
Abstract
Concentration gradients in calcium are common in metamorphic or magmatic garnets and can be used to determine the timescales of geological processes. However, the kinetics of Ca diffusion in garnet is poorly constrained and experimental studies have to date yielded widely varying diffusion coefficients. In this paper, we describe a new method for generating diffusion profiles in garnet. We incorporated polished and compositionally homogeneous garnet seeds in a finely ground powder of clinopyroxene and garnet. During the experiments (1.3 GPa, 1,050–1,250°C, and ƒO2 ≤ the graphite-O2 buffer), the mineral powder partially melted, recrystallized, and formed a 10–50 μm wide overgrowth zone of compositionally distinct garnet around the seeds. Long duration experiments generated measurable relaxation profiles at these seed/overgrowth interfaces. We performed analytical transmission electron microscope traverses across the interfaces in each experiment. Thirteen usable compositional profiles were obtained with characteristic distances of diffusion ranging from 300 to 1,000 nm. From these profiles, Ca–(Fe, Mg) interdiffusion coefficients were retrieved using an analytical solution for the diffusion equation and the data were cast in an Arrhenius relation. Linear regression of the data yields an activation energy Q Ca–(Fe, Mg) equal to 188 ± 48 kJ mol−1 and a frequency factor D 0 equal to 6.6 × 10−14 m2 s−1. Within the compositional range studied, the composition of garnet has no major effect on the Ca–(Fe, Mg) interdiffusion coefficient. The very slow diffusion rate of Ca is in agreement with natural observations indicating that Ca diffuses more slowly than Fe and Mg. The Ca diffusion coefficients derived from this study are not model-dependent and can be used to determine the durations of geological events from Ca relaxation profiles in natural garnets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Minerals are markers of geological processes, whose time constants vary from a few hours, in volcanic conduits for instance, to millions of years in orogenic belts. The characteristics of these mineralogical records include compositional zoning, the presence of mineral or melt inclusions, and reaction rims. In this respect, garnet is of special interest: it has long been regarded as a sensor of pressure (P), temperature (T), and mineral reactions during metamorphism (e.g. Tracy 1982; Loomis 1983; Chakraborty and Ganguly 1991). In addition, experiments and petrological studies demonstrate that garnet grows also during partial melting of crustal rocks and in response to compositional (X) changes of the system (Vielzeuf et al. 2005c). Garnet growth under changing P–T–X conditions leads to compositional zoning in major elements (Ca, Fe, Mg, and Mn). Whatever the origin of the initial zoning, if the crystal is held at high temperature for a long time, chemical diffusion through the crystal lattice (volume diffusion) will, at the limit, produce a compositionally homogeneous garnet. Incomplete relaxation leads to diffusion profiles that can be used as a quantitative chronometer if the original profile can be inferred and if the relevant diffusion coefficients are known (see Perchuk and Philippot 2000; Dachs and Proyer 2002; Faryad and Chakraborty 2005 for references). This kinetic approach represents an interesting complement to radiometric measurements it allows for determining the duration of specific processes, it can be applied to rocks of any age, and it covers a wide range of timescales (e.g. Philippot et al. 2001; Costa and Chakraborty 2004). For this purpose, garnet is attractive because: (1) slow diffusion rates enable garnet to retain the chemical signatures of its initial growth, (2) diffusion in garnet is expected to be isotropic owing to its atomic structure, and (3) natural alumino-silicate garnets contain four divalent cations (Fe, Mg, Mn, and Ca) with potentially different diffusion rates. To date, the large uncertainties in the reported diffusion coefficients of calcium in garnet limit the use of Ca-zoning to accurately model timescales of geological processes.
Only four self-diffusion coefficientsFootnote 1 (D*Fe, D*Mg, D*Mn, and D*Ca) need to be known as a function of P and T to calculate diffusion profiles for a specific garnet couple (Lasaga 1979; Brady 1995). In recent years, self-diffusion coefficients for Fe, Mg, and Mn have been evaluated either directly by isotopic tracer diffusion experiments (Cygan and Lasaga 1985; Chakraborty and Rubie 1996; Schwandt et al. 1995), or indirectly from interdiffusion couples (Loomis et al. 1985; Gerasimov 1987; Chakraborty and Ganguly 1992; Ganguly et al. 1998). In contrast, the diffusivity of Ca is largely unconstrained and is still a matter of debate. Volume diffusion of Ca is expected to be slower than volume diffusion of other divalent cations because the effective ionic radius (r) of Ca is larger than that of Mn2+, Fe2+, and Mg2+ (r = 1.12 Å, compared to 0.96, 0.92, and 0.89 Å, respectively; Shannon 1976). This inference is consistent with the extremely slow Ca self-diffusion coefficients determined experimentally at 1 atm by Schwandt et al. (1996). However, these results are not compatible with the Ca–(Fe, Mg) interdiffusion coefficientsFootnote 2 determined by Freer and Edwards (1999). In this latter study, the authors concluded that Ca–(Fe, Mg) interdiffusion coefficients are significantly higher than those estimated previously (e.g. Chakraborty and Ganguly 1992), and even higher than the rate of Fe–Mg and Fe–Mn exchange in garnet (e.g. Chakraborty and Ganguly 1992). Obviously, new experiments are required to provide constraints on Ca diffusivity, especially for relatively Ca-poor garnets (<20% grossular, grs). In an effort to provide these constraints, we carried out a series of interdiffusion experiments at 1,050–1,250°C and 1.3 GPa. In this paper, we present the experimental details and describe some characteristic run products. We also discuss the analytical techniques used to obtain high spatial resolution chemical profiles, present representative results, and analyse the data in terms of Ca–(Fe, Mg) interdiffusion. In a subsequent article, we will present a multicomponent simulation of these experimental profiles.
Experimental methods
A common way to measure interdiffusion coefficients is to conduct experiments using a garnet–garnet couple made of two gem-quality crystals with different compositions and highly polished surfaces. Elphick et al. (1985) describe in detail experiments of this type. Zoning profiles induced in such couples contain, in theory, more information than self-diffusion experiments because of the possibility to constrain both the interdiffusion coefficients and their compositional dependencies, as well as the self-diffusion coefficients. The application of this technique has produced a wealth of valuable data (Elphick et al. 1985; Chakraborty and Ganguly 1992; Ganguly et al. 1998; Freer and Edwards 1999). However, these authors note that the approach is not without experimental difficulties. For instance, the mechanical integrity of the garnet–garnet couple is difficult to preserve during the experiment (Elphick et al. 1985; Chakraborty and Rubie 1996; Ganguly et al. 1998). Even if the contact between the two garnets is perfect, there is no crystallographic continuity at the interface and damage at each surface due to mechanical polishing can affect the profile of slow-diffusing species (Chakraborty and Rubie 1996). Finally, large supplies of gem-quality crystals of constant composition are difficult to find (Ganguly et al. 1998). We have developed a novel method for generating garnet–garnet diffusion couples that is well suited for conducting interdiffusion experiments.
