
Abstract
The effect of TiO2 and P2O5 on the ferric/ferrous ratio in silicate melts was investigated in model silicate melts at air conditions in the temperature range 1,400–1,550 °C at 1-atm total pressure. The base composition of the anorthite–diopside eutectic composition was modified with 10 wt % Fe2O3 and variable amounts of TiO2 (up to 30 wt %) or P2O5 (up to 20 wt %). Some compositions also contained higher SiO2 concentrations to compare the role of SiO2, TiO2, and P2O5 on the Fe3+/Fe2+ ratio. The ferric/ferrous ratio in experimental glasses was analyzed using a wet chemical technique with colorimetric detection of ferrous iron. It is shown that at constant temperature, an increase in SiO2, TiO2, and P2O5 content results in a decrease in the ferric/ferrous ratio. The effects of TiO2 and SiO2 on the Fe3+/Fe2+ ratio was found to be almost identical. In contrast, adding P2O5 was found to decrease ferric/ferrous ratio much more effectively than adding silica. The results were compared with the predictions from the published empirical equations forecasting Fe3+/Fe2+ ratio. It was demonstrated that the effects of TiO2 are minor but that the effects of P2O5 should be included in models to better describe ferric/ferrous ratio in phosphorus-bearing silicate melts. Based on our observations, the determination of the prevailing fO2 in magmas from the Fe3+/Fe2+ ratio in natural glasses using empirical equations published so far is discussed critically.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Iron in silicate melts is stable in two oxidation states, Fe3+ and Fe2+, with the first species prevailing at high fO2 and the second one at low fO2 values. The ferric/ferrous equilibrium in melts can be described by the reaction:
with the reaction constant:
where a i and γ i are the activities and activity coefficients, respectively, of iron oxides in silicate melts, X i refers to the mole fraction on oxide basis, and fO2 is the oxygen fugacity. The Eq. (2) may be further converted into
In this equation, the coefficient (k) at logfO2 is equal to 1/4 in an ideal case, when γ FeO1.5/γ FeO is approximately constant in the fO2 range under consideration. However, small deviations of k from the ideal value have been reported and are the subject of discussion (e.g., Kress and Carmichael 1988, 1991; Jayasuriya et al. 2004; Borisov 2010).
At constant T-fO2 conditions, log(X FeO1.5/X FeO) must be proportional to log(γ FeO1.5/γ FeO) in the silicate liquid, which reflects the compositional dependence of the ferric/ferrous ratio. In a simple case such a dependence may be approximated as a linear function of the mole fractions of the main oxide component in silicate melts (SiO2, Al2O3, MgO, CaO, Na2O, and K2O), Σd i X i, where d i and X i are empirical coefficients and mole fractions of individual oxides in the melt, respectively. This results in empirical equations in which Fe3+/Fe2+ ratio is a function of T, fO2, and melt composition in the form (e.g., Sack et al. 1980; Kilinc et al. 1983):
where T is in K, and k, h, and c are constants. The type (4) empirical equations are important from two points of view:
-
1.
For given experimental or suggested natural T-fO2 conditions, such equations allow to recalculate Fe3+ and Fe2+ species in glasses in which only the total Fe (Fet) is known, typically determined with EPMA. For example, the determination of the Fe–Mg partition coefficient between olivine and melt (Roeder and Emslie 1970; Toplis 2005) requires prior calculation of the proportions Fe3+/Fet. The different size and charge of Fe3+ and Fe2+ imply that physical and chemical properties of silicate melts are affected by the Fe3+/Fe2+ ratio and also explain why the distribution of Fe3+ and Fe2+ is different in melts and coexisting crystalline phases.
-
2.
The Fe3+/Fe2+ ratio, similar to some other redox pairs, may be used for the estimation of the redox conditions of magmas and of the regions of magma generation. The best example is an estimation of fO2 of the mantle from which mid-ocean ridge basalt (MORB) was derived (Christie et al. 1986; Nikolaev et al. 1996; Bézos and Humler 2005; Cottrell and Kelley 2011, 2013). The approach is based on the determination of Fe3+/Fet ratios in MORB glasses by applying empirical equations such as Eq. (4). Christie et al. (1986) conducted wet chemical analyses of 78 hand-picked MORB glasses, applied Kilinc’s et al. (1983) equation and estimated fO2 to be l–2 log units below the quartz–fayalite–magnetite buffer (QFM). Cottrell and Kelley (2011) analyzed 103 MORB glasses with micro-X-ray absorption near-edge structure (μ-XANES), applied Kress and Carmichael’s (1991) equation, and estimated fO2 to be around QFM. Note that the reason for such a large discrepancy between these two fO2 estimations is primarily due to very different average Fe3+/Fet ratios found for MORB glasses (0.07 ± 0.03 in the first case and 0.16 ± 0.01 in the second one), because of the possible presence of microcrystals in the samples analyzed by wet chemistry. However, using different equations may also affect fO2 determination. For example, Nikolaev et al. (1996) re-estimated fO2 in fourteen MORB glasses already analyzed by Christie et al. (1986) using their own calibration, and fO2 was found to be higher by 0.55 log unit in average than reported in the previous paper.
In most empirical equations proposed so far to model the link between Fe3+/Fe2+ ratio and fO2, oxides such as TiO2, MnO, Cr2O3, and P2O5 are ignored because they “have very low concentrations in silicate liquids and are often not reported in the published data” (Sack et al. 1980, p. 373). However, the effects of such minor components on ferric/ferrous ratio may be not negligible.
Two of these minor components such as Mn and Cr occur in silicate melts in different valence states, Mn3+ and Mn2+ and Cr6+, Cr3+ and Cr2+, depending on fO2 (e.g., Johnston 1965; Schreiber and Haskin 1976; Pretorius et al. 1992; Schreiber et al. 1996). The investigation of the effect of these oxides on ferric/ferrous ratio is not a trivial task, because of the possible exchange of electrons between redox pairs during quenching of melts into glasses (e.g., Lahiri et al. 1974; Rüssell 1989; Borisov 2013). The study of such equilibria in situ at high temperature may be an option, but is technically difficult (Berry et al. 2003). The two other minor components such as Ti and P exist only in one valence state and Ti4+ and P5+ in silicate melts at fO2 conditions typically used in experiments on ferric/ferrous determination (from air to about QFM-2). Trivalent titanium in silicate melts is present in reasonable amount only at fO2 below iron–wüstite (IW) buffer conditions (Johnston 1965; Schreiber et al. 1978; Borisov 2012). Phosphorus as phosphide ion (P3−) was found in slags under strongly reducing conditions, more than 9 orders of magnitude below IW (Momokawa and Sano 1982). Due to the lacking redox variation of Ti and P under oxidizing to intermediate conditions (up to iron–wüstite buffer), the effect of these elements on ferric/ferrous ratio in silicate melts can be studied by “classical techniques,” i.e., by analyzing quenched samples (glasses).
TiO2 is one of the main components of lunar high-Ti basalts, which were collected in the Apollo 11 and 17 missions, with concentrations up to 15 wt % (BVSP, 1981). Terrestrial magmatic rocks are usually poor in titania, but in some cases they may contain up to 5 wt % (e.g., Pik et al. 1998). Phosphorus content in primitive mantle is estimated to be very low (86 ppm, Palme and O’Neil 2003). However, being highly incompatible, P content may increase in a melt during partial melting and fractional crystallization processes. The main P-bearing phase, apatite, has high solubility in high-temperature basaltic silicate melts (Watson 1980), and P2O5 content in such melts may reach a few weight percents before the saturation with apatite (up to 2 wt % P2O5 in Fe-rich basaltic systems, e.g., Auwera and Longhi 1994).
