
Abstract
Melt-rock reaction in the upper mantle is recorded in a variety of ultramafic rocks and is an important process in modifying melt composition on its way from the source region towards the surface. This experimental study evaluates the compositional variability of tholeiitic basalts upon reaction with depleted peridotite at uppermost-mantle conditions. Infiltration-reaction processes are simulated by employing a three-layered set-up: primitive basaltic powder (‘melt layer’) is overlain by a ‘peridotite layer’ and a layer of vitreous carbon spheres (‘melt trap’). Melt from the melt layer is forced to move through the peridotite layer into the melt trap. Experiments were conducted at 0.65 and 0.8 GPa in the temperature range 1,170–1,290°C. In this P-T range, representing conditions encountered in the transition zone (thermal boundary layer) between the asthenosphere and the lithosphere underneath oceanic spreading centres, the melt is subjected to fractionation, and the peridotite is partially melting (T s ~ 1,260°C). The effect of reaction between melt and peridotite on the melt composition was investigated across each experimental charge. Quenched melts in the peridotite layers display larger compositional variations than melt layer glasses. A difference between glasses in the melt and peridotite layer becomes more important at decreasing temperature through a combination of enrichment in incompatible elements in the melt layer and less efficient diffusive equilibration in the melt phase. At 1,290°C, preferential dissolution of pyroxenes enriches the melt in silica and dilutes it in incompatible elements. Moreover, liquids become increasingly enriched in Cr2O3 at higher temperatures due to the dissolution of spinel. Silica contents of liquids decrease at 1,260°C, whereas incompatible elements start to concentrate in the melt due to increasing levels of crystallization. At the lowest temperatures investigated, increasing alkali contents cause silica to increase as a consequence of reactive fractionation. Pervasive percolation of tholeiitic basalt through an upper-mantle thermal boundary layer can thus impose a high-Si ‘low-pressure’ signature on MORB. This could explain opx + plag enrichment in shallow plagioclase peridotites and prolonged formation of olivine gabbros.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
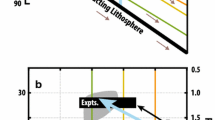
Recently, reactive porous flow features have increasingly been recognized in mantle sections of ophiolites and peridotite massifs (e.g. Dijkstra et al. 2003; Le Roux et al. 2007; Müntener et al. 2005; Rampone et al. 1997; Takahashi 2001; Van der Wal and Bodinier 1996; Zhang 2005), and in abyssal peridotites sampled at slow-spreading oceanic ridges and transform faults (e.g. Elthon 1992; Seyler and Bonatti 1997; Seyler et al. 2004). Moreover, regional-scale modification of ancient subcontinental mantle by melt-infiltration and melt-rock reaction during incipient opening of oceanic basins was recognized (Müntener and Piccardo 2003; Müntener et al. 2004). The evidence for melt-rock interactions in the mantle is thus extensive, encompassing textural relations and mineral modes, major to minor element-phase compositions, melt inclusions (e.g. Kamenetsky and Crawford 1998; Kamenetsky et al. 2001; Schiano et al. 1994; Sobolev and Shimizu 1993), isotope systematics, and local to regional variations in MORB chemistry (e.g. Langmuir et al. 1992; le Roux et al. 2002b). Nonetheless, numerous questions regarding the controls on precise reaction processes between ascending partial melts and peridotite, and the consequent modes of melt transport and melt evolution remain to be addressed adequately. This is particularly the case for the transitional region or ‘thermal boundary layer’ (‘TBL’; cf. White 1988) between the asthenospheric and the lithospheric mantle. Here, the local geotherm deviates from the adiabatic (mantle) geotherm, but conductive heat loss to the surface does not yet dominate over advection of heat from below (as is the case for the lithosphere). The potential cases where a significant TBL can exist, thus promoting widespread melt-rock reaction, are schematically illustrated in Fig. 1. Obviously, direct observation of upper-mantle processes is impractical, and both the reacted mantle rocks and percolating or trapped melts will undergo significant alterations and modifications on their way from upper-mantle levels to the surface. It is, therefore, crucial to perform experimental investigations to attain better understanding of the first-order reaction processes and their effects on the liquid lines of descent of migrating magmas.

Schematic illustration of geodynamic settings (ultra-slow oceanic spreading, top left, and continental rifting, top right) where ascending melts may encounter a thermal boundary layer (TBL) in the uppermost mantle (inspired by Cannat et al. 1997; Ildefonse et al. 2007; Müntener and Piccardo 2003; Niu 2004). Partial melting will cease in the TBL, as beginning conductive heat loss to the surface reduces the temperature to below the peridotite solidus. Basaltic liquids produced in the upwelling upper mantle will undergo progressive cooling as they rise into the TBL, leading to fractionation of the liquids and reaction with the host peridotites. The lower panels show hypothetical sketches of (modal and chemical) ‘depletion’ of the host rocks at the base of the TBL (Panel ①), followed by progressive enrichment upon further ascent/cooling of the liquid (Panel ②) and finally stagnation and ‘freezing’ of all liquids (Panel ③). Such rocks are described as ‘refertilized’ peridotites and can later be exhumed at the sea-floor by detachment faulting (e.g. Fig. 17 of Müntener et al. 2010)
A number of experimental studies broadly related to upper-mantle melt-rock reaction processes have been carried out (Beck et al. 2006; Daines and Kohlstedt 1994; Kelemen et al. 1990; Lambart et al. 2009; Lundstrom 2000, 2003; Morgan and Liang 2003, 2005). Furthermore, there is a larger body of studies dealing with systematics of and processes related to partial melting in the upper mantle (e.g. Baker and Stolper 1994; Baker et al. 1995; Doukhan et al. 1993; Draper and Green 1999; Falloon et al. 1997, 1999, 2001; Hirschmann et al. 1998, 2003; Johnston and Schwab 2004; Kinzler and Grove 1992; Lo Cascio et al. 2004; Pickering-Witter and Johnston 2000; Robinson et al. 1998; Schiano et al. 2006; Schwab and Johnston 2001; Wasylenki et al. 2003; Yaxley 2000). These experimental studies, combined with a number of numerical studies dealing with melt-rock reaction and reactive melt flow (e.g. Aharonov et al. 1995; Asimow and Stolper 1999; Liang 2003; Liang and Guo 2003; Ortoleva et al. 1987; Spiegelman et al. 2001; Wark and Watson 2000), have contributed significantly to the understanding of melt-rock reaction. One general outcome of these studies is the recognition of the complexity of upper-mantle partial melting and melt-rock reaction processes, which are mutually dependant on a number of intensive (e.g. P-T, fO2 conditions, and liquid properties) and non-intensive parameters (e.g. grain size, interconnected porosity, and other textural relationships).
To our knowledge, melt-infiltration experiments have not yet been performed with primitive upper-mantle partial melts at P-T conditions encountered in TBL’s of the upper mantle. In a recent paper, Lambart et al. (2009) have investigated basalt–peridotite interactions at P-T conditions plausible for the TBL. However, these authors performed direct-mixing experiments at relatively high (for the TBL) temperatures, thereby producing high melt/rock ratios, as the rationale of their study was to investigate focused magma transport. The lack of melt-infiltration experiments applied to the TBL is particularly surprising as tholeiitic basalt is the most common basaltic magma erupted at the Earth’s surface and therefore, is often studied to deduce information on upper-mantle melting and (reactive) melt migration processes, as well as mantle compositions and oceanic crust formation. It is also well established that various processes (e.g. fractional and/or equilibrium crystallization, assimilation of crustal rocks, mingling and/or mixing of magmas in lower-crustal magma chambers) influence initial melt compositions. Therefore, magmatic processes occurring en route from the melting regions in the upper mantle to the transition from asthenosphere to mantle lithosphere and lower crust are often obscured and difficult to assess from MORB compositions alone.
This contribution presents results of a comprehensive experimental study with applications to reactive melt transport in the upper mantle in ocean–continent transition zones and beneath oceanic spreading centres. The general objective is to derive a consistent set of data to evaluate the compositional and time-dependent evolution of migrating magma over a range of temperatures and pressures as a function of initial melt and peridotite composition. The grain-scale reaction processes related to melt-rock reaction at upper-mantle conditions are documented in an additional publication (Van den Bleeken et al. 2010). The present paper addresses the compositional evolution of tholeiitic basalt as it infiltrates and reacts with depleted peridotite, as a function of P, T and starting liquid composition. Rather than focusing only on km-scale pervasive flow in high-porosity melt conduits, we target to put constraints on melt variability during reactive porous flow, melt impregnation and eventual melt stagnation in an upper-mantle TBL (Fig. 1).
Experimental methods
Melt-peridotite reaction processes were studied in a series of experiments where a layer of primitive basaltic powder (the ‘melt layer’; ‘ML’) is overlain by a peridotite layer (‘PL’) and a layer of ~40-μm-large vitreous carbon spheres (Fig. 2). The porosity of the melt trap (‘MT’) enhances flow of melt into the peridotite layer, thereby promoting reactive melt percolation. In addition, the melt trap conserves reacted melts for analyses. The three-layered configuration is contained in a graphite container that tightly fits into a Pt capsule (Fig. 2). Furthermore, two direct-mixing experiments were performed to bracket the results of the aforementioned infiltration-reaction runs. Experiments were carried out at P-T conditions that are typically encountered in the thermal boundary layer underneath oceanic spreading centres, i.e. at 1,170–1,290°C and 0.65–0.8 GPa. The starting materials employed in this study are listed in Table 1.

Representative BSE images relative to the experimental set-up, illustrating the textural evolution of ML’s and PL’s of 0.65 GPa experiments as a function of run temperature. The initial starting mix texture of irregular angular grains stays recognizable throughout the entire temperature range but is progressively obscured towards higher temperature due to a combined effect of diffusion and dissolution ± (re-)precipitation. Olivine and spinel show continuous (diffusive) zoning at 1,170 and 1,215°C. Spinel, cpx and opx clearly undergo partial dissolution with or without (re-)precipitation of sub-/euhedral overgrowths and melt pocket precipitates (e.g. reprecipitated ‘Sp2’ enveloping a remnant ‘Sp1’ core). At 1,170°C (and to a lesser degree at 1,215°C), the ML has undergone severe fractionation with almost no melt left. Deeper into the PL at 1,170°C, and with increasing run temperature, melt pockets become larger and the melt fraction increases. Note that the capsules are actually run with the MT-side facing upwards in the piston cylinder apparatus
Starting materials
As melt analogue, we initially used the primitive basaltic glass composition HK#19.1 (Villiger et al. 2004) that is in equilibrium with a lherzolitic residue (ol, opx, cpx) at 1.5 GPa and 1,350°C (Hirose and Kushiro 1993). Villiger and co-workers performed a comprehensive set of fractional and equilibrium crystallization experiments on this composition at P and T conditions relevant for our study (Villiger et al. 2004, 2007). This microscopically heterogeneous starting material composed of oxide and silicate powders proved to be prone to disequilibrium melting during the initial stages of heating, producing ‘exotic melts’ as they are referred to hereafter. Therefore, we vitrified HK#19.1 under controlled fO2 in a gas-mixing furnace, producing the starting composition “HK19.v”. The vitrification process lead to a minor decrease in the FeO content, whereas the subsequent grinding in an agate mortar to obtain a very fine-grained powder of the recovered glass resulted in a marginal increase in the silica content (see Table 1). A second melt analogue was subsequently mixed from oxides, carbonates and silicates and vitrified to test the influence of melt composition on melt-rock reaction. This second analogue is in equilibrium with a harzburgitic residue (ol, opx) at 1.5 GPa and 1,400°C corresponding to the melt composition in run 20 of Hirose and Kushiro (1993) and was accordingly named HK#20.v. To improve readability, we use the acronyms “HK#19” and “HK#20” hereafter, unless we specifically want to distinguish between “HK#19.1” and “HK#19.v”. Due to a higher Ca/Si ratio in HK#19 compared to HK#20, cpx fractionation is favoured in HK#19, whereas opx fractionation is favoured in HK#20.