Starting materials and experimental design
We prepared small polished garnet seeds (Ø 250–350 μm) using the air abrasion technique developed by Krogh (1982). Garnets from a metapelitic granulite found as a xenolith in the late Tertiary alkaline volcano from Bournac—Massif Central (Leyreloup et al. 1977) were used as starting material for the seeds. We performed numerous electron microprobe (EMP) analyses on randomly selected grains in order to determine the chemical composition and homogeneity of this garnet (alm50.8 prp45.3 spe0.5 grs3.4 —Vielzeuf et al. 2005a; Table 1; endmember garnet abbreviations are listed in the footnote to Table 1). We mixed the polished garnet seeds in a finely ground (≤5 μm) mineral powder made of 44.4 wt.% clinopyroxene (SY5) and 55.6 wt.% garnet (M6-401) (both compositions are listed in Table 1); we will refer to this fine-grained mineral powder as the matrix. The mass ratio of garnet seeds to matrix material in this mixture is ≈1/10 by mass. Note that the garnet in the matrix has a different composition than that of the garnet seeds (Table 1).
For each experiment, we put splits of the mixture of garnet seeds and matrix in a graphite container that in turn was sealed within a platinum capsule (Fig. 1b). At the beginning of an experiment, the minute garnet grains in the matrix react with the clinopyroxene and result in the growth of a new garnet zone of distinct composition around the spheroid seeds. This experimental design is based on the observation that relict garnet cores in high-pressure runs are often surrounded by newly formed garnet rims with a dissimilar composition (Perchuk and Lavrent’eva 1983; Vielzeuf and Holloway 1988; Patiño Douce and Johnston 1991).

a Piston–cylinder pressure cell for high pressure/high temperature experiments. b Enlargement of the capsule and design of the seed/overgrowth interdiffusion experiment (left hand side: before the experiment, right hand side: after the experiment). The setup is designed to allow the development of a garnet overgrowth of distinct composition on the seed. Long duration experiments allow diffusion to proceed at the seed/overgrowth interface
The choice of minerals in the matrix comes from an experimental study carried out by Alaoui et al. (1997) on the Fe–Mg partitioning between clinopyroxene and garnet. This study showed that the garnet that formed during the experiments tended towards a Ca-rich composition (∼grs18), and that the partial melt that formed at temperatures as low as 1,000°C served as an efficient flux that aided the approach to equilibrium. Thus, in our kinetic Seed/Overgrowth Interdiffusion (SOI) experiments the rapid growth of a garnet rim over a garnet seed generates a step-like compositional gradient, which then relaxes via chemical diffusion over the course of the isothermal experiment. This SOI experiment is possible only because crystal growth is orders of magnitude faster than chemical diffusion (Haase et al. 1980).
Experimental apparatus
All experiments were carried out at 1.3 GPa using a 1.91-cm piston–cylinder apparatus at LMV (Clermont-Ferrand). The pressure assembly (Fig. 1a) is nearly the same as that described by Vielzeuf and Clemens (1992). Independent temperature calibrations suggest that the temperature difference between the thermocouple tip and the centre of the capsule is less than 5°C. To perform an experiment, the assembly was cold pressed to 0.3 GPa and the temperature was raised to 650°C at 50°C/min. At 650°C, the assembly softened and over the course of 5 min the nominal pressure was raised to 1.3 GPa. The temperature was then raised to the nominal temperature of the experiment at 50°C/min. The final temperature of the experiment was usually reached in less than 30 min; this time interval is considered negligeable in comparison to the duration of the experiment. No friction correction was applied to the nominal pressure and no pressure correction was applied to the thermocouple emf.
Analytical techniques
Electron microprobe
Electron microprobe analyses were performed using a four spectrometer Cameca SX100 instrument at LMV. Operating conditions were a 15 kV accelerating voltage and a 10 nA sample current. The counting times for each element were 10 s on the peak and 5 s on the background. The following standards were used: natural albite (Si, Na), orthoclase (K), corundum (Al), synthetic hematite (Fe), olivine (Mg), wollastonite (Ca), MnTiO3 (Mn, Ti), Cr2O3 (Cr), and a glass with 1,000 ppm P2O5 (P); data were reduced using a ZAF correction. For both the EMP and the analytical transmission electron microscopy (ATEM) analyses discussed below, total iron was treated as Fe2+ and the molar proportions of the garnet end-members (alm, prp, grs, and spe) were calculated as the percentage of the number of atoms of Fe or Mg or Ca or Mn over the sum of the number of divalent cations: Fe + Mg + Ca + Mn.
Focused ion beam
High spatial resolution TEM analyses using an Energy Dispersive Spectrometer require a sample transparent to electrons, i.e. not thicker than ∼300 nm. We prepared TEM garnet foils using the Focused Ion Beam (FIB) technique. FIB milling was performed with an FEI Model 200 TEM FIB system at CP2M (Marseille, France). A thin layer of platinum (1–2 μm) is deposited on the specimen at the location that is to be milled to protect the sample from abrasion and reduce gallium implantation in the garnet. A 30 kV Ga+ beam operating at ∼20 nA excavates garnet from both sides of the Pt layer to a depth of 5–7 μm. On one side of the platinum strip, the trench is sputtered to produce a broad angle to allow the foil to be cut along its edges by tilting the sample to about 45°. Thinning is achieved by the repeated milling of steps on both sides of the foil. When the foil is about 500 nm thick, it is cut almost free along both sides with only two narrow strips left to hold the section in place. This step prevents possible tension and bending of the foil. Thinning to the final thickness (ideally 100 nm) necessitates careful observation, slight tilting of the sample during milling, and a progressive reduction of the beam current (to 100 pA) to minimize amorphization of, and gallium implantation into the sample. Finally, the stabilizing strips are cut and the foil (∼15 × 5 × 0.2 μm) is lifted out by micromanipulator and deposited on a carbon coated TEM copper grid. In the early stages of this study, milling a section across the interface between garnet seed and its overgrowth proved difficult because of the limited imaging capabilities of the FEI Model 200 TEM FIB. We solved this problem by carrying out preliminary SEM observations of each sample and by ‘marking’ domains of interest at the garnet–garnet interfaces by performing long duration (∼30 min) analytical traverses with the SEM on these regions. The marks left by these traverses could be detected under the FIB and greatly facilitated the preparation of the strips. This marking method necessitates keeping the same carbon coating on each sample for both the SEM observation and the FIB preparation.