Even such small P2O5 contents may be essential in affecting iron redox ratio but, in the absence of experimental calibrations, such effects are not well constraint. Jayasuriya et al. (2004) stressed “the extraordinarily high” ability of P2O5 to decrease Fe3+/Fe2+. Mysen (1992) found two opposite trends in changing ferric/ferrous ratio by diluting an initial Ca silicate melt either with P2O5 or with Ca3(PO4)2. At last, Gwinn and Hess (1993) revealed a significant increase in Fe3+/Fe2+ with the addition of P in very silicic melts with SiO2 concentrations up to 78 wt %.
Although the effect of melt composition on the Fe3+/Fe2+ ratio is small in comparison with the effects of temperature and, especially, oxygen fugacity, it cannot be ignored. To clarify the compositional dependence of the ferric/ferrous ratio in silicate melts, investigations in rather simple systems have been conducted, e.g., Na2O–FeO–Fe2O3–SiO2 (Lange and Carmichael 1989), K2O–FeO–Fe2O3–SiO2 (Tangeman et al. 2001), and CaO–FeO–Fe2O3–SiO2 (Kress and Carmichael 1989). Alternatively, natural melts may be modified by adding the components of interest. For example, Thornber et al. (1980) modified an initial diabase composition by increasing Al2O3 up to 28.7 wt %, CaO up to 24.3 wt %, K2O up to 7.6 wt %, which are the concentrations well above that of typical basaltic melts. One may argue that the effect on Fe3+/Fe2+ may be different for low and high amounts of components. However, often variation over a large compositional range is required to properly resolve such effects. Following this approach, we have added up to 30 wt % TiO2 and up to 20 wt % P2O5 to a haplobasaltic melt to quantify the effect of these components on the redox state of iron.
Experimental procedure
A synthetic iron-free melt of diopside–anorthite eutectic composition (DA) was chosen as the base composition for the experiments. This composition was modified by adding about 10 wt % Fe2O3 (total iron expressed as Fe2O3) and different amounts of TiO2 or P2O5 to obtain two sets of melt compositions: DAFT series (DAF, DAFT10, DAFT20, and DAFT30) with a total formula of 10Fe2O3·xTiO2·(90-x)DA and DAFP series (DAF, DAFP05, DAFP10, DAFP15, and DAFP20) with a total formula of 10Fe2O3·xP2O5·(90-x)DA. Some Ti-containing mixtures were additionally modified by adding silica (DAFST series: DAFS, DAFST15, and DAFST25) with a total formula of 10Fe2O3·xTiO2·(90-x)·(0.5DA·0.5SiO2). One P-containing mixture (DAFP10) was also modified by adding silica (marked as DAFSP08). In all cases, the two-digit numbers in the mixture label denote the rough amount of TiO2 or P2O5 (wt %) and x in formulas refer to wt % of the respective components.
The experiments were conducted in a one-atmosphere vertical tube furnace with the loop technique (e.g., Donaldson et al. 1975). Platinum wire (0.125 mm thick and 99.9 % pure, Chempur®) was used to prepare loops about 2 or 3 mm in diameter. Silicate powder mixed with weak polyvinyl alcohol–water solution was inserted into the loops that were then suspended on the sample holder and transferred into the furnace. The investigated temperature range was 1,400–1,550 °C. Temperatures were determined with a Pt/PtRh10 thermocouple that was calibrated against the melting point of Au (1,064 °C) and expected to be accurate within ±2 °C. All experiments were conducted in air.
Two sample sizes were used in this study: 2-mm loops with average sample weight of about 14 mg for preliminary investigations of phosphorus loss from DAFP10 composition (Table 1) and 3-mm loops with average sample weight of about 30 mg for the main series of experiments (Table 2). In the last case, up to 7 samples of different composition were simultaneously placed into the furnace at a given temperature.
In order to determine equilibration times, experimental series with different run duration were conducted with basic (DAF) and more silicic (DAFS) compositions at 1,500 °C. Although redox equilibrium was found to be reached quickly (see below), most of the experiments with Ti-containing compositions were carried out with relatively long duration (e.g., 70 h at 1,450 °C).
The loss of phosphorus from the melts even at air conditions (see below) forced us to reduce experimental duration with P-containing compositions to a minimum needed to reach equilibrium Fe3+/Fe2+ ratio. However, DAFSP08 composition was found to be very viscous in comparison with all other mixtures and thus potentially to be most problematic in reaching equilibrium. Thus, a few longer runs with DAFSP08 were performed to have a time series at 1,500 °C.
After the experiments, the samples were quenched by quickly withdrawing the sample holder from the furnace. The small weight of the loop samples (30–35 mg) guarantees fast quenching. The samples were then crushed, and at least two small glass fragments of each sample were mounted in epoxy, polished, and carbon-coated for electron microprobe analysis (EMPA).
Bulk compositions of experimental glasses were determined by EMPA using a Cameca SX100. Conditions of measurements were 15 keV accelerating voltage, 15 nA beam current, 10 s counting time, and focused beam. At least twenty points were analyzed in each glass sample, and the average composition of the run products is given in Table 2.
The ferric/ferrous ratio in experimental glasses was analyzed using a wet chemical technique, based on the colorimetric method of Wilson (1960) and modified following the procedure given by Schuessler et al. (2008). In brief, the analytical procedure is as follows: 5–7 mg of powdered sample was weighed into a 15-mL Teflon beaker containing 1 mL of an ammonium vanadate solution dissolved in sulfuric acid (5 M H2SO4). After the addition of 0.5–1 mL concentrated HF, the beaker was tightly sealed and left overnight at room temperature. It is assumed that V5+ in vanadate solution oxidizes Fe2+ as soon as it is released from the sample according to the reaction:
with V4+ in solution being much more resistant to oxidation when compared to Fe2+. After sample dissolution, 5 mL saturated boric acid solution was added to neutralize the excess HF. The content of the beaker was then quantitatively transferred into a 100-mL volumetric flask, containing 10 mL ammonium acetate solution and 5 mL 2:2′ bipyridyl solution, and the remaining volume was filled with distilled water. The ammonium acetate buffer adjusted the pH value to about 5, which brings the reaction (5) to the left and regenerates Fe2+ from Fe3+. The complex of Fe2+ with 2:2′ bipyridyl shows an intensive absorption band at about 523 nm. Measurements of Fe2+ and Fetot were taken with Shimadzu UV-1800 spectrometer on the same solution before and after adding 5–10 mg solid hydroxylamine hydrochloride to an aliquant of about 10 mL. This reducing agent converts all Fe3+ into Fe2+. The advantage of such modification is that uncertainties in ferric/ferrous ratios arise mainly from the spectrometric measurements, whereas weighing and dilution errors cancel out.
All experimental glasses were analyzed in three sessions (Table 2). An in-house standard PU-3 (see Schuessler et al. 2008 for details) with known Fe2+ and Fet was included in every session. The Fe3+/Fe2+ ratios were found to be 1.47 ± 0.08, 1.53 ± 0.08, and 1.52 ± 0.08, which are identical within error limits to the values 1.60 ± 0.11 (1σ for the average of 33 measurements collected over a time period of about 1 year) determined by Schuessler et al. (2008).