The peridotite analogues consist of recombined powders obtained from hand-picked separates of natural grains. Although the use of natural grains is debatable for melting studies (e.g. Falloon et al. 1999), we opted for natural grains in this melt-rock reaction study. Indeed, our goal is not to reach complete equilibration for all grains, but to perform experiments that allow the study of melt-rock reactions as they could occur in the upper mantle. As shown in Van den Bleeken et al. (2010), newly formed phases have compositions that are very similar to those of equilibrium melting studies, suggesting that they precipitated from geologically meaningful liquids. Low-Ca orthopyroxene, high-Ca clinopyroxene and Cr-spinel were separated from the Balmuccia peridotite (Ivrea-Verbano Zone, Northern Italy), whereas olivine originates from the San Carlos Volcanic Field (Arizona). Final equilibration of the Balmuccia spinel lherzolite took place at 850–950°C and ~1.0 GPa (Shervais 1979). San Carlos spinel lherzolite xenoliths retain two-pyroxene equilibration temperatures of 1,050°C (Brey and Köhler 1990) and a pressure of 1.3 GPa (Köhler and Brey 1990). Averaged compositions for the different minerals and calculated starting mix compositions are listed in Table 1. The mineral separates were ground separately under ethanol and sieved in order to obtain well-defined grain-size fractions. From these separates, two starting fractions were mixed: BaSC1 (200–80 μm) and BaSC2 (80–30 μm), with “BaSC” derived from “Balmuccia—San Carlos”. A third starting material, BaSC3, was produced by grinding BaSC1 under ethanol for half an hour. This resulted in a “porphyric” mix of larger grains (up to 50 μm) embedded in a fine-grained groundmass (<10 μm). BaSC1/3 has a composition (in wt%) of 65 ol, 24 opx, 9 cpx and 2 spl, whereas BaSC2 contains 63 ol, 25 opx, 10 cpx and 2 spl. Both compositions represent depleted lherzolite, with a solidus temperature of ~1,260°C (inferred from forward melting of BaSC2 at 1,260°C and 0.8 GPa, displaying incipient melting).
Since liquid compositions in the infiltration-reaction experiments potentially depend on a multitude of processes related to the experimental set-up, additional experiments were performed in which HK#19.v was mixed homogeneously with BaSC2. Two such mixes were produced, Mix1 and Mix2, containing 7 and 20 wt% starting liquid, respectively. Two direct-mixing experiments were performed at 1,200°C and 0.8 GPa, one run for 15 h (GB51) and the other for 93 h (GB52). In both runs, the charge consisted of an outer Pt capsule containing two inner graphite capsules, filled with Mix1 and Mix2, respectively. The direct-mixing experiments were devised to constrain the influence of run duration and amount of infiltrating liquid on the reacted liquid compositions in the infiltration-reaction runs.
All starting materials were dried at 220°C for at least 24 h to drive off adsorbed water and subsequently stored in an oven at 110°C. For the more hygroscopic melt analogues, the heat treatment at 220°C was always repeated prior to loading of the capsules. Starting materials were taken out of the oven only moments before loading of a capsule.
Experimental strategy
To minimize Fe loss to the noble metal capsule and to constrain fO2 near the C–CO2–CO equilibrium, the Pt-graphite double-capsule technique was applied (Ulmer and Luth 1991). Layers of vitreous carbon, peridotite and basaltic powder are subsequently loaded into a graphite container (3.4 mm OD, 2 mm ID) that is closed with a tight-fitting lid. The graphite container was placed in a Pt capsule (4 mm OD) that was welded shut only after additional drying for several hours at 110°C. Pt capsules were checked for leaking by weighing prior to and after submersion in acetone.
All experiments were conducted in a 14-mm bore end-loaded Boyd and England (1960)-type solid-media piston cylinder apparatus at the high-pressure laboratory at ETH Zürich. Initially, salt–Pyrex–MgO, and later talc–Pyrex–MgO assemblies were employed, with a friction correction of ~3 and ~10%, respectively, applied to the nominal pressure. Temperature was measured with Pt94Rh06–Pt70Rh30 (B-type) thermocouples with an estimated accuracy of ±10°C, without accounting for pressure effects on the EMF. Temperature increments of 30°C were used for successive experiments, considering temperature reproducibility of ±15°C in piston cylinder experiments. Runs were gently cold pressurized to 0.4 GPa, before temperature was raised at a rate of 80°C/min. From ~700°C, the pressure was gradually increased along with the temperature until annealing conditions were attained. To ensure that no melt migration and subsequent reaction takes place, annealing has to be performed in the sub-solidus domain. In earlier experiments (until GB17), we annealed runs using the HK#19.1 composition at 1,100°C, resulting in percolation of a small fraction of exotic melts into the PL and MT. Therefore, later experiments were annealed at 1,000°C, but comparison of the two sets of runs revealed no discernable influence of early melt percolation in the earlier runs. After 5 h of annealing (referred to hereafter as the stage of hot isostatic pressing, or HIP-stage), after which ~6% porosity remained (Van den Bleeken et al. 2010), temperature was increased to the desired values at rates between 150 and 200°C/min. Experiments were quenched by shutting off the power supply, resulting in quenching rates in the order of 200–300°C/s for the first 600°C. After the experiment, the recovered charge was sectioned longitudinally, mounted in epoxy and polished for microanalysis. Most experiments were kept at run temperature for 15 h, but several shorter (0–0.5 h) and a few longer runs (92 h) were performed to bracket the results of the 15-h runs (see also Van den Bleeken et al. 2010). Here, we focus on the results of the 15-h runs. This run time was chosen to allow enough reaction to install local equilibria (see also Table 5 and Figs. 7, 10 and 11 of Van den Bleeken et al. 2010), while preserving compositional gradients from ML to PL. By preventing achievement of complete equilibrium across the reaction couple, we investigated the largest possible compositional variations upon reactive infiltration of MORB-type basalt in the TBL. Comparison with the direct-mixing experiments constrains potential influences of run duration and amount of ‘infiltrated’ melt on the reacted liquid compositions (Table 4, “Direct-mixing experiments”).
Microprobe analysis
Wavelength-dispersive analyses as well as backscattered electron (BSE) images and X-ray concentration maps were acquired on two JEOL JXA-8200 Superprobes. Most analyses were performed at the Geological Institute of the University of Bern (Switzerland). Some additional measurements were carried out in the Institute of Mineralogy and Geochemistry of the University of Lausanne (Switzerland). An accelerating voltage of 15 kV was applied for all analyses. Crystalline phases were measured with a beam current of 20 nA and a focused beam, whereas quenched liquids were analysed with 7 nA beam current and 1–10 μm beam size, depending on the sizes of the melt pools. Well-characterized minerals and synthetic compounds were used as standards, and the PRZ (CITZAF) correction procedure was applied to the raw data. Peak position counting times were 20 s for all elements, except for Na that was counted for only 6 s to avoid Na migration, and Ni that was counted for 30 s. Averaged measurements (n = 10) of the homogeneous glass SV75 (from Villiger et al. 2007) did not reveal any dependence of element concentrations as a function of spot size (i.e. 1, 5 or 10 μm). The totals of glass analyses usually varied between 96 and 99 wt%, depending on the quality of the polished surface. The detection limit for the various elements was typically between 0.01 and 0.03 wt%, except for NiO with a detection limit of ~0.12 wt%.
Results
Three new infiltration-reaction experiments were performed at 0.65 GPa and temperatures of 1,170°C (GB43), 1,215°C (GB42) and 1,260°C (GB44). This ‘low-pressure’ data complement our 0.8 GPa data (Van den Bleeken et al. 2010) and permit the evaluation of reactive liquid evolution in the TBL. Furthermore, direct-mixing experiments were carried out at 0.8 GPa and 1,200°C, with run durations of 15 h (GB51) and 93 h (GB52). Run conditions and phase assemblages for selected experiments are given in Table 2. In Van den Bleeken et al. (2010), average glass compositions were listed in Table 5 and used for mineral–liquid element partition calculations. In the present study, we provide a list of representative glass analyses from 0.65 to 0.8 GPa runs in Table 3 and present the complete data sets of glasses from across the reaction couples of selected runs (e.g. Fig. 4). This allows the evaluation of melt evolution upon reactive infiltration as a function of P, T and starting liquid composition. Moreover, the full data sets are provided in a supplementary online data file (Online Resource 1).
Run products
A general textural overview of the 0.65 GPa runs as a function of temperature and position in the reaction couple is given in Fig. 2. At 1,260°C, the only phases present are olivine and opx. Olivine occurs as euhedral crystals in the ML and PL, although deeper within the PL, some grains maintain initial (irregular) BaSC textures. Opx occurs as rounded inherited cores in the PL, but only from distances of ~400 μm deep in the PL, thereby defining a dunitic reactive boundary layer (RBL). The rounded cores often display 5- to 10-μm-thick sub to euhedral (faceted) rims, resulting from dissolution–reprecipitation (see Van den Bleeken et al. 2010). At 1,215 and 1,170°C, initial BaSC textures are mostly retained. Inherited grains of pyroxenes and spinel commonly feature thin dissolution–reprecipitation rims, and olivine grains and spinel cores exhibit diffusional zoning. These grain-scale observations are similar to those made in 1,200–1,230°C runs at 0.8 GPa (Van den Bleeken et al. 2010). At both temperatures, plagioclase is present in the PL. However, at 1,215°C, it is constrained to a 250 μm area adjoining the ML, whereas it occurs throughout the PL at 1,170°C. The phase assemblage of the ML at 1,215°C is identical to that of the 0.8 GPa 1,230°C run GB41, as it contains (in decreasing abundance) cpx, prismatic plagioclase, melt and olivine. At 1,170°C, the ML consists almost exclusively of fine-grained (≤10 μm) subhedral cpx and anhedral plagioclase, with subordinate amounts of melt and olivine being present. Furthermore, a few μm-scale bright grains of probable Cr-spinel (Cr# = ~0.4) occur. Textural, modal and chemical variations of mineral phases in 0.8 GPa runs were reported in Van den Bleeken et al. (2010).
Reaction textures in the direct-mixing runs are very similar to those in the infiltration-reaction experiments. Olivine and spinel display continuous zoning patterns, whereas inherited cores of pyroxenes and, far less frequently, spinel cores are surrounded by dissolution–reprecipitation rims. The overall texture appears more equilibrated with increasing run duration and HK#19 proportions. Concordantly, the proportion of inherited cores over newly formed rims of cpx decreases significantly, up to a point were hardly any cores of cpx can be observed after 93 h of reaction in the Mix2-containing inner capsule of GB52. Anhedral to euhedral plagioclase occurs in the Mix2 inner capsule of GB51, but is absent in the Mix1 capsule of that run, indicating a sensitive compositional control around plagioclase-in. It does not occur at all in GB52, suggesting a slightly higher effective run temperature,
Assessment of and correction for quench modification of glasses
Quench modification of melt compositions by rapid metastable crystallization on rims of primary crystal phases during quenching of experimental runs is one of the major hurdles facing experimental studies (e.g. Falloon et al. 2001). Quench olivine will nucleate as thin rims on olivine crystal faces, whereas feathery quench cpx will nucleate on both cpx and opx crystals. One way to circumvent this problem, developed by Laporte et al. (2004), utilizes microfractures in the inner graphite capsule itself to extract very low fractions of melt safe from quench crystallization. More commonly, researchers have employed a layer of material in the experimental capsule that maintains an open porosity at high P-T, thereby serving as a melt trap where quench crystals cannot nucleate (e.g. Baker and Stolper 1994; Falloon et al. 1999, 2001; Hirose and Kushiro 1993; Wasylenki et al. 2003). Following the latter method, we have employed the use of a layer of vitreous carbon spheres on top of the ML–PL reaction couples. However, this only provides us glass compositions unaffected by quench crystallization for the top-most part in any experimental charge (see Fig. 2). Compositions of glasses in ML’s and melt pockets in PL’s most likely underwent varying degrees of quench modification (Fig. 3) and, thus, cannot be used directly to access compositional variations related to melt-rock reaction in the experiments.