Analytical transmission electron microscopy (ATEM)
Analytical transmission electron microscopy analyses on the garnet FIB foils were performed in the French TEM facility at Marseille and Lille. At Lille, TEM observations and X-ray microanalyses were carried on a Philips CM30 transmission electron microscope operating at 300 kV. This TEM is equipped with an energy-dispersive X-ray spectrometer (EDS Tracor-Voyager) composed of a Ge detector and an ultra-thin window. This configuration allows the detection and quantification of light elements. The K-emission line of oxygen is detected with a high sensitivity but quantitative microanalysis requires precise knowledge of the thickness of the analysed area to apply absorption corrections. We used the absorption correction method developed by van Cappellen and Doukhan (1994) for silicate minerals. This method yields precise and reproducible results consistent with the rules of cation-site occupancy. The relative errors based on counting statistics are ∼2% for major elements. For each compositional profile, we introduced a further correction by normalizing the TEM analyses to those collected with the EMP on either side of the interface. Individual spot analyses within a traverse were selected using the scanning transmission electron mode, a technique that allows for precise spot location, regular spacing of spots along a traverse, and the possibility to store and retrieve any spot position along a traverse. Measuring a compositional profile involved four steps: first, we performed fast qualitative traverses to locate the interface which is not visible; second, we positioned the traverse normal to the interface; third, we measured the compositional profile with a relatively wide spacing between spots (∼500 nm) to locate the interdiffusion zone; and forth, we performed additional analyses in the zone of interest with a narrower spacing (usually 100 nm) to improve the resolution of the profile. For most measurements, we used a highly focused electron beam (∼6 nm) with an electron beam current of 1.2 nA. Each analysis lasted about 3 min, the time required to reach 6,000 counts on oxygen.
We performed another series of measurements at CRMCN (Marseille, France) using a 200 kV JEOL-2000FX electron microscope. This TEM is equipped with a double, ±30° tilt, side-entry, high-resolution specimen holder and a Tracor-Northern energy-dispersive X-ray spectrometer (TN 5502, series II) with a Si–Li diode detector and a thin window allowing the analysis of light elements. An interesting feature of this instrument is the high take-off angle of the X-rays (72°) collected by the diode through the upper objective polar piece. Since no tilt of the specimen is required for the analysis, the incident electron beam remains parallel to the garnet seed-overgrowth interface and that minimizes profile spreading. A high signal-to-noise spectrum is collected and no absorption correction is necessary. We used beam diameters of 30–40 nm, a constant beam current of 1.2 nA, and a 100 s counting time throughout the study. We processed the raw data (peaks minus fitted backgrounds) using a Semi-quantitative Microanalysis for Thin Films (SMTF) data reduction procedure that is close to the original Cliff and Lorimer method (Cliff and Lorimer 1975; Lorimer and Cliff 1976). The Kx,Si “K-factors” for X-ray Kα emissions of x = Ca, Mg, and Al were pre-calibrated against stoichiometric silicate standards, and built-in values were used for Fe and Ti. We checked that electron channeling (the Borrman effect) had not corrupted the recorded data.
Results
Description and characterization of the run products at the microscale (400–2 μm)
We carried out seven experiments at 1.3 GPa and temperatures of 1,050–1,250°C. The duration of the experiments varied from 5 to 42 days (Table 2).
The main features of the garnet seeds and the garnet–clinopyroxene–glass matrix after annealing are shown in photomicrographs in Fig. 2.

Backscattered electron images of the run products (scanning electron microscope and electron microprobe). a Low magnification image of a run product showing the garnet seeds in the recrystallized matrix (exp. Dif2, 1,150°C, 1.3 GPa for ∼22 days). b Recrystallized matrix and interstitial melt. In this back-scattered electron (BSE) image, glass is darker than clinopyroxene, which in turn is darker than garnet. Some garnet-inherited cores are still visible (lighter than the rim and denoted by black arrows) indicating that diffusion was unable to erase the garnet heterogeneities in the matrix at a 5 μm scale (exp. Dif2). c Enlargement of Fig. 2a showing a garnet seed and its overgrowth. Note the euhedral shape of the garnet. d Another example of a garnet seed and euhedral overgrowth. Note that the difference in grey level in this BSE image is indicative of a compositional contrast between the garnet seed and the overgrowth rim. In this experiment (Dif3, 1,250°C, 1.3 GPa for ∼5 days), the proportion of melt is high. e Garnet overgrowth in experiment Dif4 (1,200°C, 1.3 GPa for ∼36 days). Note the presence of rounded inclusions of clinopyroxene. f Garnet overgrowth in experiment Dif3; an enlargement of Fig. 2d. Note the quench crystals at the glass/overgrowth interface. The darker lines at the seed/overgrowth interface are marks left after electron microprobe traverses
Except in one experiment (Dif3, 1,250°C), garnet and clinopyroxene crystals in the matrix are evenly distributed, small (∼10 μm, e.g. Fig. 2b), euhedral, and commonly associated with melt that tends to segregate around the garnet overgrowths (Fig. 2c). In the Dif3 experiment at 1,250°C, the proportion of melt is high, most garnet crystals in the matrix have dissolved, and clinopyroxene crystals have segregated to the lowest part of the capsule. However, garnet seeds with overgrowth rims are still present (Fig. 2d, f). The back-scattered electron (BSE) images of the charges indicate that, as intended, garnet overgrowths formed around the garnet seeds (Fig. 2). In addition, the BSE images show that the seeds and overgrowths have different grey levels indicating that they are compositionally distinct. The total range of overgrowth widths is 10–40 μm. Unfortunately, the geometry of the experiment (overgrowth around a spheroid seed) does not allow the determination of a relationship between the width of the overgrowth, the temperature, and the duration of the experiment. In some experiments, the overgrowths exhibit euhedral faces suggesting that the chemically composite garnets are single crystals. Small rounded inclusions of clinopyroxene (and sometimes melt) are present in the overgrowths but it is quite easy to find regions that are inclusion-free.
We performed a series of EMP analytical profiles across the overgrowth/seed interface for each experiment. One representative profile per experiment is shown in Fig. 3.

