
Abstract
Hydrothermal volatile-solubility and partitioning experiments were conducted with fluid-saturated haplogranitic melt, H2O, CO2, and S in an internally heated pressure vessel at 900°C and 200 MPa; three additional experiments were conducted with iron-bearing melt. The run-product glasses were analyzed by electron microprobe, FTIR, and SIMS; and they contain ≤0.12 wt% S, ≤0.097 wt% CO2, and ≤6.4 wt% H2O. Apparent values of log f O2 for the experiments at run conditions were computed from the [(S6+)/(S6++S2−)] ratio of the glasses, and they range from NNO −0.4 to NNO + 1.4. The C–O–H–S fluid compositions at run conditions were computed by mass balance, and they contained 22–99 mol% H2O, 0–78 mol% CO2, 0–12 mol% S, and <3 wt% alkalis. Eight S-free experiments were conducted to determine the H2O and CO2 concentrations of melt and fluid compositions and to compare them with prior experimental results for C–O–H fluid-saturated rhyolite melt, and the agreement is excellent. Sulfur partitions very strongly in favor of fluid in all experiments, and the presence of S modifies the fluid compositions, and hence, the CO2 solubilities in coexisting felsic melt. The square of the mole fraction of H2O in melt increases in a linear fashion, from 0.05 to 0.25, with the H2O concentration of the fluid. The mole fraction of CO2 in melt increases linearly, from 0.0003 to 0.0045, with the CO2 concentration of C–O–H–S fluids. Interestingly, the CO2 concentration in melts, involving relatively reduced runs (log f O2 ≤ NNO + 0.3) that contain 2.5–7 mol% S in the fluid, decreases significantly with increasing S in the system. This response to the changing fluid composition causes the H2O and CO2 solubility curve for C–O–H–S fluid-saturated haplogranitic melts at 200 MPa to shift to values near that modeled for C–O–H fluid-saturated, S-free rhyolite melt at 150 MPa. The concentration of S in haplogranitic melt increases in a linear fashion with increasing S in C–O–H–S fluids, but these data show significant dispersion that likely reflects the strong influence of f O2 on S speciation in melt and fluid. Importantly, the partitioning of S between fluid and melt does not vary with the (H2O/H2O + CO2) ratio of the fluid. The fluid-melt partition coefficients for H2O, CO2, and S and the atomic (C/S) ratios of the run-product fluids are virtually identical to thermodynamic constraints on volatile partitioning and the H, S, and C contents of pre-eruptive magmatic fluids and volcanic gases for subduction-related magmatic systems thus confirming our experiments are relevant to natural eruptive systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Explosive volcanism typically involves fluid-saturated, intermediate-silica-content to felsic magmas largely enriched in H2O and containing lesser but variable concentrations of CO2, S, halogens, and other volatile and fluxing constituents. Interpretation of processes of magmatic fluid exsolution, fluid evolution, and subsequent surficial degassing through geochemical research calls for experimental and thermodynamic constraints on the solubilities of H2O, CO2, and S in a variety of silicate melts over a range of mantle and crustal pressures and temperatures. At present, systematic experimental studies of solubilities and partitioning behavior of volatile components in fluid-saturated felsic melts are limited to individual and binary volatile pseudosystems involving melt plus H2O ± CO2 (Holloway and Blank 1994; Tamic et al. 2001; Moore 2008), H2O ± S (Scaillet et al. 1998; Keppler 1999, 2010); Clemente et al. 2004; Burgisser et al. 2008, or H2O-Cl (Webster 1992a, b). Some recent studies, however, offer promise because they involve experimental investigation of O–H ± C ± S ± Cl ± F-bearing fluids and a variety of aluminosilicate melts (Botcharnikov et al. 2004; Spilliaert et al. 2006; Nicholis and Rutherford 2009; Botcharnikov et al. 2005; Webster et al. 2006, 2009; Parat et al. 2008; Teague et al. 2008; Beermann 2010), but most of them have involved basaltic to intermediate-silica melts. Currently, we lack a detailed experimental analysis of volatile solubility behavior in C–O–H–S fluids coexisting with felsic melts at shallow crustal pressure. This report describes the results of experimentally constrained concentrations of H2O, CO2, and S in C–O–H–S fluids and haplogranitic melt at 200 MPa, temperatures of 895–912°C, and apparent log f O2 ranging from NNO −0.4 to NNO + 1.4. This research has also determined the partitioning behavior of H2O, CO2, and S between the melt and fluids as a function of bulk fluid composition and f O2.
Methods
Experimental
A haplogranitic glass—chemically equivalent to the haplogranite minimum-melt composition at PH2O = 200 MPa, having a molar (Al2O3/Na2O + K2O) of 1.0 and a molar (Na2O/Na2O + K2O) of 0.59, and synthesized by Corning Incorporated—was provided for this study by David London (Table 1). The glass was crushed to a fine powder and used for the silicate starting material. Carbon was added to the starting charges as C2H2O .4 2H2O, PdC2O4, or Ag2C2O4. Sulfur was added as elemental sulfur, as an aqueous solution of Na2SO4 and K2SO4 containing 2.5 wt% S and characterized by a molar (Na2O/Na2O + K2O) of 0.6, or as both. For most S-bearing experiments, the CO2 was added as oxalic acid dihydrate to avoid reaction of S with Pd or Ag, but for one S-bearing run (e.g., 1-08-05A), CO2 was added as silver oxalate. Water was added as distilled and de-ionized H2O with or without oxalic acid dihydrate and the aqueous Na2SO4 and K2SO4 solution. Three S-bearing experiments involved iron added as reagent-grade Fe3O4. The twenty-two experiments that included S were conducted in gold capsules, and the eight other S-free runs were conducted in platinum. The calculated (fluid/melt) mass ratios at run conditions ranged from 0.03 to 0.46.
The experiments were conducted in an internally heated pressure vessel (IHPV) at the American Museum of Natural History (AMNH). Pressure was monitored with Heise gauges, and temperature with two chromel–alumel thermocouples the tips of which span the length of the experimental capsules. All experiments were quenched isobarically. The IHPV provides a relatively rapid quench; run temperature decreased from 900° to <500°C in 10 s or less.
For most experiments, temperature ranged from 895 to 912°C and pressure from 199 to 204 MPa, but one S-free run was conducted at 1,100°C. The experiments ranged from 142 to 725 h in duration. During the runs, most capsules were simply exposed to the intrinsic f H2 of the steel walls of the IHPV. The log f O2 imposed by this f H2 is equivalent to NNO + 2 log units at 200 MP, 800°C, and with activity of H2O, a H2O, of unity (as determined with an oxygen sensor), but the final f O2 of most runs was likely lower because the X flH2O were less than unity and, hence, the X H2O was less than unity (described below). For three experiments involving a total of six individual capsules, the capsules were run at more reducing conditions by exposing them to H2 added to the Ar pressure medium via a Shaw membrane. As detailed in the following results description, the apparent log f O2 of most S-bearing runs was calculated from the measured [(S6+)/(S6++S2−)] ratio of the glasses, and it ranges from NNO −0.4 to NNO + 1.4 although most are between NNO and NNO + 0.9. We have no constraints on the f S2 of these experiments.
Regarding the approach to equilibrium, it should be noted that these runs began with S in the starting fluid (±elemental S) and the starting glass contained no S. Equilibrium requires sufficient diffusion of S into the melt (provided by the fluid) to generate a homogeneous melt composition and acquisition of an equilibrium distribution of S in the melt as S−2 and S+6 for the f O2 relevant to each run. Our ca. 900°C-run durations ranged from ca. 6–18 days for most experiments, but one run was conducted for 30 days. In comparison, most of the S-partitioning experiments of Keppler (2010) involving O–H–S fluid-saturated haplogranite melts were conducted for 20–40 days but at lower temperatures (750–850°C at 50–300 MPa). As shown below, our S-partitioning data generally agree with those of Keppler (2010). Moreover, four of our experiments (e.g., runs 1-08-05C, 1-08-06C, 1-08-14A, and 1-10-02B; Table 2) show remarkable consistency in their S partitioning between fluid and melt and in their apparent values of f O2 even though they were conducted for durations ranging from ca. 6 to 30 days. This consistency in the extent of S partitioning over 6–30 days and the general agreement with the data of Keppler (2010) suggest a reasonable approach to equilibrium at these conditions.