Illustration of quench modification of glasses obtained from our experiments, and the resulting reduction of the induced compositional variation by correction of the MgO contents to melt trap values (see “Assessment of and correction for quench modification of glasses”). The left column (a) shows the measured variation of Al2O3 as a function of MgO for glasses from PL’s of selected HK#19 runs, and as a function of distance to the ML/PL interface for glasses across the reaction couple. In the right column (b), the same data are displayed after correcting MgO to MT contents by addition of olivine to measured MgO contents (see text for details). Back-correction of MgO results in a decrease in the spread in Al2O3 in each run, especially for PL glasses; this results in more continuous compositional profiles. The original data from (a) are shown in grey, and tie-lines connect a selection of uncorrected values with corrected values
Correcting quench-modified glasses back to their ‘equilibrium’ composition is possible assuming that: (1) the composition of the quench mineral phase(s) is known; (2) the modal fraction of quench crystals can be assessed, or thus the initial composition prior to quenching can be adequately approximated. We have attempted to correct the glasses from ML’s and PL’s of each run by incremental addition of (equilibrium) ML olivineFootnote 1 until MgO contents correspond for each run to those of glasses unaffected by quench crystallization. The MgO content prior to quench crystallization was selected from amongst the ML glasses highest in MgO, or from the MT measurements in case we thought there might be at least some influence by quench crystallization in the ML. Exceptions are the 1,200°C (0.8 GPa) runs GB14 and GB16, and the 1,170°C (0.65 GPa) run GB43, for which the degree of compositional variation of glass compositions across the different layers is so extensive that MgO contents of MT glasses were most likely not representative for the ML/PL interface area. For these respective runs, ‘equilibrium’ MgO contents were taken from runs SV89 (1,210°C) and SV90 (1,180°C) from the 0.7 GPa equilibrium crystallization experiments of Villiger et al. (2007). Figure 3 compares measured glass compositions with back-calculated ‘quench-free’ compositions for a selection of HK#19-BaSC2 runs, spanning a 1,200–1,290°C temperature range. Correcting the MgO contents of the raw data to ‘equilibrium values’ obviously reduces Al2O3 variability and removes compositional ‘trends’ in longitudinal distance profiles that were introduced by quench crystallization. Similarly, the correction decreases variability in other components and makes trends in concentration profiles more continuous, most notably for SiO2, CaO and FeO, and Mg#. This illustrates the success of solely adding (equilibrium) olivine to measured glass compositions in order to diminish effects of quench modification. Similar results were obtained by Lundstrom (2003) and Morgan and Liang (2005).
Corrected glass compositions from runs at lower temperatures increasingly display scatter of CaO contents towards lower values, indicating increasing amounts of cpx quench crystallization. Attempts to correct glasses for cpx quench crystallization were unsuccessful, likely as quench cpx is compositionally significantly different from equilibrium cpx. Therefore, we have opted for only adjusting the raw data for quench olivine growth, followed by the careful evaluation of corrected glasses in case of notable scatter in CaO.
All data from this study reported in the text, Tables and Figures of this manuscript have been corrected for olivine quench modification, except for data from MT’s as quench crystallization does not occur here. The raw data are listed in the supplementary online table (Online Resource 1).
Liquid compositions
Table 3 reports representative liquid compositions for ML’s, PL’s and MT’s of selected runs. Table 4 lists average glass compositions for each inner capsule of the direct-mixing experiments. Compositional variations related to changes in reacting liquid composition, temperature and pressure are illustrated in Figs. 4, 5, and 6.


Glass compositions as a function of distance to the ML/PL interface (negative values for the ML domain) for glasses from the three experimental series (left, middle and right column) and colour-coded as a function of run temperature. Blue squares represent the compositions of low-degree (<2%) melts of BaSC2, at 0.8 GPa and 1,260°C (t = 15 h) and are displayed on the y-axis between the two 0.8 GPa reaction series. Shaded fields added as a background to the middle column represent average liquid compositions (2σ) from the direct-mixing experiments GB51 and GB52. See text for a detailed discussion of the systematic compositional trends observed as a function of distance and run temperature. Presented as a function of distance are Al2O3 (a–c), SiO2 (d–f), Na2O (g–i), K2O (j–l), CaO (m–o), FeO (p–r), TiO2 (s–u) and Cr2O3 (v–x)

Part of the total alkalis (Na2O + K2O; wt%) versus silica (SiO2, wt%) diagram after Le Bas et al. (1986) for glasses from PL’s of selected runs conducted at 0.65 GPa (a) and 0.8 GPa (b) employing HK#19, and at 0.8 GPa employing HK#20 (c). Included in b is the composition of the low-degree (~2%) partial melt of BaSC2 at 1,260°C and 0.8 GPa

Normative pseudo-ternary cpx–olivine–quartz diagram, illustrating liquid compositions for selected HK#19-BaSC2 reaction experiments at 0.65 GPa (a) and 0.8 GPa (b), and HK#20-BaSC2 reaction at 0.8 GPa (c). Normalization into six components in oxygen units (olivine, cpx, plagioclase, quartz, orthoclase, oxide) according to the projection scheme proposed by Grove et al. (1992); compositions are projected from plagioclase, orthoclase and oxide. 1 bar and 1 GPa phase boundaries are given for comparison and taken from Grove and Baker (1984). See text for discussion
Direct-mixing experiments
Average glass compositions from the direct-mixing runs are listed in Table 4. Although the amount of reaction increases (~proportion of unreacted cores decreases) with run duration and higher amounts of mixed-in liquid, almost all elements (the exception being Ti) display overlapping compositions on a 2σ uncertainty level. The GB51 Mix2 capsule shows slightly more internal variation than the other capsules (e.g. for Mg). This can be related to the presence of plagioclase in this capsule. The fact that plagioclase occurs in GB51 and not in GB52 suggests a slightly higher run temperature for the latter. This is also expressed by slightly lower concentrations of Al, Na and K in GB52 glasses.
The glass compositions from the direct-mixing runs are compared with glass compositions from the infiltration-reaction runs in Fig. 4 (displaying the average of all direct-mixing capsules) and Fig. 9 (displaying all measured compositions individually).
Compositional variations along longitudinal profiles
Compositional variations across reaction couples are displayed in Fig. 4, as a function of P, T and starting liquid composition. Moreover, shaded fields added as a background to the middle column represent liquid compositions from the direct-mixing experiments.
Generally, the compositional profiles are continuous across the ML/PL and PL/MT boundaries. An exception was the 1,170°C run at 0.65 GPa (GB43), where attempts to analyse the few very small (<5–10 μm) melt pockets in the ML resulted in mixed analyses with adjoining mineral grains (hence not shown in the plots). Significant compositional trends only exist in the first ~600 μm of the PL. Deeper in the PL and in the first (few) hundred(s of) micrometre within the MT, glass compositions remain constant and overlap with the direct-mixing runs. Hence, we have focused here on ML and PL glass compositions and have not shown the last part of the PL’s and the MT’s in Fig. 4. However, the MT compositions were always used to test the quality of the PL glass analyses and are listed in Table 3. As said, some components show limited upward or downward trends within the MT’s, e.g. SiO2 commonly increases by 2–3 wt% from the PL side to the top of the capsule (see supplementary online table). These trends are interpreted as resulting from melting during the initial stages of the experiment, and incomplete back-equilibration over the entire MT during the timescale of the experiments. As compositional trends are continuous over the PL/MT interface, MT glasses nearest to the PL closely approach equilibrium compositions. Therefore, the MT liquid averages listed in Table 3 were based on MT glasses analysed close to the PL. The similarity of glass compositions of direct-mixing runs and glasses in the interior of the PL of GB14 (1,200°C) and GB41 (1,230°C) testifies the robustness of glass compositions over significant time (15–93 h) and ‘infiltrated’ liquid (6–20%) intervals. Moreover, as GB14 was affected by early disequilibrium melting of HK#19.1 (see “Starting materials”), early disequilibrium melting and the potential lower-P melting induced by the initially unfilled MT seem to have no significant affect on the final outcome of our infiltration-reaction experiments.
Although PL data are more variable than ML data, many components are, within error, equal in both domains. Generally, increasing temperature leads to higher abundances of Cr2O3 and lower abundances of Al2O3, Na2O, K2O and TiO2. However, spatial trends do occur for some components, sometimes only at specific P-T conditions. Al2O3 (Fig. 4a–c) for instance decreases from ~14 wt% in the ML to ~13 wt% at the PL/MT interface for 1,290°C runs at 0.8 GPa and a 1,260°C run at 0.65 GPa. SiO2 (Fig. 4d–f) increases by 1–1.5 wt% in the first few hundred μm into the PL’s of 0.65 GPa runs GB42 and GB44, and by about 2 and 0.5 wt% in the 0.8 GPa runs GB14 and GB41, respectively. In contrast, glass compositions in the 1,170°C run at 0.65 GPa might display a decrease in silica. Alkalis (Fig. 4g–l) appear fairly constant across a given charge, except for the 1,170°C run at 0.65 GPa, where PL glasses appear enriched near the ML. Upper values for Ca are equal for ML’s and PL’s, but significant scatter to lower values may occur due to quench crystallization of cpx, especially for glasses in PL’s at temperatures below 1,260°C. Only the 1,290°C HK#20 run displays a continuous increase in CaO from ~12 wt% in the ML to ~12.8 wt% near the MT. FeO (Fig. 4p–r) is fairly constant at higher temperatures, but displays downward trends from ML values into PL’s at 1,230°C and below. This leads to FeO contents that are higher in ML’s and lower in PL’s (beyond a few hundreds of μm) for glasses from 1,170 to 1,230°C runs, in comparison with higher-temperature glasses. TiO2 (Fig. 4s–u) tends to decrease from the ML into the PL, with the amplitude of the trend increasing significantly for runs containing plagioclase. Cr2O3 (Fig. 4v–x) shows fair amounts of scatter and no clear trends, although highest values are usually recorded in PL glasses.
Overall, Fig. 4 shows that differences between experiments run at equal P-T but with the different starting liquids HK#19 and HK#20 are small. In HK#20 runs, SiO2 and K2O are slightly lower at all temperatures and Na2O is lower above 1,200°C. Furthermore, CaO at 1,290°C is clearly lower in the ML of the HK#20 run than in the equivalent HK#19 run, but this difference continuously decreases towards the MT. Differences as a function of pressure for runs employing HK#19 are also limited in general. First, there is the aforementioned slight decrease in SiO2 at 0.65 GPa and 1,170°C that was not observed at 0.8 GPa. Moreover, upper values for CaO are 0.5–1 wt% lower at 0.65 GPa than at 0.8 GPa. Finally, average Cr2O3 contents of glasses from runs at 1,260°C and 0.65 GPa are ~0.2 wt% higher than even those of 0.8 GPa glasses at 1,290°C. The generally lower CaO at 0.65 GPa indicates a change in the reaction/melting equation (e.g. due to increased destabilization of opx; see “Discussion”). Similarly, higher Cr2O3 can be related to increasing degrees of melting, hence spinel dissolution, at lower pressure.
