilst this is problematic in terms of absolute timescale determination, the true timescale of such a profile cannot exceed the timescale from a simple step profile diffused to the same width. Again, therefore, our choice of model starting condition leads us to an overestimate in timescale, and we can confidently regard the class 2 and class 3 profiles as maxima in the light of this knowledge. Whilst class 2 profiles seem to be quite reproducible in terms of their end results, giving answers close to the more ideal class 1 profiles, we are less confident about class 3, and there is no a priori method to determine how much growth versus diffusion these represent. Whilst they do represent a maximum time constraint, we regard this as very loose, and so the class 3 profiles have been excluded from our accumulated data (but can be found in Electronic Appendices 5 and 7 due to the significant proportion of sanidine crystals that have these class 3 profiles).
The recognition of all three boundary types in Sr, Ba, Ti and Fe–Mg profiles raises the question as to what processes have caused the preservation of the class 2 and class 3 profiles, and may yield insights into the precursory processes occurring in the Bishop Tuff magma chamber. Class 2 profiles, with their distinctive peak prior to the diffusion sigmoid, could represent evidence of relict growth zoning due to slow diffusion of cations to the crystal–melt interface (Magaritz and Hofmann 1978) or due to depletion in Ba/Ti/Sr/Fe/Mg in the boundary layer of melt at the crystal face. They are typically most extreme in Ba profiles, which would suggest the involvement of zone-refining and boundary depletion effects due to the extreme partition coefficient of Ba in sanidine (~28.4: Lu 1991). Class 3 profiles raise more challenging issues but, again, are most evident in Ba profiles from sanidine, and thus are likely to preserved evidence of an initial profile that had some component of growth zoning, possibly even caused by Ba retention during dissolution of the core prior to rim growth. Although these three classes of profiles have significantly different appearances, there is no systematic difference in the range of timescales modelled from each mineral–element pair. The identification of all three boundary types in all phases investigated here strongly suggests that the zoning reflects common origins, and that the timescales modelled should be roughly comparable across all phases.
Contrasts in modelled timescales between crystal phases
Modelling diffusion in sanidine
It is apparent that diffusion modelling of both Ba and Sr in sanidine yields much longer timescales, by up to 3 orders of magnitude, than modelling of Ti in quartz and Fe–Mg in orthopyroxene. This result matches other studies, for example from profiles obtained from NanoSIMS analyses (Till et al. 2012). One interpretation could be that timescales from modelling of Ba and Sr diffusion in sanidine are overestimates (Figs. 4, 6). Based on the consideration of the applicable diffusivities, the width of Sr boundaries should be ~10 times wider than those of Ba (Cherniak 2002), and had these elements been diffusing for similar amounts of time (Table 1). This is not always the case (Fig. 6; Electronic Appendices 5 and 6) and the widths of the boundaries can be similar. Given the variable nature of the relationship between Ba and Sr profile widths, and the longer timescales from Ba and Sr in sanidine diffusion modelling, there are several possible factors that could cause this discrepancy.
-
1.
Ba and Sr are not always being affected in the same way, sometimes not being correlated during crystal growth across the boundary between darker core and brighter rim, and their behaviour may not be related to the change in Ti content in quartz crystals.
-
2.
The very high activation energy and D 0 of Ba and Sr diffusion in sanidine (Table 1) is sensitive to small thermal perturbations (although variably sensitive, due to differences in their diffusion coefficients: Table 1). Short periods of time at elevated temperatures could thus allow Ba and Sr to diffuse rapidly, making their timescales appear unusually long.
-
3.
The initial Ba and Sr profiles were not step-like as assumed, and modelling requires a different initial condition with some gradient across the boundary as an initial state from which to model.
All three of these factors could contribute towards yielding longer timescales, but in practice we can rule out some of these possibilities. If Ba and Sr were unrelated in their behaviour, then the overall trends seen in graphs of Ba versus Sr in ICPMS and EPMA data would not be so clear (Fig. 7). Glass trace element data show coupled increases in Ti, Ba and Sr (Hildreth and Wilson 2007; KJ Chamberlain unpub. data) implying that the high-Ti rims on quartz are related to the high Ba and Sr rims on sanidine. These lines of evidence suggest that option 1 is unlikely.
The second option that the high activation energy of Ba and Sr could allow the diffusion behaviours to decorrelate is also demonstrably unlikely. The high-resolution EPMA data show that the steps are correlated in space, occurring across the same regions. The difference in timescales between sanidine, quartz and orthopyroxene is more reflective of differences in diffusion speed than discrepancies in profile width. For Ba diffusion to overtake Ti diffusion would require temperatures around 1,100 °C, which is wholly implausible for a rhyolitic magma in which plagioclase–sanidine pairs survived (and lacking any other mineral textural evidence for such high temperatures).
Ti-in-quartz profiles are comparable in width to Ba in sanidine profiles, again showing that these Ba profiles are artificially wide (Ti diffuses faster in quartz than Ba in sanidine at these conditions: Cherniak 2002; Cherniak et al. 2007). Given the wide profile of Ba in sanidine, it would thus seem likely that the initial assumption of a sharp step function is incorrect for the case of Ba and Sr diffusion in sanidine. The timescales modelled are therefore a combination of Ba and Sr diffusion, superposed on initially curved Ba and Sr starting profiles. This raises questions as to how initially curved Ba and Sr profiles can be generated in sanidine, but not in other phases, with the effect much more noticeable in Ba concentration profiles than those of Sr.