Results
Phosphorus evaporation from experimental samples at high temperatures
The results of the phosphorus loss from the melts of DAFP10 composition at air conditions are given in Table 1. Previous studies (Borisov, unpublished data) of phosphorus evaporation from the melts of DAP composition (DAP = Diopside–Anorthite eutectic composition + Ca2P2O7) demonstrated a severe P loss at IW buffer conditions adjusted by CO/CO2 gas mixture, and the results are also shown in Table 1. Note that the errors of P2O5 contents given in Table 1 are standard deviations for an average of ten measurements, typically made along a profile across the whole sample. The low values of these errors reflect relatively homogeneous phosphorus content even in samples that suffered substantial loss of phosphorus. One of the reasons of such homogeneity can be convection, which can occur in the melt droplets (Borisov 2001). In addition to T-fO2 conditions, the sample size (surface exposed to air or gas flux) and melt composition may also affect the extent of P loss by evaporation. Considering that the compositions are rather similar (DA as a base) and that the samples have a similar weight (13.4 ± 1.5 mg for DAFP10 vs. 15.3 ± 2.3 mg for DAP samples), we will discuss them together (Fig. 1).

Effect of temperature and redox conditions on P loss from experimental melts
Run duration and redox conditions are found to be most critical for P loss (Fig. 1). At 1,450 °C and IW buffer conditions, about 45 % of initial P content was lost from the sample after 15 h. At the same temperature in air conditions during similar duration, only 10 % of phosphorus was lost. The temperature of experiments is also important. For example, at air condition and similar duration of 15–17 h, a temperature increase from 1,450 to 1,500 °C results in an increase in P loss from 10 to 17 %.
The phosphorus loss at constant T-fO2 conditions seems to be a quadratic function of the run duration τ (h). For example, at air conditions and 1,450 °C, the phosphorus content can be predicted for the applied experimental setup with following equation (R 2 = 0.999):
It is emphasized that the phosphorus loss is less problematic in the experiments conducted to determine the Fe3+/Fe2+ ratio. As it was mentioned above, the samples of the main P series (compositions used for the experiments listed in Table 2) were melted on 3-mm loops and had an average weight of about 30 mg, which is roughly two times higher than that used for the experiments investigating the phosphorus loss. Thus, the phosphorus loss from these samples is smaller. For example, a ~30 mg sample DAFP10–30 (1,450 °C, 9 h, see Table 2) contains 10.5 ± 0.1 wt % P2O5, whereas a small sample (~15 mg) of the same initial composition melted at the same conditions would contain only 9.9 wt % P2O5 according to Eq. (6).
Equilibrium series for the investigation of the Fe3+/Fe2+ ratio
The results of the time series experiments with the basic DAF and more silicic DAFS compositions are shown on Fig. 2a. The Fe3+/Fe2+ ratio in samples of the same composition equilibrated for 2, 8, 14.5, and 24 h are the same within 1σ error limits, demonstrating fast achievement of equilibrium between melt and air at 1,500 °C. Additionally, the coincidence of the results in four independent experimental charges of the same composition validates the very good reproducibility of ferric/ferrous determination at least within the same analytical session. Fast equilibration of Fe3+/Fe2+ was previously found in natural melts equilibrated with air by the loop technique (e.g., <5 h at 1,350 °C, Kilinc et al. 1983).

Effect of equilibration time on ferric/ferrous ratio at 1,500 °C: a in basic (DAF) and more silicic (DAFS) melts; b in melts of DAFSP08 composition
The results of the time series experiments with the most viscous DAFSP08 composition at 1,500 °C are shown on Fig. 2b. Although the Fe3+/Fe2+ ratios in samples equilibrated for 2, 4, and 8 h are identical within 1σ error limits, a small trend in increasing the ratio with run duration is evident. Note that one cannot guarantee exactly the same composition in time series because P loss even from large samples at temperature as high as 1,500 °C is not negligible. Indeed, P2O5 content decreases from 8.0 wt % for the shortest experiment to 7.1 wt % for longest one. We will further demonstrate that the addition of phosphorus results in an essential decrease in Fe3+/Fe2+ ratio in silicate melts. It means that P loss should raise this ratio, and slightly different phosphorus content could explain the trend observed on Fig. 2b.
The effect of TiO2 and P2O5 contents on the Fe3+/Fe2+ ratio
The decrease in Fe3+/Fe2+ with TiO2 content is evident for the three investigated temperatures both for basic DAFT and silicic DAFST series (Fig. 3). Note that the Fe3+/Fe2+ ratio is systematically lower in the more silicic than in the basic samples (data at 1,500 and 1,450 °C) independent from TiO2 concentration. The comparison of the Ti-free samples, DAF-9 (1,500 °C) and DAFS-10 (1,450 °C) having exactly the same Fe3+/Fe2+ = 1.87 (see Table 2), indicates that increasing silica content in the melt by 20 wt % is equivalent to a temperature increase of 50 °C in affecting the ferric/ferrous ratio.

Effect of TiO2 on ferric/ferrous ratio in the experimental samples
On Fig. 4, the effect of P2O5 on ferric/ferrous ratio in the DAFP series is shown. The decrease in Fe3+/Fe2+ value upon P addition is intense for all investigated temperatures, about 1.6 times when comparing P-free DAF and DAFP10 (av. 10.5 wt % P2O5) compositions and up to 2.3 times when comparing DAF and DAFP20 (av. 19.9 % P2O5) compositions.

Effect of P2O5 on ferric/ferrous ratio in the experimental samples of DAFP series
Discussion
Phosphorus evaporation from silicate melts
The phosphorus behavior is similar to that of the sodium loss from the melt at high temperatures and low fO2 values, which is a main problem of the loop technique and which is well documented (e.g., Corrigan and Gibb 1979; Tsuchiyama et al. 1981; Sugawara 1999). Following Tsuchiyama et al. (1981), who suggested sodium evaporation as metallic species, we suggest a similar reaction for the phosphorus loss from the melt:
with the following equilibrium constant:
where f P, fO2, and a PO2.5 are the fugacities of P and O2 in a vapor phase and the activity of PO2.5 in silicate melt, respectively. The Eq. (8) may be further converted into
Thus, at constant temperature, P fugacity (and, therefore, P loss) will increase with decreasing fO2 and increasing P content in a sample.
Note that the phosphorus loss at air conditions found in the present study is substantial but not as crucial as was revealed by Kilinc et al. (1983) (see their Table 3 and Fig. 1). These authors found that natural andesite melt with an initial P2O5 of 0.25 wt % lost all phosphorus after 70 h at 1,359 °C. More data are required to construct a complete model of P evaporation from silicate melts, similar to what Sugawara (1999) has suggested for Na loss from experimental charges.
SiO2, TiO2, and P2O5 components: efficiency of network-forming cations in decreasing the Fe3+/Fe2+ ratio.
On Fig. 5, we plotted Fe3+/Fe2+ ratio as a function of (SiO2 + TiO2). At constant temperature, both DAFT and DAFST compositions lie on the same regression line, implying that adding SiO2 and TiO2 roughly equally decreases ferric/ferrous ratio.

Effect of SiO2 + TiO2 contents on ferric/ferrous ratio in the experimental samples
Following Sack’s et al. (1980) model, we approximated log ferric/ferrous of all experimental glasses as a linear function of the reciprocal absolute temperature and mole fractions of SiO2, TiO2, and P2O5, taking into account that only these four parameters are variable
The high value of R 2 = 0.988 shows that Eq. (10) is successful to model to the experimental dataset with a good accuracy. The temperature slope 5,919 ± 223 (1σ) is in reasonable agreement with values for natural melts following the empirical equations of Sack et al. (1980) and Kilinc et al. (1983): 5,730 ± 420 and 5,500 ± 390, respectively. However, it is emphasized that the temperature effect is investigated over a range of 150 °C only.