Total alkalis versus silica
Figure 5 shows total alkalis as a function of silica (cf. Le Bas et al. 1986) for glasses from PL’s of the same selection of experiments as shown in Fig. 4. All data plot in the basalt and basaltic andesite fields, with a few glasses from the 0.65 GPa 1,170°C run extending into the andesite field. The three series (BaSC2-HK#19 at 6.5 and 0.8 GPa, and BaSC2-HK#20 at 0.8 GPa) show similar evolutions as a function of decreasing temperature, although absolute values differ. Generally, total alkalis increase with decreasing temperature, whereas SiO2 first decreases and then increases again from ~1,260 to 1,230°C. The most significant difference for the two series with different starting liquid at a given pressure is that PL glasses in runs with HK#19 starting liquid are at any temperature richer in SiO2 than runs employing HK#20. For runs employing HK#19 as starting liquid, SiO2 is significantly higher at 1,260°C in the lower-P series. The same is possibly true at lower temperatures, but this cannot be concluded from the present set of experiments, as run temperatures differ.
Normative mineralogy of liquids
Glasses are plotted on the ol–cpx–qtz ternary of the basalt tetrahedron in Fig. 6. Quench-corrected glass compositions were recalculated into normative mineral end-member components (cf. Grove et al. 1992; Grove 1993) and projected from a normative oxide phase and feldspar. Figure 6a, b and c illustrates glass compositions of HK#19-BaSC2 reaction at 0.65 and 0.8 GPa, and HK#20-BaSC2 reaction at 0.8 GPa, as a function of temperature.
Glasses from all three series vary from olivine-normative to quartz-normative compositions, with the bulk of the data scattering fairly close to the opx–cpx tie-line that separates both fields. This is in agreement with the experimental data from anhydrous melting experiments (see Fig. 7a of Ulmer 2001), which reveals that partial melts coexisting with (cpx)–opx–ol residues start to become quartz-normative at 1.0 GPa and that normative quartz increases with decreasing pressure. The fact that especially our 0.65 GPa glasses plot to the right of the 1.0 GPa ol-opx phase boundary from Grove and Baker (1984) can also be explained as such. Apart for glasses from experiments ran at less than 1,215°C, compositional variation is fairly small. Moreover, some of the spread directed away from the cpx apex is due to cpx quench crystallization. The 1,170°C run at 0.65 GPa (Fig. 6a) and both 1,200°C 0.8 GPa runs (Fig. 6b, c) plot furthest away from the 1.0 GPa multiple saturation boundary. This is most significant for glasses of the HK#19-BaSC2 runs GB43 (0.65 GPa) and GB14 (0.8 GPa). PL and MT glasses for these runs show most deviation from the 1.0 GPa multiple saturation point. Aside from high overall variability, significant divergence of PL and MT glasses is observed in GB43, with the bulk of the glasses broadly scattering along an olivine control line (Fig. 6a).
Discussion
In the current experiments, we have juxtaposed 1.5 GPa partial melts of fertile lherzolite with depleted peridotite at lower pressures, thus ‘melt-rock’ reaction must occur. The grain-scale processes by which the starting liquid and the peridotite layer equilibrate, are described in Van den Bleeken et al. (2010). Mineral phases were documented to react with infiltrating liquid by cation-diffusion, dissolution ± reprecipitation, or a combination of both. The key factor controlling the reaction mechanism was shown to be the local melt composition, which is dependent on starting compositions, P-T conditions (fractionation of the ML, ‘melting’ of PL phases) and distance from the ML/PL interface (Van den Bleeken et al. 2010). The effects upon reaction on the liquid phase throughout the tholeiitic basalt-depleted peridotite reaction couple are discussed below. Furthermore, geological implications will be assessed by a comparison with natural glass compositions and a variety of observations made in case studies of natural upper-mantle rocks.
Comparison with liquid lines of descent of tholeiitic basalt and peridotite partial melting
Data from experimental studies on the crystallization of tholeiitic basalt and peridotite partial melting can serve as ‘end-member analogues’ for evolution of liquids in ML’s and PL’s of our experiments. A comprehensive experimental study investigating anhydrous fractional and equilibrium liquid lines of descent of tholeiitic liquids was performed by Villiger and co-workers on HK#19 (Villiger et al. 2004, 2007). Thus, we are able to compare HK#19 ML’s directly with the modal and compositional evolution of the tholeiitic fractionation ‘end-member’. Partial melting of a ‘depleted peridotite end-member’ was studied by Wasylenki et al. (2003), whose starting composition is very similar to that of BaSC, making results from their study a valuable point of reference for the PL domains in our runs.
Comparison of modal abundances of mineral and melt phases
On Fig. 7, modal abundances of mineral and melt phases for ML’s from 0.8 GPa BaSC2-HK#19 experiments are compared with modal abundances obtained in the Villiger et al. (2004, 2007) experiments.

Modal compositions as a function of temperature for ML’s from 0.8 GPa HK#19-BaSC2 reaction experiments (middle panel) and HK#19 equilibrium crystallization at 1.0 and 0.7 GPa (left and right panels, respectively; data from Villiger et al. 2004, 2007). Note that spinel also occurs in runs GB14 and GB08, but their abundances were too small to be included
Although modal abundances differ between ML’s in this study and the literature data, the phase assemblages as a function of temperature are fairly similar, especially considering the ±15°C temperature uncertainty of piston cylinder experiments. The ML domain thus largely evolves along the tholeiitic fractionation trend as a function of temperature, although differences occur due to compositional exchange with the PL. One indication for the latter is the extended olivine stability to lower temperatures in our reaction experiments compared to Villiger et al.’s HK#19 crystallization experiments, thereby leading to the formation of olivine gabbro. Moreover, a significant increase in olivine abundance results in a significant reduction in liquid mass above 1,230°C.
Comparison with quenched glasses in associated PL’s provides further information of the effect of melt-rock reaction on the reacting liquid. Modal abundances of PL’s (see Van den Bleeken et al. 2010) are evidently dominated by the initial BaSC mineralogy. However, an important qualitative observation is that the mineral assemblage precipitated from interstitial melt pools in the PL does not always fully correspond to the mineral mode in the adjacent ML’s. For instance, plagioclase occurs in the ML’s of one 1,230°C run (GB41) and all runs at lower temperature, but it only crystallized throughout melt pockets in the PL in the lowest-temperature 0.65 GPa run (GB43). Quantitatively, for the PL domain, we can only compare melt fractions with data from peridotite melting studies. Figure 8 illustrates melt percentages in PL’s from our 0.8 GPa runs, together with data from partial melting studies on depleted peridotite analogue DMM1 and a compilation of data on fertile peridotite MM3 (see caption of Fig. 9 of Wasylenki et al. 2003). Melt proportions for the present study were determined by image analysis on Mg, Al and Ca X-ray maps of 400 × 400 μm PL areas in close proximity to the ML (see Van den Bleeken et al. 2010). Consequently, they cover the area most affected by melt-infiltration and melt-rock reaction. Thus, not surprisingly, Fig. 8 shows that melt proportions as a function of temperature for these areas are significantly higher than for DMM1 and MM3. In other words, the apparent ‘solidus’ of the depleted peridotite has been shifted to significantly lower temperatures, as expected upon ‘fertilization’ of BaSC2 with tholeiitic melt (e.g. Hirschmann 2000; Yaxley 2000). It has to be noted though that, as ~6% of initial porosity remains after the HIP-stage, this accounts for part of the excess melt fraction at any given temperature through advective infiltration of ML liquid. Additionally, we compare melt proportions from our 0.8 GPa experiments with 1.0 GPa melting studies, and according to Asimow et al. (1997), the pressure difference could account for ~4% of excess melt at any given temperature (see their Fig. 7). A linear best-fit reveals fairly good correlation for all the 0.8 GPa data (R 2 = 0.89), with the slope representing an average melt productivity of 0.18%/°C. A second melt productivity ‘fit’ was plotted parallel with but to 10% lower melt proportions at any given temperature to account for the aforementioned initial porosity and P difference with the literature data. The calculated melt productivity is comparable to the productivities reported by Wasylenki et al. (2003) for cpx-saturated melting of DMM1 (0.3 ± 0.2%/°C) and MM3 (0.21 ± 0.02%/°C), and higher than the respective productivities above cpx-out (0.08 ± 0.01%/°C and 0.12 ± 0.01%/°C, respectively). In our experiments, we do not observe decreased melt productivity above cpx-out. However, the good correlation of the data over the investigated temperature range in combination with a realistic melt productivity rate strongly suggests that the obtained values truly constrain increased melting by reactive melt infiltration.

Melt fraction (F in wt%) as a function of temperature for PL’s from 0.8 GPa melt-rock reaction experiments and from a forward melting experiment on BaSC2 (both this study). For comparison, the results from (1.0 GPa) melting experiments on depleted (DMM1) and fertile (MM3) peridotite (after Wasylenki et al. 2003) are shown. Also plotted is a ‘corrected fit’ to our data that takes into account the initial porosity of the PL’s and the 0.2 GPa pressure difference with the literature data (see text for details)
Comparison of liquid compositions
To assess the compositional effects of reactive melt infiltration, oxide concentrations plotted as a function of MgO of quenched glasses from this study are compared with compositions obtained in (equilibrium) crystallization experiments on HK#19 (Villiger et al. 2004, 2007) and partial melting experiments on depleted peridotite (Wasylenki et al. 2003) in Fig. 9. MgO contents of partial melts (e.g. Baker and Stolper 1994; Falloon et al. 1999; Hirose and Kushiro 1993; Wasylenki et al. 2003) and fractionating tholeiitic liquids (e.g. Villiger et al. 2004, 2007) are positively correlated with temperature at constant pressure. Hence, as uncertainties on (inter-laboratory) applied temperatures are likely larger than those on measured MgO contents, we plotted the data as a function of MgO content. As our data were corrected to ‘equilibrium’ MgO values, they plot at distinct MgO contents for respective runs. By displaying all measured ML and PL data for every run, any spatial variation is preserved. To allow direct comparison to the HK#19 crystallization data of Villiger et al. (2004, 2007), Fig. 9 only encompasses data from HK#19-BaSC2 runs at 0.8 GPa. Furthermore, the entire set of individual glass compositions from the direct-mixing runs GB51 and GB52 are displayed for comparison.

Compositional variations of glasses from 0.8 GPa runs employing HK#19 (large circles for PL glasses, small circles for ML glasses) as a function of MgO content, compared with data from equilibrium crystallization experiments on HK#19 at 1 and 0.7 GPa (data from Villiger et al. 2004, 2007, respectively) and depleted peridotite DMM1 partial melts (Wasylenki et al. 2003). The composition of the low-degree partial melt of BaSC2 at 1,260°C and 0.8 GPa is represented in all panels by a blue square, and all measured glass compositions from the direct-mixing runs GB51 and GB52 are plotted as small yellow diamonds
A first observation is that glasses from the HK#19 crystallization trends (in particular at 1.0 GPa) and DMM1 quenched partial melts commonly overlap in the shared MgO range. More significantly, Fig. 9 reveals that glasses from this study generally scatter around the trends displayed by HK#19 equilibrium crystallization and DMM1 partial melting. PL data again show more scatter than ML data, but compositions of glasses from both domains always overlap. Al2O3 for instance shows close overlap for ML and PL hosted glasses, and good correlation with the literature data. The most significant deviations from the tholeiitic fractionation and peridotite melting trends are displayed by CaO, SiO2, TiO2, K2O and Cr2O3. It is also clear from Fig. 9 that compositions of glasses from the direct-mixing runs overlap completely with glass compositions of the 1,200–1,230°C infiltration-reaction runs. The apparent tendency of glasses from direct-mixing experiments (run at 1,200°C) to plot into the domain of the 1,230°C infiltration-reaction experiment GB41 can be the result of slightly lower amounts of melt at any given temperature in the PL’s of the infiltration-reaction experiments.