The highly compatible nature of Ba in Bishop Tuff sanidine (K d ~22: KJ Chamberlain unpub data; ~28.4: Lu 1991) means that any partial dissolution of sanidine will lead to an interfacial layer of melt that is enriched in Ba. This interfacial layer will have Ba concentrations similar to those in the crystal, rather than the lower levels found in the ambient melt, and as a consequence, the sanidine crystal cannot be in equilibrium. In response to this situation, the remaining sanidine crystal will, by diffusion, scavenge the Ba from the melt interface, leading to an enriched zone in the sanidine crystal prior to growth of the Ba-rich bright rim on sanidine, that is, a zone-refining process. Sr, with a K d of ~9 (KJ Chamberlain, unpub data) and ~11.4 (Lu 1991) will be similarly affected. The more rapid diffusion of Sr in sanidine means, however, that this initially curved Sr profile will be broader and less well preserved than for Ba by the time of eruption. The curvature of the initial starting profile will also have less effect on timescales from diffusion modelling of Sr, as the initial curvature represents less equivalent diffusion time for Sr than for Ba. From this inference, we conclude that both Ba and Sr in sanidine will yield overestimates of time for diffusion, but that for Ba the timescales recovered will be dominated by the growth curvature, whilst the Sr profile would contain appreciable components of both diffusion and growth. This conclusion agrees with the trends seen in timescales from Ba and Sr diffusion compared with those timescales from Fe–Mg interdiffusion in orthopyroxene and Ti in quartz (Fig. 4). Evidence for dissolution of quartz is preserved in CL imagery, where interior zones are truncated against the bright-rim overgrowth (Electronic Appendix 3; Peppard et al. 2001; Wark et al. 2007). This texture is replicated in sanidine BSE images, which commonly reveal rounded corners on grains, inferring a similar resorptive process.
In systems where partial dissolution of sanidine could have occurred prior to rim overgrowth, and until its effects on the initial profile of Ba (and to a lesser extent Sr) can be constrained, Ba and Sr geospeedometry in low-temperature rhyolitic systems (i.e. <900 °C: Till et al. 2012) yields discrepancies at ~800–840 °C and should be treated with caution. It is necessary to bear in mind that a step change in composition is a model assumption, and that at one extreme, diffusion and growth are comparable processes at sub-micron lengthscales, placing a limit on how sharp a profile junction can be. This limit in turn controls how far spatial resolution can reasonably be pushed before the assumptions of diffusion modelling have to be considered for their reliability.
Removing the ‘growth’ effect
In order to derive a viable timescale from Ba and Sr diffusion in Bishop sanidines, a model to remove the effect of growth on compositional profiles was developed. The profile widths of both Ba and Sr in sanidine are interpreted to comprise both a true diffusion component (active for the same times and temperatures whether Ba or Sr is considered) and a ‘growth’ component due to partial dissolution prior to overgrowth of the bright rim. Given that for individual profiles of Ba and Sr across the same boundary within the same crystal, the modelled timescale due to diffusion alone should be the same, and thus by exploiting differential diffusion speeds, the combined modelling of both Ba and Sr diffusion simultaneously should allow for the removal of the variable growth component. Twelve paired profiles of Ba and Sr were used to test this model, with all the modelled timescales being significantly reduced when compared to the timescales for either Ba or Sr alone (Table 4). Modelled timescales range from 24 to 1,540 years, with ten of the twelve modelled boundaries yielding timescales of <500 years. These values are within an order of magnitude of those from Ti-in-quartz modelling (Figs. 9, 10). Prior to the combined modelling, the average model timescales in these crystals were 1,500 (Ba) and 540 years (Sr), ~63 and ~23 times longer (respectively) than the average Ti-in-quartz model timescales, respectively. The average from combined modelling is now 440 years (~19 times longer than the Ti-in-quartz average), but with the two longest timescales removed this is shortened to only 230 years (~10 times longer than the Ti-in-quartz average).

Cumulative frequency plot comparing the timescales modelled from Ti diffusion in quartz (diamonds), and the combined modelling of Ba and Sr in sanidine (red stars). The 1 SD error window is shown as grey (Ti in quartz) or red (Ba and Sr in sanidine) fields. The original timescales from Sr diffusion in sanidine (crosses) and Ba diffusion in sanidine (triangles) from the crystals used in the combined modelling are also plotted to show the significant reduction in Ba timescales once combined modelling is used

a Histogram of all combined diffusion timescales including those from Fe–Mg interdiffusion in orthopyroxene, Ti diffusion in quartz, and combined Ba–Sr diffusion in sanidine with b cumulative frequency chart showing the timescales and their uncertainties for each method. All uncertainty bars are ±1 SD
Given the very high activation energy of both Ba and Sr diffusion in sanidine when compared with Ti diffusion in quartz, small temperature changes from those at which diffusion is modelled could allow for closer agreement between the combined Ba and Sr timescales and those modelled from Ti in quartz. As temperature is increased, the Ba and Sr in sanidine combined timescales will decrease faster than, and thus approach those of the Ti diffusion in quartz data. It is not, however, possible to completely close the gap with reasonable temperature estimates. The ratio in the timescales falls to a factor of 6 at ~827 °C, an uppermost temperature limit taken from the work of Hildreth and Wilson (2007) and one that would likely not represent the entire magma body.