The values of the coefficients d SiO2 = −0.674 ± 0.037 (1σ) and dTiO2 = −0.673 ± 0.044 are identical within error. This implies that adding SiO2 and TiO2 to a melt is equally effective (on a molar basis) in decreasing Fe3+/Fe2+ ratio. On the other hand, the value of the coefficient d P2O5 of −4.377 ± 0.113 (1σ) is much lower than that of d SiO2 or d TiO2. This implies that adding P2O5 to a melt is much more efficient in decreasing Fe3+/Fe2+ ratio than adding silica or titania.
The form of Eq. (10) implies that the relative effect of any two components on the Fe3+/Fe2+ ratio is given by the difference in those empirical coefficients, di. Indeed, from Eq. (10), it can be deduced that Δlog(X FeO1.5/X FeO) at exchanging 10 mol % SiO2 in a melt by 10 mol % P2O5 is equal to (d P2O5–d SiO2)/10 = −0.37. Such an effect is equivalent to a decrease in oxygen fugacity by approximately 1.5 log unit, assuming a slope of log (X FeO1.5/X FeO) versus logfO2 to have an ideal value of 0.25.
Thus, the present finding ranks the efficiency of network-forming oxides to decrease the ferric/ferrous ratio in CaO–MgO–FeO–Fe2O3–SiO2–TiO2–P2O5 melts in the following order: P2O5 ≫ TiO2 ≈ SiO2.
A comparison with the published data
Experiments with TiO2-bearing compositions
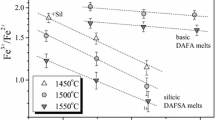
Matsuzaki and Ito (1997) conducted experiments in ternary FeO–TiO2–SiO2 melts, resembling slag compositions, with TiO2/SiO2 (wt) varying from 0.5 to 2. These experiments were carried out in equilibrium with metallic iron and fO2 was buffered with H2/CO2 gas mixtures. The authors used own data obtained at 1,400 °C along with the similar data of Ban-ya et al. (1980) in FeO–TiO2 and FeO–SiO2 binaries to plot Fe3+/Fe2+ versus XTiO2 + XSiO2 (Fig. 2 in Matsuzaki and Ito 1997). We additionally recalculated Fe3+/Fe2+ ratio in these slags to a constant oxygen fugacity of 10−10 atm (an ideal slope k = 0.25 of log Fe3+/Fe2+ vs. logfO2 was assumed) in attempt to minimize the data scattering. The results are shown on Fig. 6. The scattering is still large and reflects perhaps problems in the accuracy of the Fe3+/Fe2+ determination at these extremely low fO2 conditions where ferric content in the slags is very small. Nevertheless, the trend is evident: there is a decrease in the ferric/ferrous ratio with increasing SiO2 + TiO2 with approximately equal effects of SiO2 and TiO2.

Alberto et al. (1992) studied redox equilibria in more complex CaO–TiO2–SiO2 melts modified either with 5 or 15 wt % Fe2O3 total at 1,500 °C in air. Experimental data were not tabulated but plotted as Fe3+/ΣFe versus Ti/(Ti + Si) (Fig. 2 in Alberto et al. 1992). For the same Fe2O3 total, the most polymerized melts (more silicic, NBO/T = 1.2, X SiO2 + X TiO2 ≈ 0.63 in iron-free matrix) display much lower Fe3+/ΣFe ratio than less polymerized melts (more basic, NBO/T = 2, X SiO2 + X TiO2 ≈ 0.51 in iron-free matrix), similar to our results obtained with the DAFST and DAFT series. The exchange of Si by Ti is either negligible in terms of Fe3+/Fe2+ (series MIN(I-VI)F5) or results in a slight oxidation of iron (e.g., series CS(VII-XV)F5, see Fig. 2 in Alberto et al. 1992).
Summarizing, increasing both SiO2 and TiO2 results in a decrease in the Fe3+/Fe2+ ratio in simple slags and complex silicate melts, with TiO2 tending to be either equally (like in present experiments) or slightly less effective in comparison with SiO2. This observation is in agreement with structural studies indicating that at least part of the Ti incorporated in silicate melts is in fourfold coordination as Si (e.g., Dingwell et al. 1994; Farges et al. 1996; Henderson et al. 2002). However, part of the Ti may also be five- and sixfold coordinated.
Experiments with P2O5-bearing compositions
Toplis et al. (1994) investigated the effects of phosphorus on Fe3+/Fe2+ ratio in synthetic ferro-basaltic compositions. They revealed, for example, that at air conditions and fixed temperature of 1,446 and 1,545 °C, the ferric/ferrous ratio decreases by a factor of 1.3 as a result of the addition of 9.5 wt % P2O5 (4.4 mol %) to the initial P-free composition (see Table 2 and Fig. 1 in Toplis et al. 1994). This decrease is very similar to that observed in the present study. Note that Toplis et al. (1994) were the first who concluded that the incorporation of a P2O5 term is necessary in empirical equations to calculate Fe3+/Fe2+.
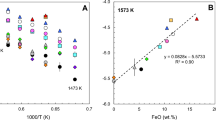
Large effects of P2O5 on ferric/ferrous ratio, much more pronounced when compared with SiO2, may also be extracted from the experimental data of Turkdogan and Bills (1957 ) in FeOt–SiO2–P2O5 melts at 1,550 °C. The authors report the CO/CO2 ratio in the buffering gas mixture [Table 1 in Turkdogan and Bills (1957)], which we converted into fO2 values using the thermodynamic data of Deines et al. (1974). The Fe3+/Fe2+ ratios in these slags (49 experiments in total) were then described as a function of logfO2, X SiO2, and X P2O5 to obtain the following equation:
The parameter k = 0.20 ± 0.01 at logfO2 is reasonably close to the ideal value of 0.25, which, together with the high correlation coefficient (R 2 = 0.95), demonstrates the high quality of the experiments and the appropriate form of Eq. (11). Thus, one can compare the efficiency of SiO2 and P2O5 in these slags in changing ferric/ferrous ratio. The substitution of 10 mol % SiO2 by the same amount of P2O5 lowers the log(X FeO1.5/X FeO) by a value of 0.21, which is equivalent to a decrease in the oxygen fugacity of ca. 0.9 log unit.
Jayasuriya et al. (2004) were the first who included phosphorus in the empirical equation proposed by Sack et al. (1980). They fitted ln(X Fe2O3/X FeO) versus ln fO2, 1/T and mole fraction of oxides, including X P2O5 (Eq. 12 in Jayasuriya et al. 2004). Converting ln (X Fe2O3/X FeO) into log(XFeO1.5/XFeO) for a correct comparison of their results with our Eqs. (10) and (11) would lead to a value of d P2O5 = −27.3 (error was not given) and d SiO2 = 0 (silica was not included in the fitting). Here, Δlog (X FeO1.5/X FeO) after exchanging 10 mol % SiO2 by 10 mol % P2O5 is equal to −2.73 corresponding to an extremely high fO2 decrease by about 11 log unit, which seems unrealistic. The conclusion that the effect of phosphorus on Fe3+/Fe2+ ratio may be much less than suggested by Jayasuriya et al. (2004) is also confirmed by the comparison of two experimental series (PW and P series) made at 1,500 °C in air by Mysen (1992). In both cases, the author used a composition close to 44CaO·56SiO2 (wt %) as a base modified with 5 wt % Fe2O3. In the P series, P2O5 was added to the base composition and a moderate decrease in ferric/ferrous ratio was observed. In the PW series, however, phosphorus was added as Ca3(PO4)2 and this resulted in increase in ferric/ferrous ratio (see Fig. 8 in Mysen 1992). The addition of CaO to a melt in the SiO2–CaO–FeOt system is known to increase Fe3+/Fe2+ ratio (Kress and Carmichael 1989). Thus, the addition of three moles CaO for one mole P2O5 (Ca3(PO4)2 = 3CaO·P2O5) largely compensates the possible decrease in ferric/ferrous induced by the addition of phosphorus.