Highest CaO contents of ML and PL glasses overlap with the fractional crystallization and partial melting data, but (especially for PL glasses) extend to significantly lower values at T < 1,290°C (MgO content <13 wt%; see also Fig. 4m, n). This can be ascribed to quench modification by cpx. Therefore, CaO contents of glasses prior to quenching are supposed to have ‘equilibrium’ values.
TiO2 and K2O contents of quenched liquids generally decrease from the ML to the PL (see Figs. 4, 9). In comparison with literature data, lowest TiO2 values in PL’s at 1,200–1,230°C and all glasses at higher temperature correspond well with contents in DMM1 partial melts. ML and maximum PL TiO2 contents decrease from ~1.2 wt% at 1,200°C to below 0.6 wt% at 1,290°C. This decreasing trend is also found for 0.7 GPa HK#19 equilibrium crystallization (Villiger et al. 2007), but it is shifted to higher MgO contents (~T) in our runs. For instance, in equilibrium crystallization of HK#19, a value of ~1.2 wt% TiO2 is only reached at an MgO content of 7.13 wt% and a temperature of 1,180°C. Similarly, K2O measures ~0.18 wt% in the ML at 1,200°C, a value that is only reached at a temperature of 1,180°C during 0.7 kbar equilibrium crystallization of HK#19. Both the higher TiO2 and K2O can be explained by an increase in the level of fractionation in our ML’s in comparison with equilibrium and fractional HK#19 crystallization experiments at any given temperature (cf. Fig. 7). This is supported by the 1,170°C run at 0.65 GPa (GB43), where plagioclase formed throughout the reaction couple and TiO2 values are relatively high but constant. The higher amounts of incompatible elements and MgO of glasses from this study as a function of temperature, in comparison with glasses from HK#19 equilibrium crystallization, can be related to reaction with the PL. Upon melt-rock reaction within the PL, MgO will diffuse from the PL into the ML, promoting olivine fractionation. Hence, the residual melt becomes enriched in Al2O3 and CaO, consequently driving the residual liquid to plagioclase saturation. As a result, olivine and plagioclase abundances in the ML increase, as illustrated by Fig. 7. This can be quantified by calculating the portion of plagioclase in the solid fraction as plag* = [plag/(plag + cpx + opx + ol + spl)*100], with values taken from Van den Bleeken et al. (2010) and Villiger et al. (2004, 2007). Equilibrium crystallization of HK#19 at 0.7 GPa yielded 8.1% plag* at 1,180°C and 7.7% plag* at 1,210°C, whereas at 1.0 GPa, plagioclase is absent altogether at 1,210 and 1,180°C. At 1,200°C and 0.8 GPa (GB14), however, reaction between HK#19 and BaSC2 results in 28.9% plag*. Even at 1,150°C and 0.7 GPa, equilibrium crystallization of HK#19 only produces 18.9% plag*.
Cr2O3 contents of quenched liquids from this study extend to higher values than HK#19 crystallization and DMM1 melting trends for PL glasses in the 1,200–1,230°C range, and for ML and PL glasses at 1,260–1,290°C. For ML’s at 1,200 and 1,230°C, glasses have Cr2O3 contents that correspond with HK#19 crystallization. The elevated Cr2O3 contents in PL’s and ML’s at 1,260 and 1,290°C in part reflect the generally higher Cr2O3 content of 0.72 wt% in BaSC2 compared to the 0.39 wt% of DMM1 (Wasylenki et al. 2003). Moreover, the Cr2O3 content is controlled by initial Cr-bearing mineral cores (especially spinel, also cpx and opx; see Table 1), which undergo increasing dissolution with increasing temperature in reaction with the infiltrating melt (see Van den Bleeken et al. 2010).
Silica contents of glasses show at first sight ambiguous behaviour that, however, can be related to total alkali contents. SiO2 contents of glasses from the 1,230 and 1,260°C runs plot close to the 0.7 GPa HK#19 crystallization trends, but SiO2 is higher at both lower and higher temperature. At 1,200°C, most ML glasses also lie on the HK#19 fractionation trend, but PL glasses are significantly enriched in SiO2, also in comparison with low-degree BaSC melt. At 1,290°C, both ML and PL glasses plot to higher values. Possible reasons for PL glasses to plot to slightly higher values than DMM1 partial melts may be related to small modal and compositional differences of the starting compositions (e.g. Pickering-Witter and Johnston 2000; Schwab and Johnston 2001; Wasylenki et al. 2003). Additionally, the ML is a reservoir of Na2O, and melt advection and/or diffusion could increase the alkali contents in the PL enough to destabilize opx (e.g. Hirschmann et al. 1998; Lundstrom 2003) and locally increase SiO2. At 1,200°C, where the total melt fraction is small, diffusive infiltration (cf. Lundstrom 2003) would be most significant and can explain the highest SiO2 values in PL melt pockets. At 1,290°C, melt fractions are much higher, thereby increasing the level of equilibration over the reaction couple and diluting the Na excess. Because of the increasing significance of melting, probably aided by a small remaining ‘Na excess’ of the starting liquid, pyroxenes still undergo increased preferential dissolution. Although not shown, the observations and discussions from Fig. 9 can be extended to the compositional trends from 0.65 GPa BaSC2-HK#19 and 0.8 GPa BaSC2-HK#20 reaction experiments. Figure 4 reveals that, at 0.65 GPa, destabilization of opx by alkalis could be very prominent in the 1,170°C PL, especially in the first 300 μm with SiO2 >56 wt%, Na2O >3.5 wt% and K2O ~1 wt%. HK#20-BaSC runs display only slight increase in silica at 1,200 and 1,290°C, owing to the lower alkali contents of the starting liquid. For melting of dry peridotite, a minimum Na2O content for the destabilization of opx has been found to be ~3 wt% (e.g. Fig. 11 of Wasylenki et al. 2003). From this study, this threshold does not seem to apply for reactive melt infiltration in depleted peridotite. This is probably the case since any excess Na2O (compared to the peridotite partial melt at given P-T conditions) supplied by the liquid is available for reaction, whereas in equilibrium melting, it is mainly a function of cpx abundance and composition.
‘Extreme’ compositions by reactive infiltration-fractionation
Quenched liquids from 1,170 to 1,200°C runs have the largest compositional variation, both within a given run and between the different runs. K2O shows extreme enrichment in GB43 (1,170°C, 0.65 GPa) adjacent to the ML (Fig. 4j), and a general decrease with increasing temperature that is more pronounced in HK#19 runs compared to HK#20 runs (Fig. 4j–l). As K is an almost ‘perfect’ incompatible element in our runs, this variation is a good proxy for the high level of crystallization in the ML of GB43, and the higher initial concentration of K20 in HK#19 versus HK#20. Other major elements that are principally incompatible are Na and Ti. However, Na is far less incompatible than K, and Ti is to some extent taken up by cpx and spinel. This will concentrate the incompatible elements in different relative amounts in the melt phase of the plagioclase–cpx-dominated ML assemblages (e.g. Fig. 7). Moreover, infiltration of these incompatible-element-enriched melts into the PL can change the relative abundances again, both through reaction and diffusive fractionation and potentially through increased partial melting of the PL.
These processes are illustrated in Fig. 10, showing K2O/TiO2 as a function of SiO2. For the three series, K2O/TiO2 ratios generally increase with decreasing temperature, accompanied by increasing spread within a given run. K2O/TiO2 values are lowest for HK#20, since this starting composition has less than half of the K2O content of HK#19, but 2/3 of the TiO2 content (Table 1). In the 0.65 GPa 1,170°C run, the highest level of cpx + plag (see Fig. 2) fractionation across the reaction couple occurs within the ML and in the PL close to the ML. This results in the highest values of K2O/TiO2 and SiO2 in the PL near the ML. In the 0.8 GPa 1,200°C run, melt fractions are higher, and the fast diffusing K2O displays increased equilibration over the reaction couple. However, the slow diffusing TiO2 retains enrichment in and nearby the ML, resulting in increasing K2O/TiO2 from ML to PL. Moreover, as opx is now stable in the ML (Fig. 7), silica contents are buffered to lower silica in the ML and therefore mildly increase into the PL. As a result, there is a broad positive correlation between K2O/TiO2 and SiO2 for both HK#19 runs, but ML-to-PL trends are inversed (indicated by arrows on Fig. 10). It should be noted that the two runs have similar K2O/TiO2 ratios (~0.4) and SiO2 contents (~54 wt%) for PL glasses farthest away from the ML. These glasses are the least affected by reactive infiltration of fractionated ML liquids and consequently have K2O/TiO2 ratios overlapping with those of lowest-degree DMM1 melts (see Fig. 10).

K2O/TiO2 as a function of SiO2 for glasses from the present study, in comparison with data from equilibrium crystallization experiments on HK#19, depleted peridotite DMM1 partial melts, and low-degree partial melt of BaSC2 at 1,260°C and 0.8 GPa (for data sources, see caption of Fig. 9. Large circles are for PL glasses, and small diamonds are for ML glasses; all are colour-coded for temperature. Blue ellipses encircle glass compositions farthest away from the ML (‘deep PL’) for lowest-temperature runs of the 0.65 and 0.8 GPa BaSC2-HK#19 series. The compositions of glasses nearest to the PL are indicated as ‘near ML’. See text for discussion
Despite the limited length scale, and helped by the short run time, we thus observe a chromatographic decoupling of fast (e.g. K, Na) and slow (e.g. Ti) diffusers, which also depends on the compatibility of the compounds. Such processes have been described before in experiments (e.g. Lundstrom 2000, 2003) and recent numerical studies (e.g. Hauri 1997; Spiegelman and Kelemen 2003). Obviously, the compositional profiles in our experiments would have to be scaled to geological length and timescales.
Comparison with natural quenched liquids
The discussion of the experimental results above illustrates the complex influence that melt-rock reaction can exert on melt evolution in the upper-mantle TBL. At high temperatures and melt fractions, preferential dissolution of pyroxenes will increase SiO2 and lower the amount of incompatible elements in the melt phase. Moreover, as soon as cpx is consumed, CaO will decrease by dilution of olivine–opx components. At the lowest temperatures and melt fractions, the melt is a particularly complex function of initial liquid composition, local mineralogy, the amount of (reactive) fractionation, and compositional chromatography by diffusion. As such, our experiments express some features of reactive infiltration and fractionation as they are predicted in recent numerical studies, despite their small length and timescales. Generally, silica contents are high in the liquids and incompatible elements are enriched. Cr2O3 is consistently increased at all temperatures, mainly due to dissolution of spinel. These inferred systematics in reactive melt evolution potentially have profound effects on parental MORB-type liquids crossing a well-developed upper-mantle TBL. Here, we compare the newly obtained experimental results with quenched glasses found as volcanic rocks or as inclusions within phenocrysts and xenoliths contained in them.