In order to properly constrain the role of growth zoning, the thermal effect on diffusion in sanidine, and to properly quantify how closely sanidine and quartz timescales agree, further investigation using techniques such as NanoSIMS is needed.
Comparisons of modelled timescales between crystal phases
Using the current calibration of the Fe–Mg interdiffusion coefficient within orthopyroxene, diffusion timescales for the Bishop Tuff are extremely short (<20 years: Fig. 4). Given the comparable profile groups observed in orthopyroxene, quartz and sanidine (Fig. 8), and the phase stability relationships of Naney (1983), it seems valid to infer that the orthopyroxene timescales should be comparable with those from sanidine and quartz. In order for this to be the case, the value of the diffusion coefficients for Fe–Mg interdiffusion would have to be overestimated by about an order of magnitude (Fig. 10). This possibility is not surprising in the light of the large range in orthopyroxene diffusion coefficients and calibrations used in the literature (Ganguly and Tazzoli 1994; Schwandt et al. 1998). Using the Ganguly and Tazzoli (1994) calibrations, D values used here are ~1 × 10−19 to ~1 × 10−20 m2 s−1. In contrast, using the calibration of Schwandt et al. (1998), D values at the relevant temperatures used here range from ~1 × 10−22 to ~6 × 10−22 m2 s−1, two to three orders of magnitude smaller. Use of the latter figures would result in timescales that are two to three orders of magnitude longer than that calculated in this paper, i.e. up to ~14,000 years, and which are comparable with the uncorrected Ba in sanidine diffusion timescales we derive.
In order to bring the orthopyroxene timescales into alignment with those obtained from Ti diffusion in quartz, the value of D would need to be approximately one-fifth of that currently used: ~1–2 × 10−20 m2 s−1 depending on the model temperature (790 or 815 °C). Following discussion with those conducting experimental research, preliminary findings suggest that the Ganguly and Tazzoli (1994) formulation with added oxygen dependence we have used may well yield an overestimate of diffusion speed (S Chakraborty and R Dohmen pers comm). Our work suggests that regarding orthopyroxene, the quantification of D 0 and E for Fe–Mg interdiffusion in rhyolitic systems warrants further investigation, as it has significant impacts on the compatibility of timescales modelled from different phases in the Bishop Tuff.
Pyroxenes and their textural implications
The diversity of textures observed in orthopyroxene (normal-zoned, bright-rim grains, to reversely zoned, dark-rim grains, to those which are unzoned) is not matched by a diversity in timescales. In addition, the rim compositions do not converge towards a single value, unlike that seen in the Oruanui example (Allan et al. 2013). These features raise the issue as to how the process that caused the growth of Fe-richer rims was occurring over the same timescales as that which caused growth of Mg-richer rims. Orthopyroxene with contrasting rim compositions revealing similar timescales has been previously noted at Mt St. Helens (Saunders et al. 2012) where compositional differences were larger than those recorded here (8–15 mol% enstatite variation from cores to rims compared with 0.5–5.4 mol% enstatite variation in the Bishop orthopyroxenes: Electronic Appendix 2). Saunders et al. (2012) cited intrusion of a more mafic component to cause growth of both reversely zoned and normally zoned orthopyroxene, due to changes not only in melt composition, but also volatile contents and fO2. For the Bishop Tuff, there appears to be multiple options for the formation of both Fe-rich and Mg-rich rims (Fig. 11), as follows.

Cartoons illustrating three possible model options (not to scale) for the origin of orthopyroxene and its associated zoning in the Bishop Tuff. Only a segment of the lower part of the magma chamber is depicted (in green), which is intruded by the ‘bright rim’ forming magma (white) from remelting of the underlying crystal mush (grey) by a more mafic magma (red). In all three models, the end result is variably zoned orthopyroxene crystals, but the possible origins for these crystals are changeable. Note that in options 2 and 3, there must be limited convection within the melt-dominant magma body (consistent with the views of Hildreth 1979 and Hildreth and Wilson 2007) in order to preserve small-scale heterogeneities within the melt-dominant magma body prior to late-stage mingling upon eruption
Option 1. Mg-richer cores grew at depth within the Bishop magma reservoir, possibly within a crystal-rich mush zone (e.g. Hildreth and Wilson 2007) and consequently are slightly more enstatite-rich. Concurrently, a Fe-richer population of crystals was growing within the shallower melt-dominant magma chamber from the slightly more-evolved melt (Fig. 11). When the ‘bright-rim’ magma intruded the Bishop melt-dominant magma body, it either originated in, or entrained crystals from, the underlying mush zone, bringing the Mg-richer cores into the melt-dominant body. Chaotic, piecemeal mixing within the lower parts of this body (Figs. 4, 11) with this ‘bright-rim’ magma could produce rims of marginally higher or lower Mg content to crystallise simultaneously, depending on the sense of entrainment from Mg-richer to Mg-poorer melt, or vice versa.
Option 2. The ‘bright-rim’ magma was not compositionally different in Fe or Mg concentrations, but had higher fO2 values and CO2 contents (Wallace et al. 1999). In this scenario, there would be interplay between induced rapid crystallisation of Fe-rich orthopyroxene rims from melts with higher CO2 (Fig. 11) and the increased fO2 resulting in crystallisation of more Mg-rich rims (Blundy et al. 2010; Saunders et al. 2012). The interplay between these two factors and the degree of mixing would produce magma, which has orthopyroxene with both Fe- and Mg-rich rims.