Although our experiments were conducted well below the melting point of pure gold, the use of multiple gold capsules, simultaneously, typically led to sintering and adhesion of the capsules to one another, and this made it difficult to separate the capsules without causing leakage after the run. Consequently, we observed microscopic droplets of run-product liquid on the exterior of several of the capsules after the runs were quenched. The presence of liquid droplets indicates that the leakage occurred during and/or after the quench; the fluid should have boiled away if the leakage occurred at T > 100°C. For these runs the glass compositions were analyzed and the fluid compositions calculated by mass balance, even though we could not apply the freeze-and-puncture gravimetric procedure described below.
Analytical
Electron microprobe (EPMA)
The run-product glasses were analyzed for SiO2, Al2O3, Na2O, K2O, and S ± FeO at 15 keV with a Cameca SX-100 electron microprobe at the AMNH (Table 1). In the first analytical pass, Na2O, K2O, and SiO2 ± FeO were analyzed with a beam current of 2 nA, and Al2O3 and S were analyzed at 10 nA in the second pass. The standards for S analysis include troilite and barite depending on the S oxidation state (determined with the method outlined below). Troilite was used for glasses with [(S6+)/(S6++S2−)] < 0.4, and barite for all other, more oxidized glasses. Peak count times varied from 30 to 60 s. All glasses were analyzed with a defocused, 5–10 μm diameter electron beam, and the glasses were moved during analyses in order to minimize Na and K migration. The reported glass compositions represent the average of four to eight analyses of different spots in the same sample. A variety of internal “standard” glasses (e.g., the anhydrous haplogranitic starting glass; Macusani rhyolitic obsidian; and the NBS 610, 620, and 621 reference materials) were analyzed regularly to check for accuracy and precision (i.e., for instrumental drift) during the course of this experimental investigation. Repeated analyses of the NBS 620 glass, known to contain 0.112 ± 0.01 wt% S, returned a mean S concentration of 0.13 ± 0.006 wt% (1σ).
The oxidation state of S, i.e., oxidized (S6+) and reduced (S2−), was determined by measuring the wavelengths of the S K α X-rays. This method works because S6+ emits shorter wavelengths than S2− and because shifts in the wavelength (λ) of S K α X-rays relate directly to changes in oxidation state (Carroll and Rutherford 1988; Matthews et al. 1999). One can deduce the S oxidation state of an unknown glass through comparison of S peak positions for unknowns with reference materials such as BaSO4 and FeS. We analyzed the S K α radiation in the run-product glasses with EPMA wavelength-dispersive spectrometry and an LPET crystal. We counted the X-rays with a spectrometer sin θ range of 0.61158–0.61658 using sin θ steps of 0.00002 and a dwell time of 100 ms/step. We rastered a 6-μm beam diameter over a 30 × 30 μm area to avoid oxidation effects resulting from localized heating due to prolonged beam exposure (Wallace and Carmichael 1994; Métrich and Clocchiatti 1996). The electron beam was set at 15 keV and 40 nA current. We used 8 accumulations on the reference standards BaSO4 and FeS, and 30-scan accumulations per analysis to reduce the noise in the spectrum because some glasses have very low sulfur concentrations. We analyzed each glass in 4–6 locations preferably on different grains and the reference standards in at least 4 locations per minerals in each analytical session. Practically, the estimation of [(S6+)/(S6++S2−)] was difficult for glasses containing <100 ppm S; for example, the [(S6+)/(S6++S2−)] could not be determined for the glasses of runs 1-08-04A, 1-08-04B, and 1-08-05A.
To estimate the oxidation state of S from the λ shifts in the S K α X-rays, we calculated the rel% S6+ via Carroll and Rutherford (1988):
where
and
One calculates the apparent f O2 of the melt:
where \( X_{\text{S}}^{6 + } = \% \left[ {\left( {{\text{S}}^{6 + } } \right)/\left( {{\text{S}}^{6 + } + {\text{S}}^{2 - } } \right)} \right] \) and \( X_{\text{S}}^{ 2- } = \, \% \left[ {\left( {{\text{S}}^{ 2- } } \right)/\left( {{\text{S}}^{ 6+ } + {\text{S}}^{ 2- } } \right)} \right] \) and a = 1.02, b = 25,410, and c = −10.0 (Wallace and Carmichael 1994). It should be noted that the dependence of [(S6+)/(S6++S2−)] on f O2 is quite sensitive; S speciation in melt varies significantly over a very small range in f O2 (Jugo et al. 2010 and references cited therein).
Fourier-transform infrared spectroscopy
The concentrations of total H2O (molecular water + hydroxyl) and CO2 in the run-product glasses were determined with Fourier-Transform Infrared Spectroscopy (FTIR) at the AMNH (Table 1). Doubly polished chips of run-product glass were prepared, and their average thicknesses (e.g., 100–360 μm) were determined with a Mitutoyo digitometer (precision ± 2 μm) taking 6–10 measurements per chip. The measurements were conducted at room temperature in transmittance mode using a Nicolet 20SXB FTIR spectrometer equipped with a Spectra Tech IR Plan microscope. Dry nitrogen gas was added at the rate of 15 l/min. IR spectra were collected both in the mid-IR (1,400–4,000 cm−1) and near-IR (6,500–3,700 cm−1) regions using a KBr beam splitter, a MCT/A detector, and a globar source. For each spectrum, 400 scans were performed and backgrounds were collected for each glass analyzed; the analytical resolution was 4 cm−1. The height of each peak was measured from the baseline using OMNIC software. Three to six analyses were collected in different spots of each polished glass chip, and these locations were selected to avoid vesicles in the path of the infrared beam. Due to the high total water concentrations in most glasses, the 3,550 cm−1 peak was nearly always saturated so in these cases the 4,500 and 5,200 cm−1 peaks were used and the results combined to provide total H2O concentrations. Molecular CO2 was determined at the 2,349 cm−1 position, and the 1,550 cm−1 carbonate position was checked for several run-product glasses but no peaks were detected.
The volatile concentrations are related to the height of the absorbance peak by the Beer-Lambert law, which accounts for the density of the glass, molar absorptivity (i.e., the extinction coefficient) of the glass, as well as the thickness of the glass chips. Glass densities were calculated using the Gladstone-Dale rule (Gladstone and Dale 1864; Silver et al. 1990) and EPMA data; final values of glass densities were iteratively recalculated (at least twice) based on the apparent H2O concentrations used in the first calculation. The associated precision is ±0.03 g/cm3 according to Mandeville et al. (2002). The extinction coefficients that were used for the 5,200 and 4,500 cm−1 peaks are 1.79 ± 0.02 and 1.56 ± 0.02 l cm−1/mole for H2O and OH− in rhyolitic glasses, respectively (Nowak and Behrens 1997). The absorption coefficient used for molecular CO2 is 1,214 ± 78 l cm−1/mole for the 2,349 cm−1 peak in rhyolitic glasses (Behrens et al. 2004). Background subtraction was conducted with short tangential line segment approximations of the background. To test the accuracy of these analyses, two rhyolitic glasses—synthesized and originally analyzed by FTIR at the University of Hannover, Germany—were analyzed by FTIR at the AMNH. The total H2O contents determined at Hannover for these glasses were 1.32 and 2.46 wt%, and our analyses gave 1.31 and 2.41 wt%, respectively.
Secondary ion mass spectrometry
The S concentrations of 13 felsic run-product glasses were determined with SIMS (Cameca 1,280 at the Woods Hole Oceanographic Institution) to serve as a comparison and test with the S concentrations determined by EPMA (Table 1). We also analyzed and compared data for several other hydrous, S-bearing granitic glasses not generated for this study, and all data are shown in Fig. 1. A primary beam of 133Cs+ ions with currents between 1.0 and 1.5 nA was focused to a spot of ~10 μm in diameter and rastered over a 30 × 30 μm area, and a mechanical field aperture was placed at the position of the secondary ion image focal plane, such that only the central area of 15 × 15 μm was analyzed. We used a normal-incidence electron gun to compensate for the positive electrical charge deposited by the 133Cs+ ion beam.