Relicts of upper-mantle partial melts may be found in a variety of tectonic settings on the Earth’s (abyssal or aerial) surface, in the form of volcanic rocks or as minute glassy inclusions in phenocrysts and xenoliths contained in these rocks. Being the most ubiquitous natural ‘quenched partial melt’ deposited on the Earth’s surface, basalts from the global mid-oceanic spreading systems have been used to discern small-scale to regional variations in magma generation and mantle chemistry and dynamics (e.g. Elthon 1992; Kelemen et al. 1997; Korenaga and Kelemen 2000; Langmuir et al. 1992; le Roux et al. 2002a; Niu 2004). However, as parental MORB compositions can be modified by a variety of processes during melt extraction (e.g. melt-rock reaction, crystal fractionation, magma mixing and mingling), melt inclusions in phenocrysts have often been used instead as they could more closely represent primitive liquids of partial melting. These small portions of liquids, trapped during formation of the host mineral, could record directly the magma evolution upon its ascent to the surface. In the past few decades, it has become apparent though that compositions of inclusions can also be readily modified after entrapment (e.g. Danyushevsky et al. 2002; Spandler et al. 2007) or record entrapment-related reaction processes (e.g. Bedard et al. 2000; Danyushevsky et al. 2004; Yaxley et al. 2004). Such reaction processes during entrapment of melt inclusions are likely very similar to those occurring in the infiltration-reaction experiments. The same holds for glasses and reaction textures in xenoliths, which are attributed to either mantle metasomatism or reaction with the entraining melt (e.g. Shaw et al. 2006, and references therein).
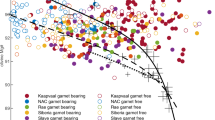
Comparison of glasses from this study with the most primitive (high-MgO) natural xenolith-hosted melt inclusions reveals large overlap between the experimental and the natural MORB data with respect to CaO and SiO2 (Fig. 11). In Fig. 11a, primitive (MgO > 8.5 wt%) olivine-, plagioclase- and spinel-hosted melt inclusions from MOR’s are plotted alongside primitive (MgO > 8.0 wt%) pillow-rim MORB glass analyses from the Smithsonian Institution catalogue (Melson and O’Hearn 2003—see also Melson et al. 2002). All glass analyses are taken from the PetDB database, accessed January 2009 (http://www.petdb.org/petdbWeb/index.jsp). For reference, fields for primitive high-CaO inclusions in phenocrysts from MOR’s and volcanic arcs, as well as the subduction-related lavas associated with the latter (after Fig. 1 of Danyushevsky et al. 2004) are shown. Primitive MORB glasses and ‘normal-CaO’ melt inclusions in MOR phenocrysts show nearly perfect overlap, reflecting a cogenetic relationship. PL glasses from our experiments are superimposed on the same fields in Fig. 11b. Except for the lowest-temperature glasses from HK#19-BaSC2 runs that plot to higher SiO2 and extend to slightly lower CaO, almost all glasses are constrained to the area taken by MORB and normal-CaO melt inclusions. The higher SiO2 and lower CaO of the lowest-temperature glasses would give rise to higher normative opx contents and thus explain some of the more silica-rich melts (~basaltic andesites, e.g. Fig. 5) at mid-ocean ridges.

CaO and SiO2 contents (wt.%) in melt inclusions from mid-ocean ridges (a) sub-divided in spinel-, plagioclase- and cpx-hosted inclusions (red, blue and yellow filled symbols, respectively); and PL glasses from the present study (b). For comparison, the compositional fields of arc- and MOR-related high-CaO melt inclusions, arc rock compositions (all after Fig. 1 of Danyushevsky et al. 2004) and MORB pillow-rim glasses with MgO >8 wt% from the Smithsonian Institution Volcanic Glass Data File (Melson and O’Hearn 2003; taken from PetDB, accessed January 2009) are given
Implications for reactive melt evolution in the TBL
Many of the liquids obtained in our tholeiitic basalt–depleted peridotite reaction experiments have fairly ‘normal’ (~N-MORB) compositions, but in detail show systematic compositional trends as a function of P-T conditions and initial starting liquid composition (e.g. Fig. 5). Translated to the Earth’s uppermost mantle, these apparent systematics can provide implications for melt migration through the TBL between asthenosphere and lithosphere.
Melt migration in the upper mantle is thought to occur by porous flow that is either diffuse or channelled in high-porosity dunitic conduits (e.g. Kelemen et al. 1997). Higher up, melt can be focused into dikes. The formation of high-porosity conduits is promoted by reactive percolation of pyroxene-undersaturated partial melts (e.g. Kelemen 1990). In our experiments, this is illustrated by the highest-temperature runs where dissolution of pyroxenes in the PL always yielded a harzburgitic residue, and sometimes a dunitic RBL adjacent to the ML (e.g. Fig. 2c, and Panel ① of Fig. 1). The liquids in these runs are close to N-MORB composition, and the small differences (e.g. elevated Cr2O3) would unlikely show up at the Earth’s surface after the various differentiation processes. However, if these SiO2-enriched and CaO-depleted liquids were to leave the high-porosity channels (e.g. Arai et al. 1997) and end up reactively percolating through the TBL, this could lead to dissolution of cpx (due to CaO-undersaturation) and formation of opx + ol as the liquid cools down, before reaching cpx ± plag saturation upon further cooling and/or reaction. Evidence for such a scenario is found in textural relations of numerous mantle peridotites, like the Lanzo peridotite body (e.g. Müntener et al. 2005; Piccardo et al. 2007). As reactive infiltration of the pyroxene-undersaturated liquids would initially increase the melt proportion and thus pore fraction (e.g. Fig. 8), this could reconcile the often more extensive than expected refertilization of many shallow peridotites.
Upon progressive cooling (hence crystallization) of the percolating liquids, the composition will become a complex function of reactive fractionation and the local lithology (~Panel ② of Fig. 1). This is illustrated in our experiments by the ‘extreme’ compositions in the lowest-temperature runs (see “Liquid compositions” and “Comparison with liquid lines of descent of tholeiitic basalt and peridotite partial melting”) and the recognition of chromatographic fractionation during reactive infiltration. Small-scale heterogeneities can also arise, as illustrated for instance in Fig. 4, by the downward trends of TiO2 and K2O from ML’s to PL’s and the large variability in Cr2O3 of PL glasses due to local reaction of Cr-spinel. Moreover, melt-rock reaction resulted in increased plagioclase fractionation. Occurrence of the latter in abyssal and ophiolitic peridotite has often been attributed to reactive porous flow (e.g. Borghini and Rampone 2007; Cannat et al. 2006; Dick 1989; Dijkstra et al. 2001; 2003; Menzies 1973; Müntener and Piccardo 2003; Müntener et al. 2004, 2005; Piccardo et al. 2004; Rampone et al. 1997; 2008; Seyler and Bonatti 1997; Takahashi 2001).
Direct evidence for the more evolved melts in the natural record would be difficult to find. In some cases, their compositions may preserve reaction processes occurring higher up in the magmatic ‘plumbing system’, as inferred for instance for high-CaO inclusions described by Danyushevsky et al. (2004). Nonetheless, it has been observed that melt inclusions in the most primitive (earliest) phenocrysts often record more profound liquid heterogeneity than later formed phenocrysts. Other potential sources of information for the nature of evolved reactive liquids are inclusions in xenoliths and modal, textural and compositional (including isotopic) observations in abyssal and ophiolitic peridotites. Many mantle xenolith-hosted glasses display far more ‘extreme’ compositions than glasses from our study (e.g. Shaw 1999). In part this reflects a combination of factors like differences in melting regimes and the presence of plume or subduction-related components (e.g. H2O and CO2), additional effects that fall well outside of the scope of the present study. However, one implication that follows from the present study is that high levels of reactive fractionation could have enlarged significantly the compositional heterogeneity of the liquids trapped in xenoliths. Moreover, it should be noted that ‘dry’ source regions of MORB contain small amounts of H2O (on average 50–250 ppm; see Hirschmann 2006) in non-hydrous minerals, reflected in the fact that MORB always contains traces of water. Hence, upon progressive cooling of parental MORB, H2O as well as other incompatible elements will concentrate in the liquid phase during shallow differentiation/reaction processes (e.g. Michael 1995; Hirschmann 2006). Therefore, in analogy to the expansion of melting to greater depths (/lower temperatures), reactive percolation of MORB-type liquids might occur to significantly shallower depths and temperatures due to increasing suppression of the solidus, with increasing fractionation/reaction and, hence, increased H2O contractions in the derivative liquids. As indicated by our intermediate- and lowest-temperature runs (~Panel ② to ③ in Fig. 1), progressive (reactive) crystallization would enrich the melt in Si, Al and incompatible elements, while Cr could be high due to reaction of spinel. In combination with the presence of H2O in the melt, the last, highly evolved melts potentially could form minerals like hornblende and phlogopite. Rare evidence for such a scenario, which could occur frequently in the TBL, is described by Matsukage and Arai (1998) and may be represented by traces of Ti-hornblende observed in many orogenic peridotites (e.g. Vannucci et al. 1995).
An additional geological implication resulting from our experimental study is the increased stability of olivine in ML’s. At P-T conditions where plagioclase is stable, reaction with a peridotitic host-rock thus promotes the formation of olivine-bearing gabbros. This in contrast to olivine-free gabbros that are predominantly formed in fractional crystallization experiments of MORB-type compositions (e.g. Villiger et al. 2004). At the same time, crystallization of olivine gabbros may release ‘enriched’ liquids into the host rocks with compositions similar to those in our 1,170–1,200°C runs. We are not aware of direct evidence (from melt inclusions) for such a scenario, but it was often proposed in case studies to explain compositional gradients of mineral phases around late-stage mafic dikes (e.g. Pertsev et al. 2009; Warren and Shimizu 2010). In our schematic Fig. 1, these are processes that could occur at the stages represented by Panels ② and ③.
Conclusions
This experimental study reveals that reaction with depleted peridotite can alter the liquid line of descent of tholeiitic melt ascending through a TBL between asthenospheric and lithospheric mantle. Although the local reactions and resulting liquid and residual rock compositions are a complex function of many variables, the experimental results allow presenting the following schematic overview of reactive melt variability in an upper-mantle TBL:
-
(1)
(Panel ① in Fig. 1) At temperatures around the ambient dry peridotite solidus, preferential dissolution of pyroxenes enriches the melt in silica and dilutes it in incompatible elements. Reactive infiltration of such melts at shallower levels results in peridotites enriched in opx and plagioclase. Reaction of spinel leads to Cr-enrichment in the melts;
-
(2)
(Panel ② in Fig. 1) At ‘intermediate’ temperatures, reaction becomes less efficient, hence silica contents decrease, whereas incompatible elements are concentrated in the melt as fractionation levels increase;
-
(3)
(Between Panels ② and ③ in Fig. 1) At ‘low’ temperatures, progressive reactive fractionation results in increased alkali contents in derivative liquids. This in turn promotes destabilization of opx, leading to increased silica contents. Adjacent to gabbro bodies and dikes, dissolution of opx from host peridotite and the resulting release of Mg can promote formation of olivine in the gabbros (opx → ol + liq).
Melt-rock reaction enhances crystallization of the infiltrating liquid, thus reduces its melt fraction. However, due to the addition of melt (e.g. lowering of the bulk Mg#) and melt-enhancing components such as alkalis (and water), the solidus of the infiltrated peridotite is lowered. Increasing levels of reactive fractionation concentrate these components in the derivative liquids, hence (progressively smaller amounts of) such liquids could migrate over long distances. Moreover, as liquids become more refractory, increasing amounts of alkalis and potentially H2O and CO2 will significantly lower the viscosity of these melts, further enhancing reactive percolation in the TBL.