Option 3. The orthopyroxene crystallised as a result of mixing between the dominant ‘normal’ Bishop Tuff magma and the ‘bright-rim’ magma (Fig. 11). This would, however, appear to be a less likely explanation, as this would require extremely rapid crystallisation of orthopyroxene (and associated clinopyroxene). Although the timescales for Fe–Mg interdiffusion in orthopyroxene are relatively short (using the Ganguly and Tazzoli 1994 calibration), current investigation suggests that the inaccuracies in constraining the diffusion coefficients could be the cause of these apparently short timescales (S Chakraborty pers comm). In order to distinguish between options 1 and 2, more detailed investigation into the composition (major and trace elements) of orthopyroxene and other phases in the Bishop Tuff is required.
A major question has been raised in previous studies over the occurrence of pyroxene (both ortho- and clino-) in the Bishop Tuff, with some suggesting that the pyroxenes are not in equilibrium with other crystal phases (e.g. Ghiorso and Sack 1991; Frost and Lindsley 1992; Evans and Bachmann 2013). The euhedral appearance of orthopyroxene (and clinopyroxene) crystals (Fig. 5), the similar compositions of inclusions within the orthopyroxenes when compared with free crystals (Hildreth 1977, 1979) and phase stability relations (Naney 1983) based on the two-feldspar thermometry presented here counts against them being ‘foreign’. In addition, the similarity in groups of diffusion profiles (Fig. 8) from all three crystal phases modelled here could be used to suggest that Bishop pyroxene, sanidine and quartz crystals experienced similar controls on growth and diffusion.
Rejuvenation of the Bishop magma body
Many explanations have been put forward for the bright-rim overgrowths on quartz and sanidine. Initially, it was proposed that these rims grew from sinking of the crystals into less-evolved, Ba-, Sr- and Ti-richer magma within the magma chamber (Anderson et al. 2000; Peppard et al. 2001). Studies on melt inclusion entrapment pressures and compositions have however shown that core and rim inclusions were crystallised at comparable pressures, but within varying melt compositions (Wallace et al. 1999; Roberge et al. 2013). These observations imply that introduction of a melt with a slightly different composition underlies the growth of bright rims on quartz and sanidine (Wark et al. 2007; Roberge et al. 2013). The short timescales presented here support this hypothesis of shortly pre-eruptive rejuvenation of lower parts of the Bishop magma chamber by interaction with a melt enriched in Ti, Ba, Sr and CO2 (Wallace et al. 1999; Hildreth and Wilson 2007; Wark et al. 2007; Roberge et al. 2013).
Our timescales from quartz, sanidine and orthopyroxene diffusion profiles do not provide any evidence for long-lived (>1,000 years) interactions between the ‘bright-rim’ magma and the resident melt-dominant body. However, zircons from the same samples or eruptive units as those studied in this paper have parallel records of the presence or absence of bright-rim overgrowths, plus sparse numbers of grains wholly crystallised from the ‘bright-rim’ magma (Chamberlain et al. 2014). Age data from sectors of the crystal cores just inside the bright rims suggest that growth of these rims could have commenced up to ~10 kyr before eruption. It is plausible that the longer history of zircon interaction with the ‘bright-rim’ magma may be tracing crystal growth within the ‘bright-rim’ magma prior to its later interaction with the main Bishop magma body, and thus provides a maximum time constraint for the interaction of the ‘bright-rim’ magma in the Bishop magma system. In addition, the contrasting timescales recorded of interaction between the zircon, sanidine, quartz and orthopyroxene and the ‘bright-rim’ magma may reflect a more gradual sequence of events than any immediately pre-eruptive triggering event.
In the sanidine, quartz and orthopyroxene records, intrusion of the ‘bright-rim’ magma into the main Bishop melt-dominant magma body seems to have consisted of multiple events (at least three, from preserved crystal zonation: Electronic Appendices 2–6) which began ~500 years prior to eruption, as shown by the fairly continuous distribution of timescales that can be calculated from sequentially higher Ba/Sr/Ti profiles within sanidine and quartz crystals (e.g. Fig. 4; Electronic Appendices 4 and 5). The modelled timescales appear to cluster towards eruption age (Figs. 4, 10), with 50 % of the timescales being <18 years (Ti in quartz), <250 years (combined Ba and Sr in sanidine) and <2.5 years (Fe–Mg in orthopyroxene). Although the three suites of model ages have varying absolute values, their cumulative frequency distributions and textural boundary features are closely comparable.
The apparent clustering of timescales closer to eruption could be due to exponential increases in the volume and frequency of magmatic intrusion into the lower magma chamber immediately prior to eruption, which would favour the concept of mixing as an eruptive trigger (Wark et al. 2007). Another possibility, however, is that although rates of ingress of the external ‘bright-rim’ magma stayed relatively constant, magmatic overturn and increased thermal contrasts between the less-evolved magma and the ‘normal’ Bishop magma caused increases in mixing within the lower regions of the magma chamber. This would then expose increasing numbers of crystals to a less-evolved (and slightly hotter) melt than that which they initially resided in, generating the shorter timescales and apparent increases in mixing intensity, even if there was not an increasing rate of intrusion into the magma body. Whatever the cause, it is still apparent that rejuvenation (recorded from diffusion modelling in the major mineral phases) and mixing with a melt that was marginally hotter, but enriched in Ti, Sr and Ba, occurred over a short timescale (<500 years) and continued to cause compositional variation within the Bishop magma chamber until quenched by eruption.