Representative electron microprobe compositional profiles for each experiment. Note that the composition of the overgrowth (LHS) changes as a function of the temperature of the experiment. Due to the spatial averaging effect of the beam, these EMP profiles cannot be directly interpreted in terms of diffusion profiles
These profiles confirm that the garnet seeds are compositionally homogeneous (RHS of the profiles; Fig. 3) and show that, in general, the overgrowth zones have fairly constant compositions (LHS of the profiles; Fig. 3c, f, g). In some of the overgrowth zones, the almandine content increases monotonically while the pyrope content decreases outwards (LHS of the profiles; e.g. Fig. 3a, b, d, e). These variations may reflect a shift in the Fe/Mg ratio of the matrix with increasing thickness of the garnet overgrowth. Nevertheless, these compositional variations are relatively small (2–5 mol% over a distance of 10–20 μm) and do not affect the grossular content across the overgrowth zone.
Multiple traverses performed on garnets in a given experiment yield compositional profiles that are identical within analytical error. However, two different experiments run at the same nominal temperature and pressure often generate slightly different overgrowth compositions (see Fig. 3e, f). This variation can be ascribed to a slight change in the bulk composition reflecting very small changes in the mass ratio of garnet seeds to matrix material. Figure 3 shows that the compositions of the overgrowth zones are significantly richer in grossular (grs17–20) than the garnet seeds (grs3). Another noticeable feature is that the garnet composition of the overgrowth zone depends on the temperature of the experiment (Figs. 3, 4; Table 3); i.e. while the change in grossular content is limited (from grs20 at 1,050°C to grs16 at 1,250°C), the changes in alm and prp contents are more dramatic (from prp34–alm44 at 1,050°C to prp53–alm30 at 1,250°C). These changes reflect the temperature dependence of Fe–Mg partitioning among the phases in the experimental charge.

Compositions of the garnet overgrowths as a function of temperature. The duplicated data at 1,150 and 1,200°C with different durations of the experiments have been offset for clarity
In summary, Fig. 3 shows that the compositions of the overgrowth zones are different from the compositions of the garnet seeds. The plateaus on the LHS of the profiles (Fig. 3) are either flat or display patterns that are regular enough to determine initial conditions at the interface. However, regardless of the temperature or duration of the experiment the diffusion profile for Ca, Mg, or Fe is inevitably ∼3 μm wide. Since the width of the diffusion zone should scale with temperature and run duration, we infer that the EMP profiles are not simple diffusion profiles but reflect strong spatial averaging due to the large diameter of the microprobe’s electron beam (and analytical volume) relative to the true width of the diffusion zone (Ganguly et al. 1998). To avoid this problem we used an ATEM, which has a spatial resolution several orders of magnitude smaller than the EMP to measure the concentration gradients within our garnet couples (see Meissner et al. 1998).
Description and characterization of the run products at the micro to nanoscale (15 μm–10 nm)
As discussed above, FIB foils were cut perpendicular to the trace of the interface between the seed and the overgrowth (Fig. 5a). A foil at its final stage of preparation is shown in Fig. 5b.

Observations of the samples at micro and nanoscales. a Preparation site of a FIB foil at the seed (lower left)/overgrowth (upper right) interface. Note the clinopyroxene inclusions in the overgrowth (exp. Dif3). b FIB foil (black arrow) at a final stage of preparation (Dif3). c TEM bright-field image of a FIB foil recorded with parallel electron beam (Dif4). The dashed line marks the seed/overgrowth boundary determined by chemical profiling. Note the similarity of the electron diffraction patterns recorded from two selected areas in the seed (upper right) and the overgrowth. d Large-angle convergent beam electron diffraction image of a FIB foil (same sample as Fig. 5c). Note the sharpness of the Bragg fringes and the fact that they cross the seed/overgrowth interface without disruption or deviation. This is indicative of good crystal quality and same crystallographic orientation of the seed and the overgrowth. e TEM bright-field image of another FIB foil (Dif4). Note the absence of a physically distinctive boundary between the seed/overgrowth interface (arrows and light dashed line). The 500 nm–spaced ‘graduations’(white arrows) are the result of the superposition of two consecutive raster areas (500 × 250 nm, enhanced by the dashed rectangles) from a first traverse; see text for details. The regularly spaced and aligned black dots are the marks of the impacts of the highly focused TEM beam (∼6 nm) from a second traverse. The approximate size of an electron microprobe beam is shown for a comparison (lighter circle on the right). f Enlargement of Fig. 5d showing the dumb-bell shaped doublets of the highly focused electron beam of the ATEM. Each dark spot of the doublet (black arrows) corresponds to the entrance or the exit locus of the electron beam on the foil. The white aureole around each doublet (white arrow) is due to amorphization of the garnet under the beam. Doublets are visible because the FIB foil was tilted while the photograph was taken
Figure 5c is a TEM bright-field image of a FIB foil collected with a parallel electron beam. The image shows a low density of dislocation lines while the presence of strain contrast along the Bragg fringes indicates the presence of inclusions. Large-angle convergent beam electron diffraction (LACBED) is a TEM technique that provides crystallographic information (Bragg lines) superimposed on a defocused image of the sample (Tanaka et al. 1980). It provides simultaneous information on the real and the reciprocal space of the crystal and provides a characterization of crystal defects (Cherns and Morniroli 1994; Cordier et al. 1995; Morniroli et al. 1997). Figure 5d shows a LACBED image obtained on a FIB section from experiment Dif4 (1,200°C; 36 days). The Bragg fringes indicate good crystal quality for both the seed and the overgrowth and show that neither grain nor subgrain boundaries are present. The image shows that as the Bragg fringes cross the seed/overgrowth interface they are neither disrupted nor modified. This indicates that the seed and the overgrowth have the same crystallographic orientation and are in fact a single crystal. The similarity of the electron diffraction patterns recorded from two selected areas in the seed and the overgrowth (Fig. 5c) confirm this conclusion. Since there is no physical evidence of the interface, its position can only be determined by chemical methods. Figure 5e shows the trace of the impacts of the TEM electron beam on the FIB foil. An initial traverse was done using a 500 × 250 nm rastered beam. The regularly spaced linear graduations in Fig. 5e are the result of the superposition of the boundaries of two consecutive rastered areas. A second traverse with a highly focused beam (~6 nm) was superposed upon the first defocused one. It generated the succession of black dots at 100 nm intervals. The approximate size of an EMP beam is shown for comparison. Figure 5f is an enlargement of Fig. 5d and shows the traces of the impacts along two traverses. Due to the tilt of the FIB foil while the photograph was taken, the analytical beam traces are no longer seen as spots but as dumb-bell shaped doublets. Each dark spot of the doublet corresponds to the entrance or the exit locus of the TEM beam on the foil. The white aureole around each doublet may be due to minor amorphization of the garnet structure under the beam. Concerning the ATEM traverses, it is important to note that we cut the FIB foils perpendicular to the trace of the interface (Fig. 5a) and that, during ATEM sessions, we paid attention on locating the interface on the FIB foil so as to perform traverses perpendicular to the interface (Fig. 5c). This double orientation, normal to the successive traces of the interface, minimizes the overestimation of the diffusivities that would occur if traverses were non-perpendicular. In this study we measured 16 ATEM profiles. Of these, one did not reach the Ca-rich plateau, one did not reach the Ca-poor plateau due to technical TEM problems, and one proved unacceptable due to complicated patterns generated by topographic defects at the surface of the garnet seed. This left a total of 13 useable experimental profiles four of which are shown in Fig. 6 and include data on Fe, Mg, Ca, and Mn.