Plot exhibiting good agreement between the sulfur concentration (wt%) of haplogranite run-product glasses of this study (circles) and other granitic glasses (triangles) measured by secondary ion mass spectrometry (SIMS) versus electron microprobe (EPMA). See text for description of “Methods”; precision shown is 1−σ
For each analytical cycle, the glasses were pre-cleaned by sputtering for a period of 4 min, secondary ion intensities of 30Si− and 32S− were measured, and this cycle repeated 10 times with a total analysis time of ~20 min. Intensity ratios of 32S− against 30Si− were averaged for 10 cycles. The NBS610 standard glass was analyzed 4 times, and a linear working curve was constructed through these data and the origin. This curve involves normalization of the (32S/30Si) for differences in bulk-silica content of the glasses and provides the calibration equation:
Using these instrument conditions, prior work indicates a detection limit for S of at least 7 ppb (Rose-Koga in review). The agreement for S content between the EPMA and SIMS measurements is good. In addition, SIMS analyses accumulate tens of thousands of counts for S in a 5-second analysis of a glass containing several hundred ppm S, and with the number of S counts, n = 10,000, the (n 1/2/n) estimate for precision based on count statistics gives 1% relative precision. This degree of precision contrasts with that determined by EPMA. For example, the total S counts range from 100 to 300 for glasses containing 100 to 600 ppm S, respectively, using the EPMA conditions described previously. With n = 300 S counts, the associated precision is 6 rel%, and with n = 100, the estimated precision for S by EPMA is 10 rel%, so the theoretical analytical variability for an individual analysis is considerably less for SIMS (summarized by Ripley et al. in press).
Estimating the fluid compositions
We assume that each experiment involved only one fluid phase in addition to silicate melt, and we computed the fluid composition at run conditions (Table 2) using the mass-balance approach of Webster (1992a, b) and Webster et al. (2009). This method involves computing the mass of the melt using the quantity of starting glass powder and accounting for the dissolution of H2O, S, Na2SO4, and K2SO4 into the melt from the elemental S and fluids in the starting charge. It assumes that all S added to the starting charges dissolved in the melt and fluid. The mass of bulk fluid was calculated by accounting for the masses of H2O, S, Na2SO4, and K2SO4 not sequestered by the melt and thus available for the fluid. The method also involves an iterative approach in computing the melt and fluid masses until the errors on bulk-system mass approach acceptable minima (i.e., when the observed change in fluid composition, between successive calculations, was <1 rel%).
Raman spectra of the dominant volatile-component species in the aqueous fluid-bearing vesicles of nine of the quenched, S-bearing run-product glasses were collected in the laboratory of Robert Bodnar at the Virginia Polytechnic Institute and State University (see Berkesi et al. 2009 for methods). Raman spectroscopy shows H2O in all vesicles, and the fluid-bearing vesicles of seven of these run products contained CO2. Six of the run-product glasses showed detectable H2S in vesicles, and vesicles of two glasses showed measurable SO2. None of the vesicles contained detectable CO or CH4 at the estimated limit of detection of approximately one mol% (Robert Bodnar, personal communication).
The presence of CO and CH4 in experimentally generated fluids can have a significant on volatile solubilities in melt at reducing f O2. A recent study of C–O–H fluids and haplobasaltic melts at 200–300 MPa and 1,250°C observed that the CO2 solubilities in melt were reduced by the presence of CO and CH4 in the fluid phase (Morizet et al. 2010). It is noteworthy that those experiments of Morizet et al. (2010) showing reduced CO2 solubility in melt were conducted at relatively low log f O2 (ca. NNO −3.2 to NNO −1.6), and that their runs at relatively higher f O2 did not demonstrate this relationship. The lack of CO or CH4 in the quenched run-product fluids in vesicles of our runs, determined by Raman analysis, suggests that neither of these species may have been present (at this limit of detection) at run conditions, but this argument assumes that no back reactions occur in the fluids during or after the quench. In this regard, Lin and Bodnar (2010) demonstrated that S-free, CH4-bearing aqueous fluids can be quenched successfully without detectable CH4 breakdown. Our run-product fluids, however, contained oxidized and reduced S species, and conceivably the reduction of SO2 by CO and/or CH4 could consume these reduces C species during the quench, via:
Since we do not know if reactions like these consumed any CO and/or CH4 that may, conceivably, have been present prior to the quench, we must rely on other evidence, discussed below, about the likelihood of these reduced C species in our relatively reduced runs.
To calculate fluid compositions, we have assumed that the only quantitatively significant C-bearing species in the fluid at run conditions was CO2. Our experiments were conducted at significantly higher f O2 and at significantly lower temperatures than those of Morizet et al. (2010). It has been demonstrated that the (CO/CO2) of the fluid correlates with temperature (Holloway and Blank 1994), so the lower temperature runs of our study means that they were less likely to contain CO (than the runs of Morizet et al. 2010). Moreover, we calculated the maximum concentrations of CO and CH4 in the fluids of our runs using PELE (available at: http://www.nicholas.duke.edu/people/faculty/boudreau/DownLoads.html) for a COHS fluid in equilibrium with a felsic melt at the pressure–temperature conditions of our S-bearing runs. The maximum concentration of CO in these modelled fluids was 0.6 mol% with a molar (CO2/H2O) of 1.1 in the fluid and at log f O2 of NNO −1.6 (below that of our runs), and was 0.3 mol% at log f O2 of NNO with a molar (CO2/H2O) of 1.2. The CH4 content of all of the modelled fluids was less than 0.01 mol% with log f O2 of NNO −1.6 to NNO. Given that our S-bearing runs were conducted with the log f O2 varying from NNO −0.4 to NNO + 1.4, the concentrations of CO and CH4 are deemed negligible. In addition, it should be noted that the resulting data addressed herein do not account for the N2 in the air present in the starting charge and capsule, but calculations by Tamic et al. (2001) estimate the X fluidN2 < 0.04 for experimental conditions similar to those of this study.
At present, there is no simple, readily available, and dependably accurate means of directly measuring the quantity of S in quenched run-product fluids (see summary in Webster and Botcharnikov in press). It is also worthy of mention that few prior experimental investigations have attempted to address the solubility behavior of H2O, CO2, and volatile S compounds in aluminosilicate melts simultaneously. Consequently, in order to evaluate the mass-balance calculations on these run fluids, we estimated the quantities of CO2 ± other gaseous compounds and quantity of liquid H2O in the run products at ambient conditions with the following gravimetric procedure. After each run, the exteriors of capsules were cleaned with reagent-grade alcohol and the capsule masses checked to confirm that the capsules had not lost volatiles by leakage during the experiment. Those capsules that had not leaked were cooled overnight to <−10°C in order to freeze the aqueous liquid still containing minor quantities of alkali sulfates, and the capsules were punctured and their masses recorded. The difference between the capsule mass after the run and the mass after the capsule freeze-and-puncture represents the quantity of CO2 ± other volatile compounds (e.g., potentially H2, SO2, H2S, SO3) that did not solidify at the freezing temperature of salt-bearing H2O and, hence, were released with the CO2-dominated vapor when the capsule was punctured. Recall that the fluid of the S-bearing runs must contain some Na, K, and S as well as H2O with or without dissolved CO2. Subsequently, the opened capsules were heated to 115°C for at least 30 min, and the masses of the capsules recorded. This second mass difference (between that after the freeze and puncture and that after heating) represents the quantity of H2O in the residual liquid that was vaporized from the capsule. These mass-difference measurements do not include the small quantity of volatiles trapped in fluid inclusions (vesicles in glass) nor the small masses of solid, residual Na2SO4 and K2SO4 in the heated run products. We note that trace coatings of salts were observed on the surfaces of run-product glass when the heated-and-dried capsules were subsequently cut open and the residual glass extracted for analysis.
In this regard, we have compared the total masses of H2O and CO2 added to the starting charges (based on the masses of compounds added to the capsules prior to welding) with these post-run gravimetric measurements (Fig. 2). Figure 2a compares the former value for H2O with the sum of the mass-difference-measured-after-heating and the computed mass of H2O in the run-product glass (based on the measured concentration of H2O in the glass and the calculated mass of melt at run conditions). This latter value should be a good indication of the total quantity of H2O in the run products (not accounting for the H2O in vesicles/fluid inclusions), and the agreement is good for the great majority of the experiments. Similarly, the total mass of CO2 added to the starting charges is compared with the sum of the mass-difference-after-freeze-and-puncture of the capsule and the computed mass of CO2 in the run-product glass (Fig. 2b), and here the comparison is quite good. It should be noted this agreement also indicates that very little S was released as volatile compounds in vapor while the frozen capsule was punctured, and therefore, that most of the S was contained in the silicate glass and the frozen liquid portion of the run products. In other words, these measurements show that measurably little of the S in the run products occurred as SO2, H2S, or SO3 in vapor and that S was largely contained by the frozen run-product liquid after the quench. We do note that some capsules emitted a strong H2S odor when punctured.