Implications and conclusions
We show that timescales calculated from simplistic diffusion modelling of Sr and Ba in sanidine are 1–3 orders of magnitude longer than Ti diffusion in quartz, which are in turn an order of magnitude longer than timescale from Fe–Mg interdiffusion in orthopyroxene. There are no a priori reasons as such for supposing which of the three systems yields the ‘correct’ result. Recognition of the role of growth zoning in sanidine highlights an incorrect initial assumption of an initially sharp step in Ba and Sr contents in this mineral. We have used combined Ba and Sr diffusion modelling to remove this growth effect, and the resulting timescales are shortened to become more comparable (to within roughly an order of magnitude) of those modelled from Ti diffusion in quartz. Given the similar nature of diffusion profiles extracted from orthopyroxene images to those from quartz and sanidine images, we suggest that modelling of Fe–Mg interdiffusion in orthopyroxene may produce artificially short timescales. In order for orthopyroxene timescales to align with those in modelled from Ti diffusion in quartz, the calculated values of D would have to be approximately an order of magnitude smaller than those using the Ganguly and Tazzoli (1994) calibration with fO2 dependence at T = ~800 °C, but 1–2 orders of magnitude larger than that of the Schwandt et al. (1998)’s calibration. Adoption of the latter values would then align the orthopyroxene timescales more closely with those derived from the growth-corrected profiles for Ba and Sr in sanidine. These gross disparities highlight the very pressing need in particular for the calibration of Fe–Mg interdiffusion at temperatures and pressures representative of evolved (particularly rhyolitic) compositions, in order to extract viable and consistent timescales.
Two-feldspar thermometry shows that a temperature stratification of ~80 °C existed between the upper and lower reaches of the Bishop Tuff magma chamber, and that this stratification was not simply a result of the late-stage intrusion at the base of the magma chamber. This temperature range is similar to that suggested by Fe–Ti oxide and oxygen-isotopic thermometry, and supports the model of Hildreth and Wilson (2007) for a unitary zoned magma chamber for the Bishop Tuff. An important implication arising from the similarity of the temperature estimates is that the intruding ‘bright-rim’ magma was not significantly hotter than the basal parts of the Bishop magma chamber. Instead, it would appear that the intruding magma was enriched in Ba, Ti, Sr and CO2 (e.g. Wallace et al. 1999; Wark et al. 2007), which has affected crystallisation conditions to cause growth of the bright rims on orthopyroxene, quartz and sanidine (and zircon: Chamberlain et al. 2014).
The similarity of diffusion profile shapes between quartz, orthopyroxene and sanidine and the order of magnitude agreement between sanidine and quartz geospeedometry indicates that for at least ~500 years prior to eruption, these phases shared a common history and experienced magmatic intrusion causing the overgrowth of compositionally contrasting rims. It remains an open question as to whether the three major phases are actually recording events with closely similar timings, and that the diversity in model-age ranges reflects systematic errors in the diffusion parameters available at present. An important conclusion of our work is that the application of diffusion modelling over multiple co-erupted mineral species is restricted by uncertainties over values for diffusion coefficients in such evolved, relatively low-temperature magmatic systems. The alternative end-member inference to explain the diverse zoning patterns and contrasting diffusive timescales is that the crystals are recording different timescales because gradational interaction with the ‘bright-rim’ magma affected different mineral species now found together within single pumices over different timescales. The great ranges in proportions of zircon crystals with bright rims, and the diversity of development of zircon overgrowths from the ‘bright-rim’ magma (Chamberlain et al. 2014), support this alternative inference to some extent. These observations would imply that the zircons found within single pumices are mixtures that did not all grow in situ from the host melt represented in that pumice (cf. Hildreth 1979), and such inferences may also apply to the major mineral phases, at least in the lower portion of the magma chamber which is pyroxene-bearing and has other crystal species with bright rims.
We show from textural proportions that rejuvenation of the Bishop magma chamber had noticeable effects on mineral phases only found in the northern and north-western ignimbrite packages, inferred to represent the deeper, less-evolved parts of the magma body. Rejuvenation by the ‘bright-rim’ magma was not a single event and, from the timescales measured here in the common mineral phases, occurred over a period of ~500 years prior to eruption. The longer timescale for the interaction of the ‘bright-rim’ magma (up to ~10 ky) indicated by zircon age spectra from cores to bright-rimmed crystals would imply that the intruding magma was in itself probably not the trigger for the eruption. Instead, its rise into and interaction with the Bishop magma chamber may have reflected other processes, such as changes in the regional stress conditions (cf. Allan et al. 2013) or disturbance of the mush zone by extraction of large volumes of melt. As such, the ‘bright-rim’ magma could be regarded as the marker for processes leading to eruption, rather than the trigger. Our study has important implications not only for models of the Bishop Tuff magma chamber, but also raises significant issues regarding the application of systematic diffusion chronometry to other rhyolitic systems.