Four representative sets of Fe, Mg, Ca, and Mn diffusion profiles obtained at 1,050, 1,100, 1,150, 1,200°C, and 1.3 GPa. In all profiles the seed is on the right and the overgrowth on the left, as in Fig. 3. The titanium distribution along the profiles is also shown (Ti* and Ti**: concentrations multiplied by 50 and 100, respectively). In most cases, Ti can be used as a marker of the original interface. The smooth lines correspond to simulated curves using Ca–Fe, Mg) interdiffusion coefficients of 3 × 10−21, 4 × 10−21, 8 × 10−21 and 2 × 10−20 m2 s−1, from a to d, respectively. Note that in three cases (a, b, and d), Fe, Mg, and Ca profiles can be simulated with a single interdiffusion coefficient
Because of the low concentrations of manganese in both the seeds and the overgrowth rims, our experiments cannot be used to calculate Mn diffusivities. However, Fe, Mg, and Ca profiles are well defined and provide important constraints on the diffusivities of these elements. The most important observation is that the width of the diffusion zone is extremely small in all of the sets: from about 300 nm in experiment Dif1 (1,150°C and 5 days; Fig. 6c) to about 1,000 nm in experiment Dif4 (1,200°C and 36 days; Fig. 6d). In the long duration experiments, the width of the Fe, Mg, and Ca diffusion zones in a given ATEM traverse are identical. This is not the case in the short duration experiment (Dif1) where the width of the Ca diffusion zone is narrower than those for Fe and Mg (e.g. Fig. 6c). At present, we do not have an explanation for this observation. Figure 6 shows that there is a significant difference in titanium concentrations between the seeds and the overgrowths, with a sharp transition between the two domains. Unpublished observations in natural garnets (plutonic rocks and lavas) by one of us (D.V.) indicate that, like P, Ti provides a marker of the original interface between growth zones (see Vielzeuf et al. 2005c for images showing phosphorus). This apparent lack of P and Ti diffusion is consistent with these elements being coupled with silicon and having extremely low diffusivities. The presence of Fe, Mg, and Ca diffusion zones combined with a sharp Ti transition in our experiments indicates that the Ti diffusion coefficient is many orders of magnitude lower than those for Fe, Mg, and Ca, and thus we consider titanium as a reliable marker of the original interface. In Fig. 6, we observe some irregularities along certain elemental profiles, in particular those of calcium (e.g. Fig. 6a at 3.8 μm and Fig. 6d at 9.7 μm). We ascribe these compositional irregularities to either the presence of tiny clinopyroxene or melt inclusions in the garnet or irregularities at the original surface of the garnet seeds due to imperfect air abrasion polishing. Numerical modelling of multicomponent diffusion, not discussed further in this article, indicates that these Ca spikes are not the result of uphill diffusion. However, the wavy shape along the Mg profile (Fig. 6c) is indicative of uphill diffusion, a specific feature of multicomponent diffusion that to date has not been unambiguously observed in natural or experimental garnets (Ganguly 2002, p. 282). These features are discussed in more detail by Vielzeuf et al. (2005b) and will be further developed in a subsequent paper. Finally, in some TEM profiles we observe an intermediate plateau close in composition to the garnet seed (e.g. Fig. 6d between 9.7 and 11.5 μm). We tentatively ascribe these plateaus to a recrystallization process that occurs at the surface of the garnet seed early in the experiment.
Determination of Ca–(Fe, Mg) interdiffusion coefficients
Smooth concentration profiles in a diffusion couple can be used in different ways to derive diffusion coefficients (see Chakraborty et al. 1995, p. 263 for a discussion). Although diffusion in garnet is a multicomponent process (Loomis 1978), we defer such a detailed analysis for a subsequent paper and here simply treat the data in terms of an interdiffusion coefficient that is independent of composition. A similar approach was taken by Elphick et al. (1985) and Freer and Edwards (1999). The advantage of such an approach is that it combines kinetic and thermodynamic information into a phenomenological coefficient well suited for determining the duration of geological events. In addition, numerical simulations indicate that the determination of “pseudobinary” interdiffusion coefficients in garnet instead of multicomponent interdiffusion coefficients introduces a maximum error factor of 3, well within the error bar that one can accept for diffusion coefficients. However, care should be taken to apply these Ca diffusivities to compositions within the range of those used to derive the coefficients (∼grs3–grs20). In this study, the Ca–(Fe, Mg) interdiffusion coefficient (D) was determined using the analytical solution for diffusion given by Crank (1975):
where C is the element concentration as a function of distance; C max and C min the initial values of the upper and lower plateau; x is distance (x = 0 at the interface); and t is time. In the present case, the diffusion coefficient is the unknown and we determined it by visual adjustment of the calculated curve to the experimental data. Errors on Ds were estimated by searching the upper and lowermost D-values bracketing the experimental data; most of them are within the range of ±0.4 log units. To calculate the Ca–(Fe, Mg) interdiffusion coefficients, we primarily used the grossular profile. Since the Mn content of both the core and rim garnets is low, the sum of Fe + Mg is necessarily close to (100−X grs) and thus a curve that fits the grossular profile is also valid for (Fe + Mg). Examples of calculated curves fitting the experimental data are shown in Fig. 6. In most cases (Fig. 6a, b, d), the profiles for Ca, Fe, and Mg in a particular diffusion couple can be fit by a single interdiffusion coefficient. This is in agreement with an observation made earlier by Elphick et al. (1985). However, the Fe and Mg profiles in Fig. 6c cannot be fit with the interdiffusion coefficient determined for Ca. This is due to the uphill diffusion pattern that appears in the Mg profile. Such a pattern cannot be generated by binary diffusion. Nevertheless, this does not prevent us from using the calcium profile to determine a Ca–(Fe–Mg) interdiffusion coefficient so long as uphill diffusion does not affect the Ca profile. We checked the results of our analytical solution for Ca with numerical calculations based on a finite difference algorithm for the diffusion equation \( \partial C{\text{/}}\partial t = \partial {\text{/}}\partial x{\text{[}}D \times \partial C{\text{/}}\partial x{\text{]}} \) (Kirkaldy and Young 1987, p. 502). Both methods yield identical results which are summarized in Table 4.