Plot showing a the sum of the measured mass change after previously frozen and punctured run-product capsules of C–O–H–S-haplogranitic melt experiments were heated to 115°C (equivalent to mass of H2O in run-product fluid) plus the mass of H2O sequestered by the melt versus the total mass of H2O added to the starting charge in the capsule. Squares represent C–O–H experiments and circles represent C–O–H–S runs. This comparison shows agreement for most experiments which is consistent with good analytical accuracy for H2O in the glass, small weighing errors, and correct compositions of H2O-bearing compounds in the starting charges. Plot b shows the sum of the measured mass change after run-product capsules were frozen at <−10°C and punctured (equivalent to mass of CO2 in run-product fluid) plus the mass of CO2 sequestered by the melt versus the total mass of CO2 added to starting charge. This step preceded the procedure described for a. This comparison shows good agreement for most experiments which is consistent with good analytical accuracy for CO2 in the glass, small weighing errors, and correct compositions of CO2-bearing compounds in the starting charges. See text for details on “Methods”; precision shown is 1−σ
Prior research on the behavior of S in high-temperature experiments indicates that S may be incorporated into gold capsules for run durations ≥120 h. Botcharnikov et al. (2004) observed that the distribution of S is heterogeneous in gold capsules after the runs are quenched, and they report that the maximum concentrations of S in the capsules are <500 ppm. Conversely, Keppler (2010) ran tests with S-bearing, fluid-only runs in gold capsules to evaluate the extent of S loss to gold during experiments as long as 15 days, and he noted a small reduction in the S contents of the fluids but interpreted this change to difficulty in sampling the run-product fluids and not to loss of S to the gold capsules. Given the inherent difficulties associated with quantitatively estimating the quantity of S lost from the charges to the capsule material of our experiments, our calculations do not account for the potential loss of S to the capsules and, hence, the reported S concentrations of fluids may represent maximum values. Prior study (Mysen and Popp 1980) has also determined that the activity of S, a S, in runs can be controlled or buffered by the presence of Me-MeS x assemblages in which Me = Cu, Ag, or Pt. Hence, for run 1-08-05A it is likely that the a S was controlled at some unconstrained value because of the presence of Ag (from the silver oxalate) and S (from the added Na2SO4 and K2SO4), and similarly for the Fe-bearing runs 1-10-15 A,B,C the a S was likely modified by the reaction of Fe with reduced S. As a result, we report only the starting composition of the fluid for these runs (Table 2).
Results
The volatile-bearing run-product glasses are variably vesicular and are treated as compositionally equivalent to the melts in equilibrium with fluid at run conditions (Fig. 3). All experiments conducted, except for the three Fe-bearing runs and one unreported experiment, contain no crystals. The former runs include magnetite in the run products. The latter unreported run was conducted with a pure-CO2 fluid, was crystal rich and melt poor, and hence, analysis of the glass was problematic. The molar (Al2O3/Na2O + K2O) ratio of the glasses ranges from 0.95 to 1.06 and the molar (Na2O/Na2O + K2O) ranges from 0.55 to 0.61. These subtle compositional variations are due to the variable extraction of alkalis from the melt by S-bearing fluids (B. Evans, personal communication.; Webster et al. 2009; Beermann 2010).

Schematic representation of heterogeneous phase equilibria of this study of the pseudo-ternary system involving haplogranitic melt and C–O–H ± S-bearing fluids (expressed by tie lines shown as faint straight curves) projected on S-free basal plane for melt + H2O + CO2 based on 200-MPa molar abundances of volatiles in fluids and melt (i.e., [H2O + CO2]-bearing melt shown as solid curves, [H2O + CO2 + relatively oxidized S as dot-dashed curve], and [H2O + CO2 + relatively reduced S] as dotted curve). Arrows express the observed shift in the topological saturation surface for melt + COH-fluid with addition of reduced S to system and a potential shift in the saturation surface suggested by data of this study and indicated by those of Moretti and Baker (2008)
The glasses contain ≤0.12 wt% S, ≤0.097 wt% CO2, and ≤6.4 wt% H2O. The abundances of S determined by SIMS agree well with the EPMA data (Fig. 1), and all references to the S contents of run-product glasses in the discussion that follow refer to S determined by EPMA. The [(S6+)/(S6++S2−)] of these glasses ranges from 0.12 to 0.91, and the log f O2 of most S-bearing runs computed for pressure of 200 MPa varies from NNO −0.4 to NNO + 1.4. It should be noted that recent modeling of sulfide-saturated rhyolitic melts at these P–T conditions (Moretti and Baker 2008) shows that the relationship involving f O2 and [(S6+)/(S6++S2−)] (Carroll and Rutherford 1988) varies with pressure and a H2O in the system, but we cannot account for the influence of changing a H2O on the apparent f O2 values reported herein given that the a H2O of the melts is unconstrained.
Calculations show the fluids contained from ca. 10–99 wt% H2O (22–99 mol%), 0–90 wt% CO2 (0–78 mol%), 0–18 wt% S (0–12 mol%), and <3 wt% alkalis at 200 MPa and 900°C (Table 2). The associated volatile-solubility behavior in the felsic melt ranges from simple to complex (Fig. 4). For all S-free runs, the square of the mole fractions of H2O of the glasses increases linearly with the modeled H2O content of the fluid. Moreover, the H2O contents of nearly all S-bearing glasses show greater dispersion (Fig. 4a). The solubility behavior of CO2 in melt is more complex (Fig. 4b). For H2O–CO2-only runs and for the relatively oxidized C–O–H–S experiments having [(S6+)/(S6++S2−)] ≥ 0.4, the molar concentration of CO2 in the glasses increases with the modeled molar CO2 content of the fluid and this correlation is quite robust. Comparison of these data with those for the relatively reduced S-bearing experiments having [(S6+)/(S6++S2−)]<0.4, however, indicates that there is distinctly less CO2 dissolved in these glasses for given concentrations of CO2 in the S-bearing fluids. Thus, S dissolved primarily as S2− in melt is associated with reduced CO2 contents of C–O–H–S fluid-saturated felsic melts. The behavior of S in melt is similar to that of H2O, because increasing concentrations of S in the fluid cause increasing S in the melt (Fig. 4c), but the relationship for S exhibits greater dispersion than that shown for H2O (Fig. 4a).

Plot showing a the computed mol% H2O of the fluid versus the square of the computed mole fraction of H2O in haplogranitic melt (X meltH2O )2 based on measured H2O in run-product glass) at 200 MPa and 900°C. Squares represent C–O–H experiments, solid diamonds represent C–O–H–S runs at unconstrained f O2, open circles represent C–O–H–S runs at ca. log f O2 > NNO + 0.3, and filled circles show C–O–H–S runs at ca. log f O2 ≤ NNO + 0.3. Data show that S-free C–O–H runs are consistent with linear increase in the (X meltH2O )2with increasing H2O in the fluid, and the partitioning of H2O between C–O–H–S fluid and melt is independent of the (S/S + CO2) of most runs. Plot b shows the computed mol% CO2 of the fluid versus the computed X meltCO2 (based on measured CO2 in run-product glass) at run conditions. Data show S-free runs are consistent with a linear increase in CO2 concentration of melt with increasing CO2 in the fluid, and the partitioning of CO2 between fluid and melt is independent of the (S/S + CO2 + H2O) of most relatively oxidized runs. Plot c shows the computed S concentration of the fluid versus the computed X meltS (based on measured S in run-product glass) at run conditions. Data show more dispersion than (a) or (b), that most runs are consistent with increasing S in melt resulting from increasing S in fluid, and that the distribution of S between most melts and fluids does not vary detectably with (CO2/CO2 + H2O). Precision shown is 1−σ
Discussion
Degassing of felsic magmas: volatile partitioning between C–O–H–S fluids and melt
The abundances of the primary volatile components C, H, and S and their ratios in pre-eruptive magmatic fluids and eruptive volcanic gases provide important geochemical constraints on processes of degassing and information valuable for interpreting eruptive behavior, and our data represent results of the first experimental investigation of volatile dissolution behavior in C–O–H–S fluid-saturated felsic melts. The granitic-rhyolitic composition of the melts of this study is particularly relevant to volcanic processes because most explosive activity involves relatively evolved, silica-enriched, highly polymerized aluminosilicate melts (Cioni et al. 2000).