References
Allan ASR, Wilson CJN, Millet M-A, Wysoczanski RJ (2012) The invisible hand: tectonic triggering and modulation of a rhyolitic supereruption. Geology 40:563–566
Allan ASR, Morgan DJ, Wilson CJN, Millet M-A (2013) From mush to eruption in centuries: assembly of the super-sized Oruanui magma body. Contrib Mineral Petrol 166:143–164
Anderson AT, Davis AM, Lu F (2000) Evolution of Bishop Tuff rhyolitic magma based on melt and magnetite inclusions and zoned phenocrysts. J Petrol 41:449–473
Arienzo I, Heumann A, Wörner G, Civetta L, Orsi G (2011) Processes and timescales of magma evolution prior to the Campanian Ignimbrite eruption (Campi Flegrei, Italy). Earth Planet Sci Lett 306:217–228
Bailey RA, Dalrymple GB, Lanphere MA (1976) Volcanism, structure, and geochronology of Long Valley caldera, Mono County, California. J Geophys Res 81:725–744
Bindeman IN, Valley JW (2002) Oxygen isotope study of the Long Valley magmatic system, California: isotope thermometry and convection in large silicic magma bodies. Contrib Mineral Petrol 144:185–205
Blundy J, Cashman KV, Rust A, Witham F (2010) A case for CO2-rich arc magmas. Earth Planet Sci Lett 290:289–301
Buddington AF, Lindsley DH (1964) Iron-titanium oxide minerals and synthetic equivalents. J Petrol 5:310–357
Caricchi L, Annen C, Blundy J, Simpson G, Pinel V (2014) Frequency and magnitude of volcanic eruptions controlled by magma injection and buoyancy. Nat Geosci 7:126–130
Chamberlain KJ, Wilson CJN, Wooden JL, Charlier BLA, Ireland TR (2014) New perspectives on the Bishop Tuff from zircon textures, ages and trace elements. J Petrol 55:395–426
Cherniak DJ (1996) Strontium diffusion in sanidine and albite, and general comments on Sr diffusion in alkali feldspars. Geochim Cosmochim Acta 60:5037–5043
Cherniak DJ (2002) Ba diffusion in feldspar. Geochim Cosmochim Acta 66:1641–1650
Cherniak DJ (2010) Cation diffusion in feldspars. Rev Mineral Geochem 72:691–733
Cherniak DJ, Watson EB (1992) A study of strontium diffusion in K-feldspar, Na-K feldspar and anorthite using Rutherford backscattering spectroscopy. Earth Planet Sci Lett 113:411–425
Cherniak DJ, Watson EB, Wark DA (2007) Ti diffusion in quartz. Chem Geol 236:65–74
Christensen JN, Halliday AN (1996) Rb-Sr ages and Nd isotopic compositions of melt inclusions from the Bishop Tuff and the generation of silicic magma. Earth Planet Sci Lett 144:547–561
Costa F, Dungan M (2005) Short time scales of magmatic assimilation from diffusion modeling of multiple elements in olivine. Geology 33:837–840
Costa F, Morgan D (2010) Time constraints from chemical equilibration in magmatic crystals. In: Dosseto A, Turner SP, Van Orman JA (eds) Timescales of magmatic processes: from core to atmosphere. Wiley, Chichester, pp 125–159
Costa F, Chakraborty S, Dohmen R (2003) Diffusion coupling between trace and major elements and a model for calculation of magma residence times using plagioclase. Geochim Cosmochim Acta 67:2189–2200
Crank J (1975) The mathematics of diffusion, 2nd edn. Oxford University Press, Oxford, 414 pp
Davies GR, Halliday AN (1998) Development of the Long Valley rhyolitic magma system: strontium and neodymium isotope evidence from glasses and individual phenocrysts. Geochim Cosmochim Acta 62:3561–3574
Dunbar NW, Hervig RL (1992) Petrogenesis and volatile stratigraphy of the Bishop Tuff: evidence from melt inclusion analysis. J Geophys Res 97:15129–15150
Elkins LT, Grove TL (1990) Ternary feldspar experiments and thermodynamic models. Am Mineral 75:544–559
Evans BW, Bachmann O (2013) Implications of equilibrium and disequilibrium among crystal phases in the Bishop Tuff. Am Mineral 98:271–274
Fowler SJ, Spera FJ (2010) A metamodel for crustal magmatism: phase equilibria of giant ignimbrites. J Petrol 51:1783–1830
Frost BR, Lindsley DH (1992) Equilibria among Fe–Ti oxides, pyroxenes, olivine, and quartz. Part II. Application. Am Mineral 77:1004–1020
Ganguly J, Tazzoli V (1994) Fe2+-Mg interdiffusion in orthopyroxene: retrieval from the data on intracrystalline exchange reaction. Am Mineral 79:930–937
Ghiorso MS, Gualda GAR (2013) A method for estimating the activity of titania in magmatic liquids from the compositions of coexisting rhombohedral and cubic iron-titanium oxides. Contrib Mineral Petrol 165:73–81
Ghiorso MS, Sack RO (1991) Fe–Ti oxide geothermometry: thermodynamic formulation and the estimation of intensive variables in silicic magmas. Contrib Mineral Petrol 108:485–510
Ginibre C, Wörner G, Kronz A (2002) Minor- and trace-element zoning in plagioclase: implications for magma chamber processes in Parinacota volcano, northern Chile. Contrib Mineral Petrol 143:300–315
Ginibre C, Wörner G, Kronz A (2007) Crystal zoning as an archive for magma evolution. Elements 3:261–266