As anticipated from the narrow diffusion zones in the experimental profiles (Fig. 6), the diffusivity of Ca in garnet is extremely slow, from 3 × 10−21 m2 s−1 at 1,050°C to 4 × 10−20 m2 s−1 at 1,250°C. Among the retrieved diffusion coefficients listed in Table 4, two cases deserve some comments. The first case concerns profile numbers 4 and 5 measured in the same FIB foil during the same ATEM session, which yield significantly different results. Further investigations using Scanning Transmission X-ray Microscopy (STXM) and a synchrotron source indicated that profile number 4 came from a zone with an irregular interface (K. Benzerara, Personal communication), which probably led to an overestimate of the diffusion coefficient. The second case concerns profile number 9. As discussed earlier, this profile was obtained using a 550 × 250 nm rastered beam. This method induced some convolution of the signal and an overestimate of D. The very same traverse measured with a highly focused beam (profile number 10) generated a calcium diffusivity four times slower than the convoluted profile. Thus we have not included profile numbers 4 and 9 in the final linear regression of the data. In order to demonstrate that diffusion is the rate-controlling process and that the retrieved Ds are independent of time, we ran two experiments at 1,150°C for 5 and 22 days and two experiments at 1,200°C for 6 and 36 days. The inset in Fig. 6d shows the two Ca diffusion profiles (numbers 8 and 10) obtained from the 1,200°C runs for 6 and 36 days, respectively. The profile from the short duration experiment at 1,200°C (profile number 8) was slightly translated to allow a direct comparison with the profile from the 36-day run at the same temperature. The two profiles yield, within error, identical diffusion coefficients (see Table 4). We did not succeed in retrieving a complete profile for the 22-day experiment at 1,150°C. However, if we consider the portion of the profile located on the Ca-poor side of the diffusion couple (not shown here), we obtain a D of 1 × 10−20 m2 s−1, in good agreement with the value of 8 × 10−21 m2 s−1 obtained from the 5-day experiment (profile number 5).
The temperature dependence of the diffusion coefficient, D, follows an Arrhenius equation:
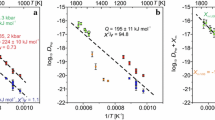
where D 0 is a frequency factor (m2 s−1) that incorporates the activation entropy of diffusion and can be seen as an empirical quantity measuring the fundamental vibrational frequency of the lattice; Q is the activation energy (J mol−1) and represents the height of the potential barrier for a jump of an atom in the lattice; R is the gas constant (8.314 J mol−1 K−1); and T is the temperature (K) (see Adda and Philibert 1966). Figure 7 shows log D-values calculated using the Ca profiles from our experiments plotted against reciprocal absolute temperature.

Arrhenius plot of experimentally determined Ca–(Fe, Mg) interdiffusion coefficients (m2 s−1) in the range 1,050–1,250°C, at 1.3 GPa in graphite capsules. Black diamonds (at 1,050 and 1,200°C) indicate two coincident data (see Table 4 )
In this diagram, the slope of the least-squares line corresponds to −Q/(2.3026R) while the intercept at 1/T = 0 gives log D 0. The best fit line through the experimental data (R = 0.94) gives Q = 188 ± 48 kJ mol−1 and log D 0 = −13.18 ± 1.74 (reported uncertainties are 1σ). These uncertainties are based on the scatter of the data in Fig. 7 and incorporate uncertainties on the individual Ds. The values of Q and D o given above are valid at 1.3 GPa and oxygen fugacities less than or equal to the graphite-O2 buffer. Assuming an activation volume of diffusion (ΔV a) independent of pressure, the value of Q at one bar can be calculated from the activation energy at the experimental pressure P (in bars), Q p, using the relation
At present there is no consensus on the activation volume of diffusion for Ca in garnet (see Ganguly et al. 1998; Freer and Edwards 1999). Following Ganguly et al. (1998) we assume that Ca has the same ΔV a as Mn (0.6 J bar−1 mol−1) since the effective ionic radius of Ca is closer to Mn than to any other common divalent cation (Chakraborty and Ganguly 1992). Using this assumption, the activation energies of Ca–(Fe, Mg) interdiffusion calculated at 1 bar and 10 kbar are 180 and 186 kJ mol−1, respectively (Table 5).
Discussion
Comparison with previous studies
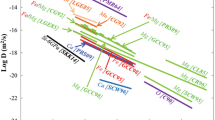
Previous experimental determinations or estimates of Ca diffusivity in garnet are listed in Table 5 and plotted in Fig. 8.

Freer and Edwards (1999) carried out Ca–(Fe, Mg) interdiffusion experiments between natural single crystals of grossular and almandine in the range 900–1,100°C and 1.5–3.25 GPa. They found an activation energy (Q) equal to 248 kJ mol−1 and a frequency factor (D 0) of 1.22 × 10−6 m2 s−1. These values generate the fastest divalent cation diffusivities for garnet in the literature and are difficult to reconcile with our results. The experiments of Freer and Edwards were done at iron-wüstite. On the basis of the work of Buening and Buseck (1973) on olivine, Chakraborty and Ganguly (1991) argue that diffusion coefficients in Fe-bearing garnets vary directly with (fO2)1/6. Adjusting the results of Freer and Edwards to the graphite–O2 buffer decrease their estimated D by ∼0.5 log units. Thus, differences in oxygen fugacity cannot account for the difference between their results and ours. Comparing the results of our study with the other datasets in the literature is complicated by the fact that the experiments of Chakraborty and Ganguly (1992), Ganguly et al. (1998), and Schwandt et al. (1995, 1996) were interpreted in terms of self-diffusion coefficients. However, as noted by Loomis et al. (1985), self-diffusivities of Fe, Mg, and Ca derived from multicomponent simulations of diffusion couples or self-diffusion experiments should bracket the range of related interdiffusion coefficients. Thus, for each considered study we plotted the fastest and slowest self-diffusion coefficient for Fe, Mg, and Ca in Fig. 8. Although the diffusivities proposed by Chakraborty and Ganguly (1992) and Ganguly et al. (1998) are slower than the ones proposed by Freer and Edwards (1999) by about three orders of magnitude, they still do not bracket our data and thus, for temperatures above 800°C, are inconsistent with our results. However, because of significant differences in the slopes of the lines in Fig. 8 (i.e. the activation energies) our mutual estimates converge at temperatures of ∼650°C. Thus, in the temperature range of 600–700°C timescales of geological events calculated using our dataset and the one provided by Ganguly et al. (1998) will be consistent within an order of magnitude. Schwandt et al. did not estimate garnet Fe self-diffusion Ds and thus it is difficult to compare our results to theirs. However, the activation energy that we determined for Ca–(Fe, Mg) interdiffusion is close to the one determined for Ca self-diffusion by Schwandt et al. (1996) and thus both datasets are consistent if we assume that Fe and Mg self-diffusivities are faster than Ca diffusivity by 1.5–2 orders of magnitude.