It is instructive to compare our experimental results with modeled and measured compositions of natural C–O–H–S-dominated fluids associated with felsic to andesitic magmas of subduction-related and other volcanic environments (Table 2). Scaillet and Pichavant (2003) determined that the X fluidH2O ranges from 0.65 to 0.95, X fluidCO2 ranges from 0 to 0.3, and that X fluidS is typically <0.05 for the pre-eruptive vapor in magmas of these geologic environments (i.e., X fluidS represents the sum of all sulfur species dissolved in the fluid in this discussion). These values are compatible with the ranges of the same parameters for most of our experiments. Eleven of our S-bearing runs have X fluidS < 0.05, four others have X fluidS between 0.05 and 0.07, and the remaining two have X fluidS of ca. 0.1–0.12. All but four experiments have X fluidCO2 < 0.3. Moreover, the range in log f O2 for felsic to intermediate-silica magmas of these natural systems is ca. NNO −0.5 to NNO + 2 (Scaillet and Pichavant 2003; Burgisser et al. 2008) which overlaps that of our experiments. In addition, if we compare the (C/S) ratios of the fluids of our experiments with those of these natural volcanic gases (Scaillet and Pichavant 2003, 2005), both sets of data are highly consistent with one another. Our results are also compatible with other constraints on natural systems. The atomic (C/S) ratios reported by Scaillet and Pichavant (2003, 2005) are consistent with the summary of Wallace and Edmonds (in press) in that the molar (C/S) of volcanic gases from volcanoes of arc environments tend to fall between 1 and 30. Our experimental molar (C/S) ranges from 0.8 to 34 (Table 2) and also shows that the bulk S content of the runs influences these parameters.
The experimentally determined fluid-melt partitioning behavior of these volatiles is also highly congruent with the modeled behavior for natural felsic to intermediate magmas (Fig. 5). The majority of these runs show similar correlations between increasing concentrations of CO2, S, and H2O in the experimental fluids and increased CO2, S, and H2O of the melts, respectively (Fig. 4), and these trends are captured by the distribution coefficients D CO2, D S, and D H2O expressing the partitioning of these volatile components between fluid and haplogranitic melt (reported in Table 2 and Fig. 5). Values of D H2O are generally near 10, and they increase with increasing H2O in the melt. This relationship implies that H2O partitioning behavior is non-Nernstian, because H2O partitions increasingly in favor of the fluid as the X H2O of the system increases. D S is generally an order-of-magnitude greater than D H2O and shows greater variability, but D S does not correlate with H2O in the melt. The values of D CO2 are generally an order-of-magnitude greater than those of D S, exhibit no clear relationship with the H2O content of the melt, and also show greater variability than D H2O. Moreover, the plots for D CO2 and D S involve outliers that differ significantly from the bulk of the data. The causes for the scatter involving D S are undetermined, but they likely reflect the variability in oxygen fugacity and may also partially reflect the effect of non-ideal mixing relationships for these volatiles in the melt and fluid. In comparison, Scaillet and Pichavant (2003) report apparent values of D CO2, D S, and D H2O that are based on the modeled fluid compositions described above and the volatile abundances of glasses in silicate melt inclusions from these systems. The general order: D CO2 > D S > D H2O is expressed by both data sets, and the approximate order-of-magnitude difference between each partition coefficient is also supported. This general degassing sequence is consistent with other well-established observations on volcanic gases (Oppenheimer 2003).

Plots of the partition coefficients (wt% in C–O–H–S fluid/wt% in melt) for CO2 (diamonds), S (triangles), and H2O (circles) as a function of the H2O concentration of haplogranitic melt at 200 MPa and 900°C showing D CO2 > D S > D H2O. D H2O increases with increasing H2O in the melt, but D CO2 and D S show no clear relationship with H2O content of melt. Arrows designate the relative partitioning behavior of these volatile components in natural pre-eruptive magmatic fluids of subduction-related magmas and magmas of other tectonic environments characterized by Scaillet and Pichavant (2003); the experimental data agree with those characterizing natural systems
At present, there are few experimental data detailing the partitioning of volatiles in C–O–H–S-bearing felsic systems, because most prior experiments only involved melt-H2O–CO2 or melt-H2O–S. We do note that Gorbachev (1990) summarizes volatile solubilities for C–O–H–S-bearing basaltic and andesitic melts and Teague et al. (2008) report S partitioning data for C–O–H–S-bearing andesitic melt at 200–800 MPa. These studies reported that S is highly soluble in mixed CO2–H2O fluids. In another experimental study, Parat et al. (2008) reacted andesite melt with C–O–H–S fluids at 400 MPa and observed that S modifies the proportions of H2O and CO2 in the melt as compared with S-free systems but they did not determine values of D S.
Given the lack of appropriate experimental constraints for this system, it is instructive to revisit the partitioning behavior for H2O, CO2, and S between fluid and haplogranitic melt (Figs. 4, 6). Figure 4a compares the H2O concentrations of our S-free haplogranitic melts with the H2O concentrations of S-bearing haplogranitic melts at 200 MPa. The latter data show a linear increase in the (X meltH2O )2 with increasing H2O in the fluid that is statistically consistent with our S-free runs, at the 1-σ level of precision. It is noteworthy that several of the S-bearing runs involving the most H2O-enriched fluids, i.e., those with the least CO2 and with (X meltH2O )2 > 0.22, suggest comparatively elevated solubility of H2O in melt compared with S-free melts. This solubility relationship was observed previously in modeling of Moretti and Papale (2004) which predicts that the 200-MPa solubility of H2O in rhyolitic melt increases by ca. 0.3 wt% as the (f SO2/f H2S) increases from 0.1 to 10 (at 827°C). Although this apparent difference in H2O solubility behavior (Fig. 4a) is not statistically valid, it is consistent with prior results and deserves additional experimental study.

Plots showing experimental results at 200 MPa and 900° C of this study a mol% CO2 in fluid versus the mole fraction of CO2 in melt for S-free, H2O–CO2 fluids (filled squares fit by fine solid line), O–H–S ± C fluids with unconstrained f O2 (filled diamonds), O–H–S ± C fluids with log f O2 > NNO + 0.3 (open circles), O–H–S–C fluids with log f O2 ≤ NNO + 0.3 (filled circles fit by bold solid curve), and—for comparison—squares containing x’s represent modeled data for S-free, C–O–H runs of Liu et al. (2005) fit by dashed line. Plot b shows wt% S in C–O–H–S fluids versus wt% S in haplogranite melts of this study fit by bold line compared with S distribution between O–H–S fluid and haplogranite melt (at 200 MPa, 850°C, and log f O2 equivalent to Co-CoO buffer [filled squares fit by fine line] and log f O2 in range of NNO + 0.5 to NNO + 1 [filled circles fit by double line] of [Keppler 2010]). Open crosses represent S-free, C–O–H runs of this study, triangles represent C–O–H–S runs of this study with log f O2 > NNO + 0.3 or unconstrained, and ellipses show O–H–S–C runs of this study with log f O2 ≤ NNO + 0.3. Precision shown is 1−σ
We also compare the partitioning of CO2 between melts and fluids of our runs with that of the S-free experiments of Liu et al. (2005) for rhyolitic melts at 200 MPa and 900°C (Fig. 6a), and the X meltCO2 increases with increasing abundances of CO2 in the coexisting C–O–H fluid for both data sets. Prior investigations involving felsic melts have either observed or assumed the CO2–H2O mixing behavior in the fluid to be fit with a linear curve (i.e., expressing ideal behavior) for S-free conditions and pressures ≤200 MPa (Blank et al. 1993; Newman and Lowenstern 2002; see summary of Moore 2008). In this context, most of our S-free runs and most of our relatively oxidized S-bearing runs are consistent, within precision, with a linear mixing curve for the fluid and Henrian-solubility behavior for CO2 in melt. Conversely, particularly strong curvature—that expresses the dramatic reduction in the CO2 concentration of these felsic melts—is associated with the addition of S at f O2 ≤ NNO + 0.3 and with <50 mol% CO2 in the fluid.
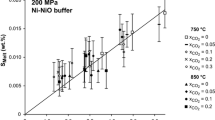
The equilibrium distribution of S between granite melt and C–O–H–S fluids shows significant dispersion that precludes robust interpretation of the mixing behavior in fluid and the corresponding solubility behavior in melt (Fig. 6b). Sulfur partitioning does vary with f O2 as reported previously for C-free granitic systems (Scaillet et al. 1998; Keppler 1999; Scaillet and Pichavant 2003, 2005; Clemente et al. 2004; Keppler 2010), but it does not vary with the (CO2/CO2 + H2O)fluid. Our data are compared in Fig. 6b with the partitioning behavior of S for O–H–S fluids and haplogranitic melts at 200 MPa and 850°C with f O2 equal to that of the Co–CoO buffer and also in the range of ca. NNO + 0.5 to NNO + 1 log units (Keppler 1999, 2010). Even though our data exhibit significant scatter, the S contents of fluid and coexisting melt for our C-free experiments are consistent with those of our C-bearing runs, so D S is indeed independent of (CO2/CO2 + H2O)fluid with felsic melts at these conditions. Thus, sulfur solubility in aqueous fluids is similar to that in aqueous-carbonic fluids at 200 MPa. This relationship has important implications for S degassing, since CO2 shows the strongest partitioning in favor of fluid (Fig. 5), and hence, CO2-enriched fluid tends to exsolve earlier (Holloway 1987) and deeper than other volatiles during magma ascent and evolution in the crust. It follows that H2O + CO2 fluids are likely to exhibit a significant capacity to dissolve and carry S at higher pressures (Oppenheimer 2003), and this is consistent with current experimental research for mafic and intermediate magmas (Teague et al. 2008; Lesne et al. in press). Interestingly, the bulk of our data lie between and show some consistency with Keppler’s (2010) data for S-free runs. Our relatively oxidized runs lie near Keppler’s NNO + 0.5 to NNO + 1 runs, and our four most-reduced runs lie nearer to the Co–CoO curve of Keppler (2010). These associations indicate a generally consistent distribution of S between melts plus aqueous fluids and melts plus aqueous-carbonic fluids as a function of apparent f O2 (even though the f O2 constrained from the [(S6+)/(S6++S2−)] of some of our glasses may involve significant error).