Gualda GAR (2007) Crystal and bubble populations in the early-erupted Bishop rhyolitic magma: microscopy, X-ray tomography and microanalysis of pumice clasts. PhD Thesis. The University of Chicago, Chicago, Illinois
Gualda GAR, Ghiorso MS (2013) The Bishop Tuff giant magma body: an alternative to the Standard Model. Contrib Mineral Petrol 166:755–775
Gualda GAR, Pamukcu AS, Ghiorso MS, Anderson AT, Sutton SR, Rivers ML (2012a) Timescales of quartz crystallization and the longevity of the Bishop giant magma body. PLoS One 7:e37492
Gualda GAR, Ghiorso MS, Lemons RV, Carley TL (2012b) Rhyolite-MELTS: a modified calibration of MELTS optimized for silica-rich, fluid-bearing magmatic systems. J Petrol 53:875–890
Hammond PA, Taylor LA (1982) The ilmenite/titano-magnetite assemblage: kinetics of re-equilibration. Earth Planet Sci Lett 61:143–150
Hildreth EW (1977) The magma chamber of the Bishop Tuff: gradients in temperature, pressure, and composition. PhD thesis. University of California, Berkeley, California
Hildreth W (1979) The Bishop Tuff: evidence for the origin of compositional zonation in silicic magma chambers. In: Chapin CE, Elston WE (eds) Ash-flow tuffs. Geol Soc Am Spec Pap 180:43–75
Hildreth W (2004) Volcanological perspectives on Long Valley, Mammoth Mountain, and Mono Craters: several contiguous but discrete systems. J Volcanol Geotherm Res 136:169–198
Hildreth W, Wilson CJN (2007) Compositional zoning of the Bishop Tuff. J Petrol 48:951–999
Huebner JS, Sato M (1970) The oxygen fugacity-temperature relationships of manganese oxide and nickel oxide buffers. Am Mineral 55:934–952
Humphreys MCS, Christopher T, Hards V (2009) Microlite transfer by disaggregation of mafic inclusions following magma mixing at Soufriere Hills volcano, Montserrat. Contrib Mineral Petrol 157:609–624
Jellinek AM, DePaolo DJ (2003) A model for the origin of large silicic magma chambers: precursors of caldera-forming eruptions. Bull Volcanol 65:363–381
Kularatne K, Audétat A (2014) Rutile solubility in hydrous rhyolite melts at 750–900 °C and 2 kbar, with application to titanium-in-quartz (TitaniQ) thermobarometry. Geochim Cosmochim Acta 125:196–209
Lu F (1991) The Bishop Tuff: origins of the high-silica rhyolite and its thermal and chemical zonations. PhD thesis. The University of Chicago, Chicago, Illinois
Lu F, Anderson AT, Davis AM (1992) Melt inclusions and crystal-liquid separation in rhyolitic magma of the Bishop Tuff. Contrib Mineral Petrol 110:113–120
Magaritz M, Hofmann AW (1978) Diffusion of Sr, Ba and Na in obsidian. Geochim Cosmochim Acta 42:595–605
Malfait WJ, Seifert R, Petitgirard S, Perrillat J-P, Mezouar M, Ota T, Nakamura E, Lerch P, Sanchez-Valle C (2014) Supervolcano eruptions driven by melt buoyancy in large silicic magma chambers. Nat Geosci 7:122–125
Martin VM, Morgan DJ, Jerram DA, Caddick MJ, Prior DJ, Davidson JP (2008) Bang! Month-scale eruption triggering at Santorini volcano. Science 321:1178
Matthews NE, Huber C, Pyle DM, Smith VC (2012a) Timescales of magma recharge and reactivation of large silicic systems from Ti diffusion in quartz. J Petrol 53:1385–1416
Matthews NE, Pyle DM, Smith VC, Wilson CJN, Huber C, van Hinsberg V (2012b) Quartz zoning and the pre-eruptive evolution of the ~340-ka Whakamaru magma systems, New Zealand. Contrib Mineral Petrol 163:87–107
Miller CF, Wark DA (2008) Supervolcanoes and their explosive supereruptions. Elements 4:11–16
Morgan DJ, Blake S (2006) Magmatic residence times of zoned phenocrysts: introduction and application of the binary element diffusion modelling (BEDM) technique. Contrib Mineral Petrol 151:58–70
Morgan DJ, Blake S, Rogers NW, De Vivo B, Rolandi G, MacDonald R, Hawkesworth CJ (2004) Time scales of crystal residence and magma chamber volumes from modelling of diffusion profiles in phenocrysts: Vesuvius 1944. Earth Planet Sci Lett 222:933–946
Morgan DJ, Blake S, Rogers NW, De Vivo B, Rolandi G, Davidson JP (2006) Magma chamber recharge at Vesuvius in the century prior to AD 79. Geology 34:845–848
Naney MT (1983) Phase equilibria of rock-forming ferromagnesian silicates in granitic systems. Am J Sci 283:993–1033
Pamukcu AS, Gualda GAL, Anderson AT (2012) Crystallization stages of the Bishop Tuff magma body recorded in crystal textures in pumice clasts. J Petrol 53:589–609
Peppard BT, Steele IM, Davis AM, Wallace PJ, Anderson AT (2001) Zoned quartz phenocrysts from the rhyolitic Bishop Tuff. Am Mineral 86:1034–1052
Reed SJB (2005) Electron microprobe analysis and scanning electron microscopy in geology. Cambridge University Press, Cambridge
Reid MR, Schmitt AK (2012) Implications of Bishop Tuff zircon U-Pb ages for rates of zircon growth and magma accumulation. Abstract V13F-07 presented at the 2012 Fall Meeting of the American Geophysical Union, San Francisco, California, 3–7 December