In summary, the results in this study and the most published data suggest a significant decrease in ferric/ferrous ratio with increasing of P2O5 content. It is worth noting however that our most SiO2-rich composition DAFSP09, being recalculated on P-free base, contains only about 55 wt % SiO2. The synthetic ferro-basalt used by Toplis et al. (1994) contains about 51 % SiO2. Mysen’s (1992) initial composition has about 53 % SiO2. Finally, the SiO2 contents of the P-containing slags of Turkdogan and Bills (1957), even after recalculation on P-free base, do not exceed 47 wt %. Thus, on the basis of the available data, a stabilizing effect of P2O5 on ferrous iron can be inferred only for the basic to intermediate melts. However, it is emphasized that the data from Gwinn and Hess (1993) indicate that the effect of P2O5 on Fe3+/Fe2+ may be the opposite in very silicic melts (>70 wt % SiO2, Fig. 7). Although there is a large data scatter for the K-rich compositions, the P-bearing liquids have systematically higher Fe3+/Fe2+ ratios than P-free melts, independently on the K/Al ratio (peraluminous or peralkaline). Note that the addition of only 0.5 mol % P2O5 at fixed K/Al ratio and SiO2 content may increase the ferric/ferrous in a melt by a factor of 2–3. The contrasting behavior of phosphorus in very silicic and more basic melts was also noted by Ryerson (1985) who calculated SiO2 activity coefficients (γSiO2) in binary and ternary melts. Ryerson (1985) showed that adding P2O5 to SiO2 results in a decrease in γSiO2, even more pronounced than adding K2O or Na2O. In forsterite/clinoenstatite or crystoballite/clinoenstatite saturated melts, the addition of P2O5 results in an increase in γSiO2.

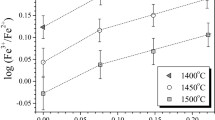
Effects of K/Al ratio on Fe3+/Fe2+ in P-free, P-poor, and P-rich silicic melts at 1,400 °C in air. Experimental data from Gwinn and Hess (1993). We present three series of experiments with zero, 0.5 mol % (1.6 wt %), and 2.6 mol % (5.4 wt %) P2O5 (data from Tables 1, 3a, b in Gwinn and Hess 1993). All melts have approximately the same SiO2 content (about 76 wt % ≈85 mol %) and total Fe2O3 (1 mol %) but with variable K/Al ratio
A detailed discussion of the structure of P-bearing aluminosilicate melts is out of the scope of this study, especially because structural information on the Fe-bearing compositions studied here is not available. However, the contrasting effects of P2O5 on the Fe3+/Fe2+ ratio in basic and felsic melts are most probably related to structural changes. Based on the experimental studies of phase relationships and applying the concepts of Korzhinskii (1959) relating mineral stability fields and melt properties, Kushiro (1975) proposed that P5+ could be a network former in basaltic and intermediate melts. However, more recent studies in basic to intermediate compositions emphasized the crucial role of alkalis in alkali-rich aluminosilicate melts in forming phosphate complexes (Toplis and Dingwell 1996; Mysen and Cody 2001). It is possible that phosphate complexes may also form with alkaline-earth elements, which would apply for the compositions investigated in this study. In contrast, in more silicic compositions, part of the phosphorus can enter the network as P5+ charge compensated by Al3+ and Fe3+ (Gwinn and Hess 1993; Toplis and Dingwell 1996).
Empirical equations including Ti and P components
As mentioned in the introduction, most of the equations developed to predict Fe3+/Fe2+ as a function of oxygen fugacity, temperature, and melt composition ignore TiO2 and P2O5. However, a few exceptions are known. Borisov and Shapkin (1990) and Moretti (2005) included titanium oxide as a component in their equations and we compare the predictions of their models with our DAFT series at 1,450 °C in air on Fig. 8a. Jayasuriya et al. (2004; Eq. 12) and Moretti (2005) included phosphorus oxide as a component in their equations, and we compare the predictions of their models with our DAFP series at the same conditions on Fig. 8b. In both cases, calculations with Kress and Carmichael’s (1991) model, where TiO2 and P2O5 are not considered, are also shown. Note that the equations under discussion were calibrated on the experiments carried out mostly with natural compositions. And the differences with our experimental results obtained with model melts can be expected, but the predicted trends of Fe3+/Fe2+ versus TiO2 or versus P2O5 contents may be informative.

Effects of “minor” components on Fe3+/Fe2+ in melts of 10Fe2O3·90DA composition modified either with TiO2 (a) or P2O5 (b) at 1,450 °C in air. A comparison of experimental results and calculation according to the published models: Moretti (2005), Kress and Carmichael (1991), Borisov and Shapkin (1990), Jayasuriya et al. (2004)
The contrasting results of Borisov and Shapkin (1990) and Moretti (2005) shown in Fig. 8a are related to the different approaches that have been used in the models. The empirical equation of Borisov and Shapkin (1990) has a form suggested by Sack et al. (1980) but with some additional complexity, the coefficients d i and X i are assumed to be T-fO2-dependent. The TiO2 was included in the regression along with other main components and no special assumption was made. The approach of Moretti (2005) follows that suggested earlier by Ottonello et al. (2001) and is based on the concept of “thermochemical models.” These models, in addition to other important assumptions, are based on the Temkin (1945) approach, subdividing the melt structure in anion and cation sublattices. Thus, either a network-forming or network-modifying behavior should be assumed a priory for every melt component. Moretti (2005), following Ottonello et al. (2001), treated Ti4+ as a network modifier.
The trend predicted by Borisov and Shapkin (1990) shows decreasing Fe3+/Fe2+ similar to the experimental data, but clearly overestimates the effect of TiO2 (see Fig. 8a). The reason is that at these conditions (1,450 °C, air), dTiO2 in the equation of Borisov and Shapkin (1990) is about 2.6 times higher than dSiO2 (−6.3 and −2.4, respectively). Note that at lower temperature, the difference between d TiO2 and d SiO2 becomes lower, at 1,330 °C, they are approximately equal, and at even lower temperatures, SiO2 is assumed to be more effective than TiO2 in decreasing the ferric/ferrous ratio. The trend predicted by Moretti (2005) is opposite to that of the experimental results. Surprisingly, the best fit between experiments and calculations is found in Kress and Carmichael’s (1991) model, which simply ignores TiO2 as a component affecting Fe3+/Fe2+ ratio. The explanation is that the small decrease in ferric/ferrous ratio with increasing TiO2 content expected by this model is simply due to a dilution effect. The equation of Kress and Carmichael (1991) includes Al2O3, FeOt, CaO, Na2O, and K2O as components affecting Fe3+/Fe2+. The alkali oxides are absent in the present melts and X FeO is approximately constant since total Fe2O3 was fixed on the level of 10 wt %. Thus, the higher the TiO2 content, the smaller X Al2O3 and X CaO in model melts are expected to be. Because d Al2O3 = −2.243 and d CaO = 3.201, the Fe3+/Fe2+ ratio in any melt with X CaO > X Al2O3 (like in present case) is expected to decrease with increasing concentration of additional components not included in the regression. Note that for a hypothetical melt with X Al2O3/X CaO > |d CaO/d Al2O3| ≈ 1.427, the effect would be the opposite. In any case, for typical basaltic melts, ignoring TiO2 seems to be the best approach.