Notes
As quench-rims of olivine are too thin to accurately measure their compositions by EMP, we have opted to use equilibrium ML olivine as a ‘best approximation’ for the olivine to be added back to the melt. We have also tested using equilibrium PL olivine instead, and this did not significantly change the results of the quench correction.
References
Aharonov E, Whitehead JA, Kelemen PB, Spiegelman M (1995) Channeling instability of upwelling melt in the mantle. J Geophys Res Solid Earth 100(B10):20433–20450
Arai S, Matsukage K, Isobe E, Vysotskiy S (1997) Concentration of incompatible elements in oceanic mantle: effect of melt/wall interaction in stagnant or failed melt conduits within peridotites. Geochemica et Cosmochimica Acta 61(3):671–675
Asimow PD, Stolper EM (1999) Steady-state mantle-melt interactions in one dimension: I. Equilibrium transport and melt focusing. J Petrol 40(3):475–494
Asimow PD, Hirschmann MM, Stolper EM (1997) An analysis of variations in isentropic melt productivity. Philos Transact A Math Phys Eng Sci 355(1723):255–281
Baker MB, Stolper EM (1994) Determining the composition of high-pressure mantle melts using diamond aggregates. Geochim Cosmochim Acta 58(13):2811–2827
Baker MB, Hirschmann MM, Ghiorso MS, Stolper EM (1995) Compositions of near-solidus peridotite melts from experiments and thermodynamic calculations. Nature 375(6529):308–311
Beck AR, Morgan ZT, Liang Y, Hess PC (2006) Dunite channels as viable pathways for mare basalt transport in the deep lunar mantle. Geophys Res Lett 33(1):L01202
Bedard JH, Hebert R, Berclaz A, Varfalvy V (2000) Syntexis and the genesis of lower oceanic crust. Ophiolites and Oceanic Crust: New Insights from Field Studies and Ocean Drilling Program (349):105–119
Borghini G, Rampone E (2007) Postcumulus processes in oceanic-type olivine-rich cumulates: the role of trapped melt crystallization versus melt/rock interaction. Contrib Mineral Petrol 154(6):619–633
Boyd FR, England JL (1960) Apparatus for phase-equilibrium measurements at pressures up to 50 kilobars and temperatures up to 1750°C. J Geophys Res 65(2):741–748
Brey GP, Köhler T (1990) Geothermobarometry in 4-phase lherzolites II: new thermobarometers, and practical assessment of existing thermobarometers. J Petrol 31(6):1353–1378
Cannat M, Lagabrielle Y, Bougault H, Casey J, de Coutures N, Dmitriev L, Fouquet Y (1997) Ultramafic and gabbroic exposures at the Mid-Atlantic Ridge: geological mapping in the 15°N region. Tectonophysics 279(1–4):193–213
Cannat M, Sauter D, Mendel V, Ruellan E, Okino K, Escartin J, Combier V, Baala M (2006) Modes of seafloor generation at a melt-poor ultraslow-spreading ridge. Geology 34(7):605–608
Daines MJ, Kohlstedt DL (1994) The transition from porous to channelized flow due to melt/rock reaction during melt migration. Geophys Res Lett 21(2):145–148
Danyushevsky LV, McNeill AW, Sobolev AV (2002) Experimental and petrological studies of melt inclusions in phenocrysts from mantle-derived magmas: an overview of techniques, advantages and complications. Chem Geol 183(1–4):PII S0009-2541(0001)00369-00362
Danyushevsky LV, Leslie RAJ, Crawford AJ, Durance P (2004) Melt inclusions in primitive olivine phenocrysts: the role of localized reaction processes in the origin of anomalous compositions. J Petrol 45(12):2531–2553
Dick HJB (1989) Abyssal peridotites, very slow spreading ridges and ocean ridge magmatism. In: Saunders AD, Norry MJ (eds) Magmatism in the ocean basins, vol 42. Geological Society of London Special Publication, London, pp 71–105
Dijkstra AH, Drury MR, Vissers RLM (2001) Structural petrology of plagioclase peridotites in the west Othris mountains (Greece): melt impregnation in mantle lithosphere. J Petrol 42(1):5–24
Dijkstra AH, Barth MG, Drury MR, Mason PRD, Vissers RLM (2003) Diffuse porous melt flow and melt-rock reaction in the mantle lithosphere at a slow-spreading ridge: a structural petrology and LA-ICP-MS study of the Othris Peridotite Massif (Greece). Geochem Geophys Geosyst 4:8613. doi:10.1029/2001GC000278
Doukhan N, Doukhan JC, Ingrin J, Jaoul O, Raterron P (1993) Early partial melting in pyroxenes. Am Mineral 78(11–12):1246–1256
Draper DS, Green TH (1999) P-T phase relations of silicic, alkaline, aluminous liquids: new results and applications to mantle melting and metasomatism. Earth Planet Sci Lett 170(3):255–268
Elthon D (1992) Chemical trends in abyssal peridotites—Refertilization of depleted suboceanic mantle. J Geophys Res Solid Earth 97(B6):9015–9025
Falloon TJ, Green DH, O’Neill HS, Hibberson WO (1997) Experimental tests of low degree peridotite partial melt compositions: implications for the nature of anhydrous near-solidus peridotite melts at 1 GPa. Earth Planet Sci Lett 152(1–4):149–162
Falloon TJ, Green DH, Danyushevsky LV, Faul UH (1999) Peridotite melting at 1.0 and 1.5 GPa: an experimental evaluation of techniques using diamond aggregates and mineral mixes for determination of near-solidus melts. J Petrol 40(9):1343–1375
Falloon TJ, Danyushevsky LV, Green DH (2001) Peridotite melting at 1 GPa: Reversal experiments on partial melt compositions produced by peridotite-basalt sandwich experiments. J Petrol 42(12):2363–2390
Grove TL (1993) Corrections to expressions for calculating mineral components in “Origin of calc-alkaline series lavas at Medicine Lake Volcano by fractionation, assimilation and mixing” and “Experimental petrology of normal MORB near the Kane Fracture Zone: 22°–25°N, mid-Atlantic ridge”. Contrib Mineral Petrol 114:422–424
Grove TL, Baker MB (1984) Phase equilibrium controls on the tholeiitic versus calc-alkaline differentiation trends. J Geophys Res 89:3253–3274
Grove TL, Kinzler RJ, Bryan WB (1992) Fractionation of mid-ocean ridge basalt (MORB). In: Phipps Morgan J, Blackman DK, Sinton JM (eds) Mantle flow and melt generation at mid-ocean ridges, vol 71. American Geophysical Union, Washington DC, pp 281–310
Hauri EH (1997) Melt migration and mantle chromatography, 1: simplified theory and conditions for chemical and isotopic decoupling. Earth Planet Sci Lett 153(1–2):1–19
Hirose K, Kushiro I (1993) Partial melting of dry peridotites at high pressures: determination of compositions of melts segregated from peridotite using aggregates of diamond. Earth Planet Sci Lett 114(4):477–489
Hirschmann MM (2000) Mantle solidus: experimental constraints and the effects of peridotite composition. Geochem Geophys Geosyst 1
Hirschmann MM (2006) Water, melting, and the deep earth H20 cycle. Annu Rev Earth Planet Sci 34:629–653
Hirschmann MM, Baker MB, Stolper EM (1998) The effect of alkalis on the silica content of mantle-derived melts. Geochim Cosmochim Acta 62(5):883–902
Hirschmann MM, Kogiso T, Baker MB, Stolper EM (2003) Alkalic magmas generated by partial melting of garnet pyroxenite. Geology 31(6):481–484
Ildefonse B, Blackman DK, John BE, Ohara Y, Miller DJ, MacLeod CJ, Integrated Ocean Drilling Program Expeditions 304/305 Science Party (2007) Oceanic core complexes and crustal accretion at slow-spreading ridges. Geology 35(7):623–626
Johnston AD, Schwab BE (2004) Constraints on clinopyroxene/melt partitioning of REE, Rb, Sr, Ti, Cr, Zr, and Nb during mantle melting: first insights from direct peridotite melting experiments at 1.0 GPa. Geochim Cosmochim Acta 68(23):4949–4962
Kamenetsky V, Crawford AJ (1998) Melt-peridotite reaction recorded in the chemistry of spinel and melt inclusions in basalt from 43 degrees N, Mid-Atlantic Ridge. Earth Planet Sci Lett 164(1–2):345–352
Kamenetsky VS, Crawford AJ, Meffre S (2001) Factors controlling chemistry of magmatic spinel: an empirical study of associated olivine, Cr-spinel and melt inclusions from primitive rocks. J Petrol 42(4):655–671
Kelemen PB (1990) Reaction between ultramafic rock and fractionating basaltic magma I. Phase-relations, the origin of calc-alkaline magma series, and the formation of discordant dunite. J Petrol 31(1):51–98
Kelemen PB, Joyce DB, Webster JD, Holloway JR (1990) Reaction between ultramafic rock and fractionating basaltic magma II. Experimental investigation of reaction between olivine tholeiite and harzburgite at 1150°C–1050°C and 5 kB. J Petrol 31(1):99–134
Kelemen PB, Hirth G, Shimizu N, Spiegelman M, Dick HJB (1997) A review of melt migration processes in the adiabatically upwelling mantle beneath oceanic spreading ridges. Philos Trans R Soc Lond Ser A Math Phys Eng Sci 355(1723):283–318
Kinzler RJ, Grove TL (1992) Primary magmas of midocean ridge basalts I. Experiments and methods. J Geophys Res Solid Earth 97(B5):6885–6906
Köhler TP, Brey GP (1990) Calcium exchange between olivine and clinopyroxene calibrated as a geothermobarometer for natural peridotites from 2 to 60 KB with applications. Geochim Cosmochim Acta 54(9):2375–2388
Korenaga J, Kelemen PB (2000) Major element heterogeneity in the mantle source of the North Atlantic igneous province. Earth Planet Sci Lett 184(1):251–268
Lambart S, Laporte D, Schiano P (2009) An experimental study of focused magma transport and basalt–peridotite interactions beneath mid-ocean ridges: implications for the generation of primitive MORB compositions. Contrib Mineral Petrol 157(4):429–451
Langmuir CH, Klein EM, Plank T (1992) Petrologic systematics of mid-ocean ridge basalts: constraints on melt generation beneath ocean ridges. In: Phipps-Morgan J, Blackman DK, Sinton JM (eds) Mantle flow and melt generation at mid-ocean ridges. Geophysical Monograph, American Geophysical Union, vol 71, pp 183–280
Laporte D, Toplis MJ, Seyler M, Devidal J-L (2004) A new experimental technique for extracting liquids from peridotite at very low degrees of melting: application to partial melting of depleted peridotite. Contrib Mineral Petrol 146(4):463–484
Le Bas MJ, Lemaitre RW, Streckeisen A, Zanettin B (1986) A chemical classification of volcanic rocks based on the total alkali silica diagram. J Petrol 27(3):745–750
le Roux PJ, le Roex AP, Schilling JG (2002a) MORB melting processes beneath the southern Mid-Atlantic Ridge (40–55 degrees S): a role for mantle plume-derived pyroxenite. Contrib Mineral Petrol 144(2):206–229
le Roux PJ, le Roex AP, Schilling JG, Shimizu N, Perkins WW, Pearce NJG (2002b) Mantle heterogeneity beneath the southern Mid-Atlantic Ridge: trace element evidence for contamination of ambient asthenospheric mantle. Earth Planet Sci Lett 203(1):479–498
Le Roux V, Bodinier JL, Tommasi A, Alard O, Dautria JM, Vauchez A, Riches AJV (2007) The Lherz spinel lherzolite: refertilized rather than pristine mantle. Earth Planet Sci Lett 259(3–4):599–612