Reid MR, Vazquez JA, Schmitt AK (2011) Zircon-scale insights into the history of a supervolcano, Bishop Tuff, Long Valley, California, with implications for the Ti-in-zircon geothermometer. Contrib Mineral Petrol 161:293–311
Rivera TA, Storey M, Zeeden C, Hilgen FJ, Kuiper K (2011) A refined astronomically calibrated 40Ar/39Ar age for Fish Canyon sanidine. Earth Planet Sci Lett 311:420–426
Roberge J, Wallace PJ, Kent AJR (2013) Magmatic processes in the Bishop Tuff rhyolitic magma based on trace elements in melt inclusions and pumice matrix glass. Contrib Mineral Petrol 165:237–257
Saunders K, Blundy J, Dohmen R, Cashman K (2012) Linking petrology and seismology at an active volcano. Science 336:1023–1027
Saunders K, Buse B, Kilburn MR, Kearns S, Blundy J (2014) Nanoscale characterisation of crystal zoning. Chem Geol 364:20–32
Schwandt CS, Cygan RT, Westrich HR (1998) Magnesium self-diffusion in orthoenstatite. Contrib Mineral Petrol 130:390–396
Self S (2006) The effects and consequences of very large explosive volcanic eruptions. Philos Trans R Soc Lond A 364:2073–2097
Self S, Blake S (2008) Consequences of explosive supereruptions. Elements 4:41–46
Snyder D (2000) Thermal effects of the intrusion of basaltic magma into a more silicic magma chamber and implications for eruption triggering. Earth Planet Sci Lett 175:257–273
Sparks RSJ, Sigurdsson H, Wilson L (1977) Magma mixing: a mechanism for triggering acid explosive eruptions. Nature 267:315–318
Thomas JB, Watson EB (2012) Application of the Ti-in-quartz thermobarometer to rutile-free systems. Reply to: a comment on “TitaniQ under pressure: the effect of pressure and temperature on the solubility of Ti in quartz’ by Thomas et al. Contrib Mineral Petrol 164:369–374
Thomas JB, Watson EB, Spear FS, Shemella PT, Nayak SK, Lanzirotti A (2010) TitaniQ under pressure: the effect of pressure and temperature on the solubility of Ti in quartz. Contrib Mineral Petrol 160:743–759
Till CB, Vazquez JA, Boyce JW, Hitzman C (2012) Quantifying the interval between rejuvenation and eruption of rhyolite at Yellowstone caldera using high-resolution NanoSIMS geospeedometry. In: Abstract V43E-01 presented at the 2012 Fall Meeting of the American Geophysical Union, San Francisco, California, 3–7 December
Wallace PJ, Anderson AT, Davis AM (1999) Gradients in H2O, CO2, and exsolved gas in a large-volume silicic magma system: interpreting the record preserved in melt inclusions from the Bishop Tuff. J Geophys Res 104:20097–20122
Wark DA, Hildreth W, Spear FS, Cherniak DJ, Watson EB (2007) Pre-eruption recharge of the Bishop magma system. Geology 35:235–238
Wilson CJN, Hildreth W (1997) The Bishop Tuff: new insights from eruptive stratigraphy. J Geol 105:407–439
Wilson CJN, Seward TM, Charlier BLA, Allan ASR, Bello L (2012) A comment on: ‘TitaniQ under pressure: the effect of pressure and temperature on the solubility of Ti in quartz’, by Jay B. Thomas, E. Bruce Watson, Frank S. Spear, Philip T. Shemella, Saroj K. Nayak and Antonio Lanzirotti. Contrib Mineral Petrol 164:359–368
Zellmer GF, Blake S, Vance D, Hawkesworth C, Turner S (1999) Plagioclase residence times at two island arc volcanoes (Kameni Islands, Santorini, and Soufriere, St. Vincent) determined by Sr diffusion systematics. Contrib Mineral Petrol 136:345–357
Zhang Y (2010) Diffusion in minerals and melts: theoretical background. Rev Mineral Geochem 72:5–59
Acknowledgements
We thank Richard Walshaw (University of Leeds) and Joe Wooden (Stanford University) for assistance during CL imaging, and Aidan Allan and George Cooper for discussions. KJC was supported by an NZ International Doctoral Research Scholarship administered by Education New Zealand, a Jack Kleinman Award administered by the U.S. Geological Survey, and two Victoria University of Wellington Faculty Strategic Research Grants. CJNW thanks the Royal Society of New Zealand for support through Marsden Fund grant VUW0813 (which also facilitated the participation of DJM) and a James Cook Fellowship. Analyses at Leeds were part-funded by DJM through School of Earth and Environment start-up funds. We thank Kate Saunders and an anonymous reviewer for their helpful reviews and Jon Blundy for editorial handling.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Jon Blundy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chamberlain, K.J., Morgan, D.J. & Wilson, C.J.N. Timescales of mixing and mobilisation in the Bishop Tuff magma body: perspectives from diffusion chronometry. Contrib Mineral Petrol 168, 1034 (2014). https://doi.org/10.1007/s00410-014-1034-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-014-1034-2