Sulfur and CO2 and H2O solubility in felsic melt
It is well established from prior experimental investigations (Holloway 1987; Fogel and Rutherford 1990; Holloway and Blank 1994; Botcharnikov et al. 2005; Moore 2008 and references therein) that the dissolution of H2O and CO2 in fluid-saturated granitic melts varies with pressure and the (H2O/H2O + CO2)fluid. Currently, there are multiple sets of FTIR extinction coefficients for the analysis of H2O and CO2 and multiple solubility models for these components in felsic glasses and melts, respectively. We analyzed the seven 900° S-free haplogranite glasses using the extinction coefficients of Behrens et al. (2004) for CO2 and Nowak and Behrens (1995) for H2O and OH− (Fig. 7). The measured H2O and CO2 concentrations of the run-product glasses are compared with modeled solubilities of these volatiles in S-free rhyolitic melt at 100–200 MPa (Newman and Lowenstern 2002), and the two data sets correspond well with one another. This consistency suggests that the IHPV quench rate and the FTIR analyses for H2O and CO2 in this investigation were adequate.

Plot showing experimental results at 200 MPa and 900°C of this study and comparing the wt% CO2 in C–O–H–S fluid-saturated haplogranite and Fe-bearing haplogranitic melt versus the wt% H2O in same melts; symbols are the same as in Fig. 6 for runs with haplogranite melt except for three iron-bearing “haplogranitic” melts shown as large filled circles. Experimental data are compared with computed CO2 and H2O concentrations of vapor-saturated rhyolite melt at 900°C and 200, 150, and 100 MPa (fine crosses) based on modeling of Newman and Lowenstern (2002). The presence of >0.02 wt% S in melt and >3 wt% S in fluid (with log f O2 ≤ NNO + 0.3) reduce CO2 solubilities in 200-MPa melts to values approximately equivalent to those for S-free, C–O–H-fluid-saturated melts at 150 MPa. Precision shown is 1−σ; see text for “Discussion”
The other experiments show that the concentration of CO2 in O–H–S ± C-bearing fluid-saturated haplogranite melts varies with the S concentration of the system and f O2 (Figs. 7, 8a). The presence of S, dissolved in melt largely in the reduced state, decreases the volatile saturation pressure for C–O–H–S-bearing melts relative to that of C–O–H-bearing melts and that of S-bearing melts at relatively oxidizing conditions (Fig. 7). For most S-bearing runs with [(S6+)/(S6++S2−)]glass < 0.4, the 200-MPa H2O and CO2 concentrations of melt are located nearer to those of the 150-MPa C–O–H volatile solubility curve of Newman and Lowenstern (2002). This contrasts with the apparent influence of oxidized S on CO2 solubilities.

Plots comparing a predicted mole fraction of CO2 in C–O–H melts and C–O–H–S melts, on a S-free basis, versus the measured mole fraction of CO2 in all melts at 200 MPa and 900°C, and b predicted mole fraction of H2O in C–O–H melts and C–O–H–S melts, on a S-free basis, versus the measured mole fraction of H2O in all melts at 200 MPa and 900°C for C–O–H ± S fluids (symbols same as in Fig. 6). Predicted CO2 and H2O concentrations derived from experiments and volatile solubility model of Liu et al. (2005) for S-free experiments at 200 MPa and 900°C as described in text. Arrows in a show range in fluid composition (e.g., mol% CO2) and allow comparison of predicted CO2 in melt for S-bearing and S-free runs. Arrows in b show fluid composition (e.g., mol% H2O) and allow comparison of predicted H2O in melt for S-bearing and S-free runs
This solubility relationship has important implications for geobarometric interpretation of the pressures of melt inclusion entrapment and for degassing behavior of C–O–H–S-bearing magmas. This application of H2O and CO2 solubilities is reviewed for S-free conditions by Moore (2008), Métrich and Wallace (2008), and references cited therein. A detailed discussion of this approach is out of scope for this investigation, but nevertheless our results for felsic melts imply that the determination of C–O–H–S solubilities for basaltic and intermediate-silica content melts should be expanded to cover a broad range of crustal pressures and temperatures (i.e., beyond the initial results of Gorbachev (1990) and Teague et al. (2008)).
Thermodynamic considerations on volatile mixing in C–O–H–S fluid-saturated melts
In systems involving heterogeneous fluid-melt equilibria, the activities of volatile components in fluid-saturated silicate melts are influenced by the concentrations of the volatiles in coexisting fluid(s) and by the ideal versus non-ideal nature of volatile mixing in the melt and fluid. For instance, increasing the abundance of CO2 in an aqueous fluid will reduce the X flH2O and X meltH2O by dilution and will, likewise, reduce the a flH2O and a meltH2O as long as there is negligible non-ideal mixing behavior in the fluid (i.e., γCO2 and γH2O change little during mixing). At 200 MPa and 900°C for example, H2O and CO2 exhibit small positive deviations from ideal mixing with S-free fluids (Kerrick and Jacobs 1981); the model of Aranovich and Newton (1999) indicates, with X flH2O and X flCO2 of 0.5, that the a flH2O is 0.56 and the a flCO2 and is 0.538.
A detailed analysis of the S-bearing fluids and coexisting melts of the present study provides additional constraints on volatile dissolution behavior. The shift in the volatile concentrations of the melts as a function of increasing reduced S2− in the melt, for example, is due primarily to reduced CO2 in the melt; the presence of reduced S in the system does not appear to alter the solubility of H2O (Fig. 8a). This graph compares the measured CO2 contents of the C–O–H melts with predicted values of CO2 in melt that account for the effects of dilution by H2O. For these S-free runs, we used the computed CO2 concentrations of the fluids and the volatile solubility data of Liu et al. (2005) to model the CO2 solubility in rhyolite melt at 200 MPa and 900°C. This accounts for dilution of CO2 in the fluid-melt system, by the addition of H2O to the starting charges, using the general solubility model of Liu et al. (2005) that is based on experiments covering a range of pressure and temperature. Figure 8a also compares the measured CO2 contents of the C–O–H–S melts with the predicted CO2 in melt for the C–O–H–S melts on a S-free basis. For this comparison, we summed the number of moles of S (i.e., total S) in the fluid with the number of moles of H2O in the fluid in order to reduce the moles of CO2 in the fluid, and then employed this modified fluid composition and the volatile solubility data of Liu et al. (2005) to model the corresponding CO2 solubility in the melt. This approach treats each mole of S in fluid as equivalent to one mole of H2O in fluid, with regard to their capacity to dilute the CO2 in the system. This comparison shows that the predicted CO2 contents of the melts in the majority of the S-free runs and in the comparatively oxidized S-bearing runs are equivalent to their measured CO2 concentrations. Conversely, the measured CO2 contents of three of the relatively reduced S-bearing melts and the two S-bearing melts with unconstrained f O2 are significantly less than those predicted for these melts on a sulfur-free basis (i.e., calculated as if in equilibrium with C–O–H-only fluids). This difference reflects the influence of reduced S in fluid-saturated systems on CO2 dissolution in melt.
The corresponding graph of measured versus predicted H2O in the melts (Fig. 8b) shows that the H2O contents of melts of the S-bearing runs (whether relatively oxidized or reduced) as well as those of the S-free, C–O–H-bearing melts either fit or lie near the 1:1 correspondence curve. In fact, except for a single outlier involving a relatively reduced S-bearing melt, the data show remarkable consistency. This general agreement implies that each mole of CO2 in the fluid behaves similarly to that of the moles of oxidized and reduced S species in the fluid with regard to dilution of H2O, and at these conditions, the S in fluid should occur primarily as SO2 plus sulfide, and potentially, sulfate species (Carroll and Webster 1994). These data also confirm that the observed changes in volatile solubility (i.e., the shifts in the 200-MPa data in Fig. 7 toward the 150-MPa solubility curve) are primarily the result of changing CO2 concentrations with little modification of the H2O contents.