However, this simplification is not recommended in case of P-containing melts (see Fig. 8b). The simple “dilution” effect resulting from the application of the model of Kress and Carmichael (1991) would not predict accurately the decrease in ferric/ferrous ratio with increasing P2O5 content. As observed for TiO2, the trend predicted by Moretti (2005) is opposite to that observed in the experimental results. The model of Jayasuriya et al. (2004) predicts a decrease in the ferric/ferrous, but dramatically overestimates the effect of phosphorus.
On Fig. 9, we plotted TiO2 and P2O5 contents in experimental glasses of three largest datasets of natural P-containing melts, which were used by Jayasuriya et al. (2004) to constrain their equation. One can note that the P-rich melts also contain large amounts of Ti. However, Jayasuriya et al. (2004) ignored titanium, following the usual practice of previous researchers, Since adding TiO2 was demonstrated to decrease Fe3+/Fe2+ ratio (see above), the apparent effect of P2O5 on ferric/ferrous ratio found by Jayasuriya et al. (2004) is most probably overestimated because it also include the effects of TiO2.

Implications for fO2 determinations of MORBs
As already mentioned in the introduction, the Fe3+/Fe2+ ratio measured in natural glasses may be used for the estimation of the redox conditions of magmas. This implies (1) an accurate determination of ferric/ferrous determination in glasses and (2) the choice of correct parameters for Eq. (4). The discussion below demonstrates that there is still a large uncertainty on the values of fO2 deduced from the interpretation of Fe3+/Fe2+ ratio in natural glasses to constrain the fO2 prevailing in natural systems.
Accuracy of the determination of Fe3+/Fe2+ ratio in natural glasses
A comprehensive review of possible wet chemical techniques and sources of error in ferric/ferrous determination is given by Bézos and Humler (2005). In most cases, if experimental glasses are free of crystalline phase and Fe2+ is determined with wet chemistry (and total iron with EPMA), the Fe3+/Fe2+ ratio is considered to have an uncertainty of about 6 % (Sack et al. 1980), which propagates to a logfO2 uncertainty of about ±0.15. In the case of natural MORBs, however, variable amounts of plagioclase microphenocrysts or microlites in glasses may be a problem (Bézos and Humler 2005; Cottrell and Kelley 2011). Thus, choosing local techniques of ferric/ferrous determination are getting mandatory for natural samples. For example, XANES measurements may give a precision on Fe3+/Fet determination of ±0.005 (about 3 %, Cottrell and Kelley 2011), which propagates to logfO2 precision of about ±0.07.
Role of compositional parameters and temperature in empirical equation
Most equations proposed in the literature reproduce the fO2 of experimental dataset obtained at 1 bar with a standard error of about 0.26 log units or higher (e.g., Kress and Carmichael 1988). However, natural glasses are quench products of high-pressure melts with a pre-eruptive temperature, which is usually poorly constrained, and the absolute errors in fO2 determination are expected to be higher.
To test the accuracy of the models for predicting fO2 from natural glasses, Eq. (4) can be formulated as follows:
where H = −h/(k·ln10). For most empirical equations, H values are rather similar (e.g., −26,251 in Sack et al. 1980, −25,183 in Kilinc et al. 1983, and −27,422 in Jayasuriya et al. 2004) and also very close to the slope of the QFM buffer in the logfO2 versus 1/T diagram (e.g., −24,442 for the calibration of Myers and Eugster 1983). Thus, by fixing arbitrary one temperature in Eq. (12) (e.g., of 1,200 °C for basaltic systems), the fO2 of various samples relative to the QFM buffer can be estimated and compared, even when the real pre-eruptive temperatures are not known accurately (e.g., Christie et al. 1986; Bézos and Humler 2005; Cottrell and Kelley 2011). Clearly, in this case, the compositional parameter Σd i X i is getting especially important.
On Fig. 10 for the same set of rocks, we compare Σd i X i/(k·ln10) (these values are in logfO2 units, see Eq. 12) calculated with two different models, Kilinc et al. (1983) and Kress and Carmichael (1991). A set of 103 MORB glasses analyzed by Cottrell and Kelley (2011) was chosen as an example. Although the glasses have a moderate variation in composition (they are all basalts), there is nearly no correlation between the two models. It implies that conclusions drawn from a fO2 variation observed for a given dataset with one model will not necessarily be confirmed with another one. For example, two arbitrary samples (a and b) are marked on Fig. 10. Let us assume them to have the same ferric/ferrous ratio. If the fO2 of these samples is calculated with Kress and Carmichael’s (1991) model, the sample b would be ~0.2 log unit more reduced than sample a. On the other hand, Kilinc’s et al. (1983) model would indicate that ample b is ~0.4 log unit more oxidized than sample a.

A comparison of Σd i X i/(k·ln10) values calculated with two different models, Kilinc et al. (1983) and Kress and Carmichael (1991), for 103 MORB glasses analyzed by Cottrell and Kelley (2011). Note that Σd i X i/(k·ln10) values are in logfO2 units. Two samples a and b are chosen arbitrary to discuss the models’ difference (see text for details)
Additional problems may arise by ignoring minor components in the empirical equation. On Fig. 11a, we plotted the difference between fO2 calculated without taking TiO2 or P2O5 and fO2 calculated considering the role of TiO2 or P2O5. As mentioned above, the effect of TiO2 is not significant, but the effect of P2O5 is more pronounced. The phosphorus variation in the range typical for MORB (0.05–0.25 wt % P2O5, Konzett and Frost 2009 ) is equivalent to 0.7 % variation in Fe3+/Fet, which is below the precision of the Fe3+/Fet determination in glasses. However, P2O5 variation of 2 wt % as observed for other basalts will be equivalent to about 7 % variation in Fe3+/Fet, which would propagate to an error of 0.16 logfO2.

Possible errors of fO2 determination, a if TiO2 or P2O5 is ignored, b if temperature slope differs from QFM buffer
Other minor problems in fO2 recalculation may arise if the pre-eruptive temperature of basaltic glasses differs from conventional 1,200 °C and if the temperature slope H of these glasses differs from QFM slope (Fig. 11b). As mentioned above, H value estimated by Kilinc et al. (1983) has only a minimal deviation from the QFM buffer slope and the resulting fO2 errors will not exceed 0.02 log unit when the difference between conventional 1,200 °C and real temperature does not exceed 50 °C (see Fig. 11b). Other equations show slightly higher but comparable errors. However, all these equations are statistically derived and a similar temperature slope for all compositions is assumed a priory. This is not the necessary case, and identical values for H for all melt compositions would be surprising. For example, Sato and Wright (1966) directly measured fO2 in a few boreholes drilled in tholeiitic basalts through the crust of Makaopuhi lava lake, Kilauea Volcano, Hawaii. They found that logfO2 changes linearly with the reciprocal absolute temperature, with slopes varying from −18,600 to −21,900. Assuming that measured fO2 values reflect fO2 of cooling basalt, the corresponding H slope is 11–31 % lower than that of QFM buffer. On the other hand, a slope of −41,327 can be calculated for a phonotephritic composition from the data given by Moore et al. (1995, composition MAS-49), which is about 70 % higher than that of the QFM buffer. More examples and the detailed discussion on the effect of temperature on the Fe3+/Fe2+ ratio in model and natural melts can be found in Borisov and McCammon (2010).