Relaxation profiles in natural garnets have been used for a long time to retrieve diffusion coefficients (Yardley 1977; Loomis 1978; Dempster 1985; Ayres and Vance 1997; Carlson 2002). The main argument stated in favour of this inverse method is that it avoids the large uncertainties that arise from extrapolating experimental data down to metamorphic temperatures. Carlson (2002) derived a set of self-diffusion coefficients for Fe, Mg, Ca, and Mn by numerical simulation of diffusion profiles in natural garnets from polymetamorphic terranes with inferred thermal histories. Figure 8 indicates that these Fe, Mg, and Ca diffusivities are faster than those derived by Chakraborty and Ganguly (1992). Other natural samples have been used to provide constraints on Ca diffusivity in garnet. Kohn (2004) described oscillatory- and sector-zoned garnets from the Main Central Thrust in central Nepal and argued that the presence of sector zoning in Ca, but not in Mn or Mg, implied maximum intracrystalline diffusion rates for Ca of ∼1 × 10−25 m2 s−1 at 0.8 GPa and 550°C, and faster rates for Fe, Mg, and Mn. Extrapolating our Ca–(Fe, Mg) interdiffusion data to these P–T conditions yields a value of 1.4 × 10−25 which is in agreement with the value derived by Kohn (2004), although it must be noted that the garnet compositions in the two studies are significantly different. Vielzeuf et al. (2005c) studied Ca and oxygen isotope diffusion profiles in metamorphic/magmatic garnets from the Pyrenees (France) and estimated that log D Ca–(Fe, Mg) (m2 s−1) lay in the range −22 ± 1 at 850°C and 0.6 GPa. The value calculated from the present experimental data at the same P and T is −21.7, in good agreement with the estimate of Vielzeuf et al. (2005c). A comparison between Ca and oxygen isotope profiles measured along the same traverse led Vielzeuf et al. (2005c) to infer that oxygen diffusion in garnet is the same order of magnitude as Ca–(Fe, Mg) interdiffusion, and thus coupled with the results of this study suggest that log D ox (m2 s−1) is close to −22 at 850°C and 0.6 GPa.
In order to visualize the differences between the different datasets for Ca diffusivities in garnet, we applied the data available in the literature to the experimental profile obtained at 1,100°C. The results are shown in Fig. 9.

Penetration curves of Ca in experiment Dif6 (1,100°C, 1.3 GPa, 42 days) simulated with diffusion coefficients available in the literature. Same abbreviations as in Fig. 8. See text for discussion
For the datasets of Freer and Edwards (1999) and Schwandt et al. (1996), we used the same analytical solution as the one described earlier in this paper (Eq. 1). The three other datasets (Chakraborty and Ganguly 1992; Ganguly et al. 1998; Carlson 2002) refer to self-diffusion coefficients and thus require multicomponent simulation. This was done following the calculation scheme described in detail by Chakraborty and Ganguly (1992) using the finite difference algorithm mentioned earlier. We tested our program against (1) analytical solutions provided by PROFILER, a program written by Morral and Brockman and available in Glicksman (2000), and (2) against the example provided by Chakraborty and Ganguly (1992, Fig. 4). As expected from the Arrhenius plot (Fig. 8), three datasets (Freer and Edwards 1999; Carlson 2002; Chakraborty and Ganguly 1992) do not reproduce our experimental data. The closest one is the dataset proposed by Ganguly et al. (1998). The penetration curve simulated with the data of Schwandt et al. (1996) is also shown. It should be kept in mind that a self-diffusion coefficient such as the one proposed by Schwandt et al. (1996) for Ca in grossular cannot be applied alone to simulate interdiffusion. However, this calculation gives a lower estimate and shows that our experimental data are consistent with very slow Ca self-diffusion.
Compositional dependence
Following Carlson (2006), we note that the variability among experimental determinations of diffusion coefficients in garnet is so large that it is difficult to judge whether the spread in values results from differences in garnet compositions or from intrinsic experimental and/or analytical difficulties. Indeed, whether or not divalent cation diffusion coefficients in garnet are compositional dependent is presently an unresolved question. Elphick et al. (1985) carried out diffusion couple experiments using two compositionally distinct pairs of natural garnet crystals (Spe–Alm and Prp–Alm). For both couples, they observed a significant asymmetry in the measured profiles and ascribed it to a dependence of the interdiffusion coefficients on composition. They argued that Fe–Mn and Fe–Mg interdiffusivities increased with increasing Fe/Mn and Fe/Mg ratios, respectively. Ganguly et al. (1998, Fig. 3) pointed out that the self-diffusion coefficients of Fe and Mn are significantly enhanced with increasing Mn/Mg, and more generally with a decreasing pyrope component in the garnet. Note that this effect would be relatively small on Mg self-diffusion. In an attempt to unify the divalent cation garnet diffusion data, Carlson (2006) formulated a comprehensive model that predicts diffusion rates for Fe, Mg, Mn, and Ca in garnets across a broad compositional spectrum and over a wide range of P, T, and ƒO2. While the model appears to reconcile much of the experimental data (excepting those of Schwandt 1995, 1996), it provides erratic predictions for some garnet compositions. For instance, application of this model to the garnets of Kohn (2004) and Vielzeuf et al. (2005c) yields diffusivity estimates several log units below the estimates originally reported by these authors. Further, Carlson’s model predicts Ca diffusivities for grossular-rich garnets that are ∼20 log units above those derived by Schwandt et al. (Carlson 2006, p. 10). Finally, the model of Carlson (2006) predicts significant variations in the diffusivities of Ca, Fe, and Mg within interdiffusion couples such as those described here—such variation should lead to large asymmetries in the experimental diffusion profiles. Since we do not observe such asymmetries in our measured diffusion profiles, we consider that divalent cation diffusivities are relatively insensitive to composition and that the model of Carlson (2006) overestimates the compositional effect on these diffusivities. At present, we ascribe the observed variability in Ca diffusivities reported in datasets to intrinsic difficulties in measuring extremely slow diffusion coefficients.