Liang Y (2003) Kinetics of crystal-melt reaction in partially molten silicates: 1. Grain scale processes. Geochem Geophys Geosyst 4:1045. doi:10.1029/2002GC000375
Liang Y, Guo Y (2003) Reactive dissolution instability driven by chemical diffusion with applications to harzburgite reactive dissolution. Geophys Res Lett 30(13)
Lo Cascio M, Liang Y, Hess PC (2004) Grain-scale processes during isothermal-isobaric melting of lherzolite. Geophys Res Lett 31(16)
Lundstrom CC (2000) Rapid diffusive infiltration of sodium into partially molten peridotite. Nature 403(6769):527–530
Lundstrom CC (2003) An experimental investigation of the diffusive infiltration of alkalis into partially molten peridotite: implications for mantle melting processes. Geochem Geophys Geosyst 4(9):8614
Matsukage K, Arai S (1998) Jadeite, albite and nepheline as inclusions in spinel of chromitite from Hess Deep, equatorial Pacific: their genesis and implications for serpentinite diapir formation. Contrib Mineral Petrol 131(2–3):111–122
Melson WG, O’Hearn (2003) Smithsonian volcanic glass file. (data file available for download on http://www.petdb.org)
Melson WG, O’Hearn T, Jarosewich E (2002) A data brief on the Smithsonian Abyssal Volcanic Glass Data File. Geochem Geophys Geosyst 3
Menzies MA (1973) Mineralogy and partial melt textures within an ultramafic-mafic body, Greece. Contrib Mineral Petrol 42:273–285
Michael PJ (1995) Regionally distinctive sources of depleted MORB: evidence from trace elements and H2O. Earth Planet Sci Lett 131:301–320
Morgan Z, Liang Y (2003) An experimental and numerical study of the kinetics of harzburgite reactive dissolution with applications to dunite dike formation. Earth Planet Sci Lett 214(1–2):59–74
Morgan Z, Liang Y (2005) An experimental study of the kinetics of lherzolite reactive dissolution with applications to melt channel formation. Contrib Mineral Petrol 150(4):369–385
Müntener O, Piccardo GB (2003) Melt migration in ophiolitic peridotites: the message from Alpine-Apennine peridotites and implications for embryonic ocean basins. In: Dilek Y, Robinson PT (eds) Ophiolites in earth history, vol 218. Geological Society of London Special Publication, pp 69–89
Müntener O, Pettke T, Desmurs L, Meier M, Schaltegger U (2004) Refertilization of mantle peridotite in embryonic ocean basins: trace element and Nd isotopic evidence and implications for crust-mantle relationships. Earth Planet Sci Lett 221(1–4):293–308
Müntener O, Piccardo GB, Polino R, Zanetti A (2005) Revisiting the Lanzo peridotite (NW-Italy): ‘Asthenospherization’ of ancient mantle lithosphere. Ofioliti 30:111–124
Müntener O, Manatschal G, Desmurs L, Pettke T (2010) Plagioclase peridotites in ocean–continent transitions: refertilized mantle domains generated by melt stagnation in the shallow mantle lithosphere. J Petrol 51(1–2):255–294
Niu YL (2004) Bulk-rock major and trace element compositions of abyssal peridotites: implications for mantle melting, melt extraction and post-melting processes beneath mid-ocean ridges. J Petrol 45(12):2423–2458
Ortoleva P, Chadam J, Merino E, Sen A (1987) Geochemical self-organization. 2. The reactive-infiltration instability. Am J Sci 287(10):1008–1040
Pertsev AN, Bortnikov NS, Aranovich LY, Vlasov EA, Beltenev VE, Ivanov VN, Simakin SG (2009) Peridotite-melt interaction under transitional conditions between the spinel and plagioclase facies beneath the Mid-Atlantic Ridge: insight from peridotites at 13°N. Petrology 17(2):124–137
Piccardo GB, Müntener O, Zanetti A, Pettke T (2004) Ophiolitic peridotites of the Alpine-Apennine system: mande processes and geodynamic relevance. Int Geol Rev 46(12):1119–1159
Piccardo GB, Zanetti A, Pruzzo A, Padovano M (2007) The North Lanzo peridotite body (NW Italy): lithospheric mantle percolated by MORB and alkaline melts. Periodico Di Mineralogia 76(2–3):199–221
Pickering-Witter J, Johnston AD (2000) The effects of variable bulk composition on the melting systematics of fertile peridotitic assemblages. Contrib Mineral Petrol 140(2):190–211
Rampone E, Piccardo GB, Vannucci R, Bottazzi P (1997) Chemistry and origin of trapped melts in ophiolitic peridotites. Geochim Cosmochim Acta 61(21):4557–4569
Rampone E, Piccardo GB, Hofmann AW (2008) Multi-stage melt-rock interaction in the Mt. Maggiore (Corsica, France) ophiolitic peridotites: microstructural and geochemical evidence. Contrib Mineral Petrol 156(4):453–475
Robinson JAC, Wood BJ, Blundy JD (1998) The beginning of melting of fertile and depleted peridotite at 1.5 GPa. Earth Planet Sci Lett 155(1–2):97–111
Schiano P, Clocchiatti R, Shimizu N, Weis D, Mattielli N (1994) Cogenetic silica-rich and carbonate-rich melts trapped in mantle minerals in Kerguelen ultramafic xenoliths: implications for metasomatism in the oceanic upper mantle. Earth Planet Sci Lett 123(1–4):167–178
Schiano P, Provost A, Clocchiatti R, Faure F (2006) Transcrystalline melt migration and Earth’s mantle. Science 314(5801):970–974
Schwab BE, Johnston AD (2001) Melting systematics of modally variable, compositionally intermediate peridotites and the effects of mineral fertility. J Petrol 42(10):1789–1811
Seyler M, Bonatti E (1997) Regional-scale melt-rock interaction in lherzolitic mantle in the Romanche Fracture Zone (Atlantic Ocean). Earth Planet Sci Lett 146(1–2):273–287
Seyler M, Lorand JP, Toplis MJ, Godard G (2004) Asthenospheric metasomatism beneath the mid-ocean ridge: evidence from depleted abyssal peridotites. Geology 32(4):301–304
Shaw CSJ (1999) Dissolution of orthopyroxene in basanitic magma between 0.4 and 2 GPa: further implications for the origin of Si-rich alkaline glass inclusions in mantle xenoliths. Contrib Mineral Petrol 135(2–3):114–132
Shaw CSJ, Heidelbach F, Dingwell DB (2006) The origin of reaction textures in mantle peridotite xenoliths from Sal Island, Cape Verde: the case for “metasomatism” by the host lava. Contrib Mineral Petrol 151(6):681–697
Shervais JW (1979) Thermal emplacement model for the Alpine lherzolite massif at Balmuccia, Italy. J Petrol 20(4):795–820
Sobolev AV, Shimizu N (1993) Ultra-depleted primary melt included in an olivine from the Mid-Atlantic Ridge. Nature 363(6425):151–154
Spandler C, O’Neill HSC, Kamenetsky VS (2007) Survival times of anomalous melt inclusions from element diffusion in olivine and chromite. Nature 447(7142):303–306
Spiegelman M, Kelemen PB (2003) Extreme chemical variability as a consequence of channelized melt transport. Geochem Geophys Geosyst 4:1055. doi:10.1029/2002GC000336
Spiegelman M, Kelemen PB, Aharonov E (2001) Causes and consequences of flow organization during melt transport: the reaction infiltration instability in compactible media. J Geophys Res Solid Earth 106(B2):2061–2077
Takahashi N (2001) Origin of plagioclase lherzolite from the Nikanbetsu peridotite complex, Hokkaido, northern Japan: implications for incipient melt migration and segregation in the partially molten upper mantle. J Petrol 42(1):39–54
Ulmer P (2001) Partial melting in the mantle wedge–the role of H2O in the genesis of mantle-derived ‘arc-related’ magmas. Phys Earth Planet Inter 127(1–4):215–232
Ulmer P, Luth RW (1991) The graphite-COH fluid equilibrium in P, T, fO2 space—an experimental determination to 30 kbar and 1600°C. Contrib Mineral Petrol 106(3):265–272
Van den Bleeken G, Müntener O, Ulmer P (2010) Reaction processes between tholeiitic melt and residual peridotite in the uppermost mantle: an experimental study at 0.8 GPa. J Petrol 51(1–2):153–183
Van der Wal D, Bodinier JL (1996) Origin of the recrystallisation front in the Ronda peridotite by km-scale pervasive porous melt flow. Contrib Mineral Petrol 122:387–405
Vannucci R, Piccardo GB, Rivalenti G, Zanetti A, Rampone E, Ottolini L, Oberti R, Mazzucchelli M, Bottazzi P (1995) Origin of LREE-depleted amphiboles in the subcontinental mantle. Geochim Cosmochim Acta 59:1763–1771
Villiger S, Ulmer P, Müntener O, Thompson AB (2004) The liquid line of descent of anhydrous, mantle-derived, tholeiitic liquids by fractional and equilibrium crystallization—an experimental study at 1.0 GPa. J Petrol 45(12):2369–2388
Villiger S, Ulmer P, Müntener O (2007) Equilibrium and fractional crystallization experiments at 0.7 GPa; the effect of pressure on phase relations and liquid compositions of tholeiitic magmas. J Petrol 48(1):159–184
Wark DA, Watson EB (2000) Effect of grain size on the distribution and transport of deep-seated fluids and melts. Geophys Res Lett 27(14):2029–2032
Warren JM, Shimizu N (2010) Cryptic variations in abyssal peridotite compositions: evidence for shallow-level melt infiltration in the oceanic lithosphere. J Petrol 51(1–2):395–423
Wasylenki LE, Baker MB, Kent AJR, Stolper EM (2003) Near-solidus melting of the shallow upper mantle: partial melting experiments on depleted peridotite. J Petrol 44(7):1163–1191
White RS (1988) The earth’s crust and lithosphere. J Petrol (Special Lithosphere Issue):1–10
Yaxley GM (2000) Experimental study of the phase and melting relations of homogeneous basalt plus peridotite mixtures and implications for the petrogenesis of flood basalts. Contrib Mineral Petrol 139(3):326–338
Yaxley GM, Kamenetsky VS, Kamenetsky M, Norman MD, Francis D (2004) Origins of compositional heterogeneity in olivine-hosted melt inclusions from the Baffin Island picrites. Contrib Mineral Petrol 148(4):426–442
Zhang HF (2005) Transformation of lithospheric mantle through peridotite-melt reaction: a case of Sino-Korean craton. Earth Planet Sci Lett 237(3–4):768–780
Acknowledgments
This work was financially supported by the Swiss National Science Foundation (SNSF Grant PP0022-102809). The Electron Microprobe Lab at the Institute of Geological Sciences at the University of Bern is supported by SNF Grant 200021-103479/1. Final preparation of the paper was supported by SNSF Grant 200021-120054. We wish to acknowledge thoughtful comments by Marc M. Hirschmann on a draft version of this contribution. Two anonymous reviewers and the editorial comments of Jon Blundy significantly improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Blundy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Van den Bleeken, G., Müntener, O. & Ulmer, P. Melt variability in percolated peridotite: an experimental study applied to reactive migration of tholeiitic basalt in the upper mantle. Contrib Mineral Petrol 161, 921–945 (2011). https://doi.org/10.1007/s00410-010-0572-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-010-0572-5