Figure 11b shows to which extent the calculated fO2 may change as a function of temperature if the H value differs from that of QFM by +30 % and −30 % (−31,775 and −17,109). A difference of 50 °C between conventional 1,200 °C and real pre-eruptive temperature results in fO2 error of about 0.17 log units.
Conclusions
-
1.
At constant temperature, the increase in TiO2 or P2O5 content in synthetic DA-Fe2O3-TiO2/P2O5 ± SiO2 compositions (DA represents the eutectic of the system Diopside-Anorthite) results in a decrease in the ferric/ferrous ratio both in basic and silicic melts. The effects of TiO2 and SiO2 on Fe3+/Fe2+ are mostly identical. In contrast, adding P2O5 was found to decrease ferric/ferrous much more effectively than adding silica. Thus, the efficiency of network-forming oxides to decrease the ferric/ferrous ratio may be ranked in the following order: P2O5 ≫ TiO2 ≈ SiO2.
-
2.
A comparison of our results with the predictions from the published empirical equations demonstrates that the effects of TiO2 are minor and that the formulation of Kress and Carmichael (1991) is sufficient to account (indirectly) for the role of TiO2 on Fe3+/Fe2+ ratio in terrestrial basaltic melts. In contrast, the effects of P2O5 should be taken into account in new formulations to better describe its role on the ferric/ferrous ratio in natural basaltic melts.
-
3.
An average ΔQFM value for MORB compositions may be estimated with reasonably good accuracy (ca. 0.3 log unit) if the ferric/ferrous ratio in glasses is determined with an uncertainty of ±3 % relative. The concentration of P2O5 is low and will not affect significantly the fO2 estimations. However, our present knowledge and published empirical equations do not allow us to account correctly for the effect of compositional changes on fO2 determination.
References
Alberto HV, Gil JM, Ayres de Campos N, Mysen BO (1992) Redox equilibria of iron in Ti-bearing calcium silicate quenched glasses. J Non-Crystalline Solids 151:39–50
Auwera JV, Longhi J (1994) Experimental study of a jotunite (hypersthene monzodiorite): constraints on the parent magma composition and crystallization conditions (P, T, fO2) of the Bjerkreim-Sokndal layered intrusion (Norway). Contrib Mineral Petrol 118:60–78
Ban-ya S, Chiba A, Hirosaka A (1980) Thermodynamics of FetO-MxOy (MxOy = CaO, SiO2, TiO2 and Al2O3) binary melts in equilibrium with solid iron. Tetsu-to-Hagane 66:1484–1493
Basaltic Volcanism Study Project (1981) Basaltic Volcanism on the Terrestrial Planets. Pergamon Press Inc., New York, p 300
Berry AJ, Shelley MG, Foran GJ, O’Neill HStC, Scott DR (2003) A furnace design for XANES spectroscopy of silicate melts under controlled oxygen fugacities and temperatures to 1,773 K. J Synchrotron Radiation 10:332–336
Bézos A, Humler E (2005) The Fe3+/∑Fe ratio of MORB glasses and their implications for mantle melting. Geochim Cosmochim Acta 69:711–725
Borisov A (2001) Loop technique: dynamic of metal/melt equilibration. Min Petrol 71:87–94
Borisov AA (2010) Ferric-ferrous ratio in liquid iron oxides: analysis and applications to natural basaltic melts. Petrology 18:471–481
Borisov AA (2012) The Ti4+/Ti3+ ratio of magmatic melts: application to the problem of the reduction of lunar basalts. Petrology 20:391–398
Borisov AA (2013) Mutual interaction of redox pairs in silicate melts: V5+/V4+/V3+/V2+ tetrad and other equilibria. Petrology 21:305–315
Borisov A, McCammon C (2010) The effect of silica on ferric/ferrous ratio in silicate melts: an experimental investigation using Mössbauer spectroscopy. Am Mineral 95:545–555
Borisov AA, Shapkin AI (1990) A new empirical equation relating Fe3+/Fe2+ in magmas to their composition, oxygen fugacity, and temperature. Geochem Int 27:111–116
Christie DM, Carmichael ISE, Langmuir CH (1986) Oxidation state of mid-ocean ridge basalt glasses. Earth Planet Sci Lett 79:397–411
Corrigan G, Gibb FGF (1979) The loss of Fe and Na from a basaltic melt during experiments using the wire-loop method. Mineral Mag 43:121–126
Cottrell E, Kelley KA (2011) The oxidation state of Fe in MORB glasses and the oxygen fugacity of the upper mantle. Earth Planet Sci Lett 305:270–282
Cottrell E, Kelley KA (2013) Redox heterogeneity in mid-ocean ridge basalts as a function of mantle source. Science 340:1314–1317
Deines PS, Nafziger RH, Ulmer GC, Woermann E (1974) Temperature-oxygen fugacity tables for selected gas mixtures in the system C-H-O at one atmosphere total pressure. Bull Earth Mineral Sciences, Experimental Station, No 88. Pennsylvania State University, USA
Dingwell DB, Paris E, Seifert F, Mottana A, Romano C (1994) X-ray absorption study of Ti-bearing silicate glasses. Phys Chem Miner 21:501–509
Donaldson CH, Williams RJ, Lofgren G (1975) A sample holding technique for study of crystal growth in silicate melts. Am Mineral 60:324–326
Farges F, Brown GE, Navrotsky A, Gan H, Rehr JJ (1996) Coordination chemistry of Ti(IV) in silicate glasses and melts: II. Glasses at ambient temperature and pressure. Geochim Cosmochim Acta 60:3039–3053
Gwinn R, Hess PC (1993) The role of phosphorus in rhyolitic liquids as determined from the homogeneous iron redox equilibrium. Contrib Mineral Petrol 113:424–435
Henderson GS, Liu X, Fleet ME (2002) A Ti L-edge X-ray absorption study of Ti-silicate glasses. Phys Chem Minerals 29:32–42
Jayasuriya KD, O’Neil HStC, Berry A, Campbell SJ (2004) A Mössbauer study of the oxidation state of Fe in silicate melts. Am Mineral 89:1597–1609
Johnston WD (1965) Oxidation-reduction equilibria in molten Na2O·2SiO2 glass. J. Americ Ceram Society 48:184–190
Kilinc A, Carmichael ISE, Rivers ML, Sack RO (1983) The ferric-ferrous ratio of natural silicate liquids equilibrated in air. Contrib Mineral Petrol 83:136–140
Konzett J, Frost DJ (2009) The high P-T stability of hydroxyl-apatite in natural and simplified MORB-an experimental study to 15 GPa with implications for transport and storage of phosphorus and halogens in subduction zones. J Petrol 50:2043–2062
Korzhinskii DS (1959) Acid-basic interaction of the components in silicate melts and the direction of the cotectic lines. Acad Sci USSR Earth Sci Sec 128:821–823
Kress VC, Carmichael ISE (1988) Stoichiometry of the iron oxidation reaction in silicate melts. Am Mineral 73:1267–1274
Kress VC, Carmichael ISE (1989) The lime-iron-silicate melt system: redox and volume systematics. Geochim Cosmochim Acta 53:2883–2893