Theoretically, the addition of S to the mixed H2O- and CO2-bearing fluids of these experiments should reduce the X flH2O and the X flCO2 by dilution (Fig. 4) just as the addition of CO2 to the fluid reduces the H2O concentration of the melt (the (X meltH2O )2 decreases in a linear fashion with the X flH2O for a decrease in the moe% H2O in the fluid). Recall that the H2O and CO2 concentrations of these fluids were diluted with as much as 12 mol% S. This behavior has been observed in prior research; for example, in hydrothermal experiments involving andesite melt-C–O–H–S (Parat et al. 2008). The addition of S to their 400-MPa runs reduced the X meltH2O , because the estimated H2O concentrations of the S-bearing run-product glasses were lower than those of the corresponding S-free runs for given amounts of H2O and CO2 in the system. We can compare, quantitatively, the relative effects of dilution by CO2 versus S in the fluids of our runs. The slope of the curve fitting the S-free runs (Fig. 4a) is:
and solving this equation for the change in X meltH2O when the H2O content of a S-free aqueous fluid is diluted by the addition of 10 mol% CO2 (i.e., which is arbitrarily chosen to be less than the maximum S content computed for the fluids of these experiments) gives the result that the X meltH2O is reduced by a factor of 0.023 (equivalent to a reduction of 0.25 wt% H2O in the melt). The apparent mixing behavior of CO2 in C–O–H–S fluid-melt systems is not as easily interpreted. The relatively oxidizing experiments show (Figs. 4b, 6a) that values of the X meltCO2 are consistent, within precision, with those determined in our S-free runs and with values predicted for CO2 partitioning between S-free melts and fluids at this pressure (Liu et al. 2005). Given the dispersion in the oxidized experiments, one cannot readily interpret the responses of γ flCO2 and γ meltCO2 as a function of increasing S in the fluid with confidence given the inherent imprecision in the computed fluid compositions. Conversely, the addition of S to relatively reduced C–O–H fluids has a distinct influence by strongly reducing X meltCO2 .
Interpretation of the influence of the mixing relationships in these C–O–H–S fluids and melts on S solubility is problematic given the strong influence of f O2 (Fig. 6b) on S solubility in silicate melts (Clemente et al. 2004; Keppler 2010). Any potential influence of the (CO2/CO2 + H2O) of our runs on the X meltS is overridden by the variable f O2. This problem is exacerbated by the fact that precise control of f O2 in experimental apparatus requires control and knowledge of the a H2O, but for these mixed C–O–H–S fluids and melts we have poor constraints on the f S2 and γH2O and hence on a H2O. Our data do imply that S solubility in C–O–H–S-bearing haplogranitic melt is generally similar to that in O–H–S-bearing granite melt, but our data also show significant dispersion. Some of the dispersion in S partitioning correlates with the apparent f O2 of the runs and is consistent with the reported change in S partitioning between haplogranite melt and O–H–S-bearing fluids with log f O2 of Co-CoO and NNO + 0.5 to NNO + 1 (Keppler 2010). We do note that the MRK modeling of Clemente et al. (2004) predicts greater non-ideal mixing behavior for SO2 than H2S, that H2S appears to be more soluble in felsic melts than SO2 (at equivalent f H2S and f S2), and that H2S solubility should follow Henry’s law at conditions like those of our experiments. Clearly, however, additional volatile solubility research—at carefully defined values of f O2—is called for.
Summary
This investigation provides the first systematic, experimental data set that constrains H2O, CO2, and S solubility and partitioning behavior for C–O–H–S fluids and felsic melt at oxygen fugacities that are geologically relevant to high-silica rhyolitic magmas and some subduction-related, calc-alkaline magmatic systems (Scaillet and Pichavant 2003). In short, the presence of S, largely as reduced S2− in melt, lowers the solubility of CO2 in felsic melt at 200 MPa. Interestingly, recent modeling of the ascent of rhyolitic magma saturated in H–O-S gas from 200 MPa to the surface (Burgisser and Scaillet 2007) indicates that the log f O2 of a S-bearing magma initially at NNO −0.5 will increase slightly and subsequently decrease—potentially to values as low as NNO −1 if the initial gas content approaches 5 wt%. It follows that the solubility and partitioning behavior of C–O–H–S volatiles will be quite complex during the ascent of fluid-saturated magma as pressure decreases, f O2 varies, and CO2 solubility in melt varies. Given the observed influence of oxidized and reduced S on H2O and CO2 solubilities in felsic melt, future experiments should search for similar relationships for felsic melts at other pressures and for intermediate and mafic melts at shallow crustal pressures. Finally, particularly important issues that are yet-to-be-resolved include: what controls f S2 in natural systems and how does iron affect S partitioning?
References
Aranovich LY, Newton RC (1999) Experimental determination of CO2–H2O activity-composition relations at 600–1000°C and 6–14 kbar by reversed decarbonation and dehydration reactions. Am Miner 84:1319–1332
Beermann O (2010) The solubility of sulfur and chlorine in H2O-bearing dacites of Krakatau and basalts of Mt. Etna. Dissertation, University of Hannover, 109 p
Behrens H, Tamic N, Holtz F (2004) Determination of the molar absorption coefficient for the infrared absorption band of CO2 in rhyolitic glasses. Am Miner 89:301–306
Berkesi M, Hidas K, Guzmics T, Dubessy J, Bodnar RJ, Szabo C, Vajna B, Tsunogae T (2009) Detection of small amounts of H2O in CO2-rich fluid inclusions using Raman spectroscopy. J Raman Spectrosc 40:1461–1463
Blank JG, Stolper EM, Carroll MR (1993) Solubilities of carbon dioxide and water in rhyolitic melt at 850°C and 750 bars. Earth Planet Sci Lett 119:27–36
Botcharnikov RE, Behrens H, Holtz F, Koepke J, Sato H (2004) Sulfur and chlorine solubility in Mt. Unzen rhyodacite melt at 850°C and 200 MPa. Chem Geol 213:207–225
Botcharnikov RE, Freise M, Holtz F, Behrens H (2005) Solubility of C-O-H mixtures in natural melts: new experimental data and application range of recent models. Ann Geophys 48:633–646
Burgisser A, Scaillet B (2007) Redox evolution of a degassing magma rising to the surface. Nature 445:194–197
Burgisser A, Scaillet B, Harshvardhan (2008) Chemical patterns of erupting silicic magmas and their influence on the amount of degassing during ascent. J Geophys Res 113(B12204):14
Carroll MR, Rutherford MJ (1988) Sulfur speciation in hydrous experimental glasses of varying oxidation state: results from measured wavelength shifts of sulfur X-rays. Am Min 73:845–849
Carroll MR, Webster JD (1994) Solubilities of sulfur, noble gases, nitrogen, chlorine, and fluorine in magmas. Rev Miner 30:231–279
Cioni R, Marianelli P, Santacroce R, Sbrana A (2000) Plinian and subplinian eruptions. In: Sigurdsson H (ed) Encyclopedia of volcanoes. Academic Press, San Diego, pp 477–494
Clemente B, Scaillet B, Pichavant M (2004) The solubility of sulphur in hydrous rhyolitic melts. J Petrol 45:2171–2196
Fogel RA, Rutherford MJ (1990) The solubility of carbon dioxide in rhyolitic melts: a quantitative FTIR study. Am Miner 75:1311–1326
Gladstone JH, Dale TP (1864) Researches on the refraction, dispersion, and sensitiveness of liquids. Phil Trans R Soc London 153:317–343
Gorbachev NS (1990) Fluid-magma interaction in sulfide-silicate systems. Int Geol Rev 32:749–836
Holloway JR (1987) Igneous fluids. Rev Miner 17:211–232
Holloway JR, Blank J (1994) Application of experimental results to C-O-H species in natural melts. Rev Miner 30:187–230
Jugo P, Wilke M, Botcharnikov RE (2010) Sulfur K-edge XANES analysis of natural and synthetic basaltic glasses: implications for S speciation and S content as function of oxygen fugacity. Geochim Cosmochim Acta 74:5926–5938
Keppler H (1999) Experimental evidence for the source of excess sulfur in explosive volcanic eruptions. Science 284:1652–1654
Keppler H (2010) The distribution of sulfur between haplogranite melts and aqueous fluids. Geochim Cosmochim Acta 74:645–660
Kerrick DM, Jacobs GK (1981) A modified Redlich-Kwong equation for H2O, CO2, and H2O-CO2 mixtures at elevated pressures and temperatures. Am J Sci 281:735–767
Lesne P, Kohn SC, Blundy J, Witham F, Botcharnikov RE, Behrens H (in press) Experimental simulation of basalt degassing at Stromboli and Masaya volcanoes. J Petrol
Lin B, Bodnar RJ (2010) Synthetic fluid inclusions XVIII: experimental determination of the PVTX properties of H2O-CH4 to 500°C, 3 kbar and XCH4 ≤ 4 mol%. Geochim Cosmochim Acta 74:3260–3273
Liu Y, Zhang Y, Behrens H (2005) Solubility of H2O in rhyolitic melts at low pressures and a new empirical model for mixed H2O-CO2 solubility in rhyolitic melts. J Volcanol Geotherm Res 143:235–291
Mandeville CW, Webster JD, Rutherford MJ, Taylor BE, Timbal A, Faure K (2002) Determination of molar aborptivities for infrared absorption bands of H2O in andesitic glasses. Am Min 87:813–821
Matthews SJ, Moncrieff DHS, Carroll MR (1999) Empirical calibration of the sulphur valence oxygen barometer from natural and experimental glasses; method and applications. Miner Mag 63(3):421–431
Métrich N, Clocchiatti R (1996) Sulfur abundance and its speciation in oxidized alkaline melts. Geochim Cosmochim Acta 60:4151–4160
Métrich N, Wallace PJ (2008) Volatile abundances in basaltic magmas and their degassing paths tracked by melt inclusions. Rev Mineral Geochem 69:363–402
Moore G (2008) Interpreting H2O and CO2 contents in melt inclusions: constraints from solubility experiments and modeling. Rev Mineral Geochem 69:333–361
Moretti R, Baker DR (2008) Modeling the interplay of f O2 and f S2 along the FeS-silicate melt equilibrium. Chem Geol 256:286–298
Moretti R, Papale P (2004) On the oxidation state and volatile behavior in multicomponent gas-melt equilibria. Chem Geol 213:265–280
Morizet Y, Paris M, Gaillard F, Scaillet B (2010) C-O-H fluid solubility in haplobasalt under reducing conditions: an experimental study. Chem Geol 279:1–16
Mysen BO, Popp RK (1980) Solubility of sulfur in CaMgSi2O6 and NaAlSi3O8 melts at high-pressure and temperature with controlled f O2 and f S2. Am J Sci 280(1):78–92
Newman S, Lowenstern JB (2002) VOLATILECALC: a silicate melt-H2O-CO2 solution model written in Visual Basic for excel. Comp Geosci 28:597–604
Nicholis MG, Rutherford MJ (2009) Graphite oxidation in the Apollo 17 orange glass magma: implications for the generation of a lunar volcanic gas phase. Geochim Cosmochim Acta 73:5905–5917
Nowak M, Behrens H (1995) The speciation of water in haplogranitic glasses and melts determined by in situ near-infrared spectroscopy. Geochim Cosmochim Acta 59:3445–3450
Nowak M, Behrens H (1997) An experimental investigation of diffusion of water in haplogranitic melts. Contrib Mineral Petrol 126:365–376
Oppenheimer C (2003) Volcanic degassing. In: Rudnick RL (ed) The crust. Treatise in geochemistry, vol 3. Elsevier, Amsterdam, pp 123–166
Parat F, Holtz F, Feig S (2008) Pre-eruptive conditions of the Huerto Andesite (Fish Canyon System, San Juan volcanic field, Colorado): influence of volatiles (C-O-H-S) on phase equilibria and mineral composition. J Petrol 49:911–935
Ripley E, Li C, Moore CH, Elswick K, Maynard JB, Paul RL, Sylvester P, Seo JH, Shimizu N (in press) Analytical methods for sulfur determination in glasses, rocks, minerals and fluid inclusions. In: Behrens H, Webster JD (eds) Sulfur in magmas and melts and its importance for natural and technical processes, vol 74. Rev Mineral Geochem
Scaillet B, Pichavant M (2003) Experimental constraints on volatile abundances in arc magmas and their implications for degassing processes. In: Oppenheimer C, Pyle DM, Barclay J (eds) Volcanic degassing, vol 213. Geol Soc Spec Pub, pp 23–52
Scaillet B, Pichavant M (2005) A model of sulphur solubility for hydrous mafic melts: application to the determination of magmatic fluid compositions of Italian volcanoes. Ann Geophys 48(4–5):671–698
Scaillet B, Clemente B, Evans BW, Pichavant M (1998) Redox control of sulfur degassing in silicic magmas. J Geophys Res 103(B10):23937–23949
Scaillet B, Luhr JF, Carroll MR (2003) Petrological and volcanological constraints on volcanic sulfur emissions to the atmosphere. AGU Geophys Mono 139:11–40
Silver LA, Ihinger PD, Stolper E (1990) The influence of bulk composition on the speciation of water in silicate glasses. Contrib Mineral Petrol 104:142–162
Spilliaert N, Metrich N, Allard P (2006) S-Cl-F degassing pattern of water-rich alkali basalt: Modelling and relationship with eruption styles on Mount Etna volcano. Earth Planet Sci Lett 248(3–4):772–786
Tamic N, Behrens H, Holtz R (2001) The solubility of H2O and CO2 in rhyolitic melts in equilibrium with a mixed CO2–H2O fluid phase. Chem Geol 174:333–347
Teague AJ, Kohn SC, Klimm K, Botcharnikov RE (2008) Sulphur solubility in Mount Hood andesites and CO2 fluids: implications for volcanic degassing EOS Trans. AGU, 89(53), (Fall Meet. Suppl. Abstract V21B-2086)
Wallace PJ, Carmichael ISE (1994) S speciation in submarine basaltic glasses as determined by measurements of SKα X-ray wavelength shifts. Am Miner 79:161–167
Wallace PJ, Edmonds M (in press) The sulfur budget in magmas: evidence from melt inclusions, submarine glasses, and volcanic gas emissions. In: Behrens H, Webster JD (eds) Sulfur in magmas and melts and its importance for natural and technical processes, vol 74. Rev Min Geochem
Webster JD (1992a) Fluid-melt interactions involving Cl-rich granites: experimental study from 2 to 8 kbar. Geochim Cosmochim Acta 56:659–678
Webster JD (1992b) Water solubility and chlorine partitioning in Cl-rich granitic systems: effects of melt composition at 2 kbar and 800°C. Geochim Cosmochim Acta 56:679–687
Webster JD, Botcharnikov RE (in press) Distribution of sulfur between melt and fluid in S-O-H-C-Cl-bearing magmatic systems at shallow crustal P-T conditions. In: Behrens H, Webster JD (eds) Sulfur in magmas and melts and its importance for natural and technical processes, vol 74. Rev Min Geochem
Webster JD, Sintoni MF, De Vivo B (2006) The role of sulfur in promoting magmatic degassing and volcanic eruption at Mt. Somma-Vesuvius. In: De Vivo B (ed) Volcanism in the Campania plain, vol 9. Elsevier, Amsterdam, pp 219–233
Webster JD, Sintoni MF, De Vivo B (2009) The partitioning behavior of Cl and S in aqueous fluid- and saline-liquid saturated phonolitic and trachytic melts at 200 MPa. Chem Geol 263:19–36
Acknowledgments
We appreciate thoughtful reviews by Editor Tim Grove and two anonymous referees. We also wish to recognize discussions on analytical methods with Charles Mandeville. Our thanks are offered to Professor David London of the University of Oklahoma who provided the haplogranite starting glass and Professor Harald Behrens of the University of Hannover, Germany, who provided two synthetic glasses with measured H2O contents that were used to test our FTIR analyses. Robert Bodnar and Charles Farley, of Virginia Tech, kindly conducted Raman analyses of 9 of the run-product glasses to determine the dominant volatile species in the vesicles. This research was supported in part by National Science Foundation awards EAR 0308866 and EAR-0836741 to J.D.W.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. L. Grove.
Rights and permissions
About this article
Cite this article
Webster, J.D., Goldoff, B. & Shimizu, N. C–O–H–S fluids and granitic magma: how S partitions and modifies CO2 concentrations of fluid-saturated felsic melt at 200 MPa. Contrib Mineral Petrol 162, 849–865 (2011). https://doi.org/10.1007/s00410-011-0628-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-011-0628-1