Kress VC, Carmichael ISE (1991) The compressibility of silicate liquids containing Fe2O3 and the effect of composition, temperature, oxygen fugacity and pressure on their redox states. Contrib Mineral Petrol 108:82–92
Kushiro I (1975) On the nature of silicate melt and its significance in magma genesis; regularities in the shift of the liquidus boundaries involving olivine, pyroxene, and silica minerals. Am J Sci 275:411–431
Lahiri D, Mukherjee B, Majumdar RN (1974) Mechanismus der wechselwirkung zweier redox-oxide in glass. Glastech Berichte 47:4–9
Lange RA, Carmichael ISE (1989) Ferric/ferrous equilibria in Na2O-FeO-Fe2O3-SiO2 melts: effects of analytical techniques on derived partial molar volumes. Geochim Cosmochim Acta 53:2195–2204
Matsuzaki K, Ito K (1997) Thermodynamics of FetO-TiO2-SiO2 melts in equilibrium with solid iron. ISIJ Int 37:562–565
Momokawa H, Sano N (1982) The effect of oxygen potential on phosphorus in the CaO-Al2O3 System. Metall Transactions 13B:643–644
Moore G, Righter K, Carmichael ISE (1995) The effect of dissolved water on the oxidation state of iron in natural silicate liquids. Contrib Mineral Petrol 120:170–179
Moretti R (2005) Polymerization, basicity, oxidation state and their role in ionic modeling of silicate melts. Annals of Geophysics 48:583–608
Myers J, Eugster HP (1983) The system Fe-Si-O: oxygen buffer calibrations to 1500 K. Contrib Mineral Petrol 82:75–90
Mysen BO (1992) Iron and phosphorus in calcium silicate quenched melts. Chem Geol 98:175–202
Mysen BO, Cody GD (2001) Silicate-phosphate interactions in silicate glasses and melts: II. Quantitative, high-temperature structure of P-bearing alkali aluminosilicate melts. Geochim Cosmochim Acta 65:2413–2431
Nikolaev GS, Borisov AA, Ariskin AA (1996) New fO2-barometers for quenched glasses of various petrochemical series. Geochem Int 34:753–756
Ottonello G, Moretti R, Marini L, Zuccolini MV (2001) Oxidation state of iron in silicate glasses and melts: a thermochemical model. Chem Geol 174:157–179
Palme H, O’Neill HStC (2003) Geochemical estimates of mantle composition, in Treatise on Geochemistry. Vol 2. Ed. R.W. Carlson, Elsevier, Oxford, 1–38
Pik R, Deniel C, Coulon C, Yirgu G, Hofmann C, Ayalew D (1998) The northwestern Ethiopian plateau flood basalts: classification and spatial distribution of magma types. J. Volcanol Geotherm Research 81:91–111
Pretorius EB, Snellgrove R, Muan A (1992) Oxidation state of chromium in CaO–Al2O3–CrOx–SiO2 melts under strongly reducing conditions at 1,500°C. J Am Ceram Soc 75:1378–1381
Roeder PL, Emslie RF (1970) Olivine-liquid equilibrium. Contrib Mineral Petrol 29:275–289
Rüssell C (1989) Redox reactions during cooling of glass melts. Glass Science Technol 62:199–203
Ryerson FJ (1985) Oxide solution mechanisms in silicate melts: systematic variations in the activity coefficients of SiO2. Geochim Cosmochim Acta 49:637–649
Sack RO, Carmichael ISE, Rivers ML, Ghiorso MS (1980) Ferric-ferrous equilibria in natural silicate liquids at 1 bar. Contrib Mineral Petrol 75:369–376
Sato M, Wright TL (1966) Oxygen fugacities directly measured in magmatic gases. Sciences 153:1103–1105
Schreiber HD, Haskin LA (1976) Chromium in basalts: experimental determination of redox states and partitioning among synthetic silicate phases. Proceedings 7th Lunar Sci Conf:1221–1259
Schreiber HD, Thanyasiri T, Lach JJ, Legere RA (1978) Redox equilibria of Ti, Cr, and Eu in silicate melts: reduction potentials and mutual interactions. Physics Chem Glasses 19:126–139
Schreiber HD, Peters LJ, Beckman JW, Schreiber CW (1996) Redox chemistry of iron-manganese and iron-chromium interactions in soda lime silicate glass melts. Glastech Ber Glass Sci Technol 69:269–277
Schuessler JA, Botcharnikov RE, Behrens H, Misiti V, Freda C (2008) Oxidation state of iron in hydrous phono-tephritic melts. Am Mineral 93:1493–1504
Sugawara T (1999) Experimental techniques to minimize Fe and Na losses in one atmosphere gas mixing furnace. J Min Petrol Econ Geology 94:425–441
Tangeman JA, Lange R, Forman L (2001) Ferric-ferrous equilibria in K2O-FeO-Fe2O3-SiO2 melts. Geochim Cosmochim Acta 65:1809–1819
Temkin M (1945) Mixtures of fused salts as ionic solutions. Acta Phys Chim URSS 20:411–420
Thornber CR, Roeder PL, Foster JR (1980) The effect of composition on the ferric-ferrous ratio in basaltic liquids at atmospheric pressure. Geochim Cosmochim Acta 44:525–532
Toplis MJ (2005) The thermodynamics of iron and magnesium partitioning between olivine and liquid: criteria for assessing and predicting equilibrium in natural and experimental systems. Contrib Mineral Petrol 149:22–39
Toplis MJ, Dingwell DB (1996) The variable influence of P2O5 on the viscosity of melts of differing alkali/aluminium ratio: implications for the structural role of phosphorus in silicate melts. Geochim Cosmochim Acta 60:4107–4121
Toplis MJ, Dingwell DB, Libourel G (1994) The effect of phosphorus on the iron redox ratio, viscosity, and density of an evolved ferro-basalt. Contrib Mineral Petrol 117:293–304
Tsuchiyama A, Nagahara H, Hushiro I (1981) Volatilization of sodium from silicate melt spheres and its application to the formation of chondrules. Geochim Cosmochim Acta 45:1357–1367
Turkdogan ET, Bills PM (1957) A Thermodynamic Study of FeO–Fe2O3–SiO2, FeO–Fe2O3–P2O5 and FeO–Fe2O3–SiO2–P2O5 Molten Systems. J Iron Steel Inst 186:329–339
Watson EB (1980) Apatite and phosphorus in mantle source region: an experimental study of apatite/melt equilibria at pressures to 25 kbar. Earth Planet Sci Lett 51:322–335
Wilson AD (1960) The micro-determination of ferrous iron in silicate minerals by a volumetric and a colorimetric method. Analyst 85:823–827
Acknowledgments
The stay of AB in Hannover was funded by the German Science Foundation (DFG, projects Ho 1337/24-1, Ho 1337/27-1 and Ho 1337/30-1). We thank Eric Wolff for the electron microprobe assistance and Anika Husen and Franziska Adams for the help in the determination of ferric/ferrous ratios in glasses. We are grateful to editor Jochen Hoefs and to the anonymous reviewers for their comments and suggestions that allowed us to improve the paper. We are also thankful to Fabrice Gaillard for his comments on an early version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Hoefs.
Rights and permissions
About this article
Cite this article
Borisov, A., Behrens, H. & Holtz, F. The effect of titanium and phosphorus on ferric/ferrous ratio in silicate melts: an experimental study. Contrib Mineral Petrol 166, 1577–1591 (2013). https://doi.org/10.1007/s00410-013-0943-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-013-0943-9