Finally, it is important to stress that the SOI experiments described here have their own set of technical problems and limitations: (1) imperfect polish at the surface of the seed may lead to overestimating the rate of very slow diffusing species; (2) mineral inclusions may be present in the overgrowth and may affect the compositional profiles; (3) the composition of the overgrowth is difficult to plan in advance; and (4) due to temperature dependent partitioning between phases, the composition of the overgrowth changes as a function of temperature making it difficult to generate diffusion couples with the same compositions over a wide range of T. Nevertheless, these disadvantages are compensated by the fact that such experiments are technically easier to execute than other experimental designs, and that the SOI design ensures excellent crystallographic continuity across the chemical interface. In combination with other experimental methods, this design can contribute to a better understanding of divalent cation diffusion in garnet and lead to better constraints on the duration of geological processes.
Notes
A self-diffusion coefficient characterizes the diffusive motion of an atom species (A) in a pure compound A, i.e. in the absence of a chemical potential gradient (for instance, D*Ca in garnet characterizes the diffusivity of calcium in pure grossular).
An interdiffusion coefficient characterizes the simultaneous diffusion of two or more atoms when chemical potential gradients are present (e.g. D Fe–Mg characterizes the interdiffusion of Fe and Mg in almandine–pyrope garnet couples).
References
Adda Y, Philibert J (1966) La diffusion dans les solides. Presses Universitaires de France, Paris, 1225 p
Alaoui HC, Kornprobst J, Laporte D (1997) Inconsistencies between cpx-grt geothermometry and field observations: example of the peraluminous eclogites from Beni Bousera (North Morocco). Terra Nova 9:83–86
Ayres M, Vance D (1997) A comparative study of diffusion profiles in Himalayan and Dalradian garnets: constraints on diffusion data and the relative duration of the metamorphic events. Contrib Mineral Petrol 128:66–80
Brady JB (1995) Diffusion data for silicate minerals, glasses, and liquids. In: Ahrens TJ (ed) Mineral physics and crystallography. A handbook of physical constants. American Geophysical Union, Washington, DC, pp 269–290
Buening DK, Buseck PR (1973) Fe–Mg lattice diffusion in olivine. J Geophys Res 78:6852–6862
Carlson WD (2002) Scales of disequilibrium and rates of equilibration during metamorphism. Am Mineral 87:185–204
Carlson WD (2006) Rates of Fe, Mg, Mn, and Ca diffusion in garnet. Am Mineral 91:1–11
Chakraborty S, Dingwell DB, Rubie DC (1995) Multicomponent diffusion in ternary silicate melts in the system K2O–Al2O3–SiO2: I. Experimental measurements. Geochim Cosmochim Acta 59:255–264
Chakraborty S, Ganguly J (1991) Compositional zoning and cation diffusion in garnets. In: Ganguly J (eds) Diffusion, atomic ordering and mass transport. Advances in physical geochemistry, vol 8. Springer, Berlin Heidelberg New York, pp 120–175
Chakraborty S, Ganguly J (1992) Cation diffusion in aluminosilicate garnets: experimental determination in spessartine-almandine diffusion couples, evaluation of effective binary diffusion coefficients, and applications. Contrib Mineral Petrol 111:74–86
Chakraborty S, Rubie DC (1996) Mg tracer diffusion in aluminosilicate garnets at 750-850°C, 1 atm. and 1300°C, 8.5 GPa. Contrib Mineral Petrol 122:406–414
Cherns D, Morniroli JP (1994) Analysis of partial and stair-rod dislocations by large-angle convergent-beam electron-diffraction. Ultramicroscopy 53:167–180
Cliff G, Lorimer GW (1975) The quantitative analysis of thin specimens. J Microsc 103:203–207
Cordier P, Morniroli JP, Cherns D (1995) Characterization of crystal defects in quartz by large-angle convergent beam electron diffraction. Philos Mag A 72:1421–1430
Costa F, Chakraborty S (2004) Decadal time gaps between mafic intrusion and silicic eruption obtained from chemical zoning patterns in olivine. Earth Planet Sci Lett 227:517–530
Crank J (1975) The mathematics of diffusion, 2nd edn. Clarendon, Oxford, UK, 414 p
Cygan RT, Lasaga AC (1985) Self-diffusion of magnesium in garnet at 750°C to 900°C. Am J Sci 285:328–350
Dachs E, Proyer A (2002) Constraints on the duration of high-pressure metamorphism in the Tauern Window from diffusion modeling of discontinuous growth zones in eclogite garnet. J Metamorphic Geol 20:769–780
Dempster TJ (1985) Garnet zoning and metamorphism of the Barrovian type area, Scotland. Contrib Mineral Petrol 89:30–38
Elphick SC, Ganguly J, Loomis TP (1985) Experimental determination of cation diffusivities in aluminosilicate garnets I. Experimental methods and interdiffusion data. Contrib Mineral Petrol 90:36–44
Faryad SW, Chakraborty S (2005) Duration of Eo-Alpine metamorphic events obtained from multicomponent diffusion modelling of garnet: a case study from the Eastern Alps. Contrib Mineral Petrol 150:306–318
Freer R, Edwards A (1999) An experimental study of Ca–(Fe,Mg) interdiffusion in silicate garnets. Contrib Mineral Petrol 134:370–379
Ganguly J (2002) Diffusion kinetics in minerals: principles and applications to tectono-metamorphic processes. EMU notes in Mineralogy, 4(chapter 10):271–309
Ganguly J, Cheng W, Chakraborty S (1998) Cation diffusion in aluminosilicate garnets: experimental determination in pyrope-almandine diffusion couples. Contrib Mineral Petrol 131:171–180
Gerasimov VY (1987) Experimental study on Mg-Fe interdiffusion in garnets. Trans USSR Acad Sci 295:684–668
Glicksman ME (2000) Diffusion in solids. Wiley, New York, 498 p
Haase CS, Chadam J, Feinn D, Ortoleva P (1980) Oscillatory zoning in plagioclase feldspar. Science 209:272–274
Kirkaldy JS, Young DJ (1987) Diffusion in the condensed state. The Institute of Metals, London, 527 p
Kohn MJ (2004) Oscillatory- and sector-zoned garnets record cyclic (?) rapid thrusting in central Nepal. Geochem Geophys Geosys, 5, 12, doi:10.1029/2004GC000737
Krogh TE (1982) Improved accuracy of U-Pb zircon ages by the creation of more concordant systems using an air abrasion technique. Geochim Cosmochim Acta 46:637–649
Lasaga AC (1979) Multicomponent exchange and diffusion in silicates. Geochim Cosmochim Acta 43:455–469
Leyreloup A, Dupuy C, Andriambololona R (1977) Catazonal xenoliths in French Neogene volcanic rocks; constitution of the lower crust; 2: chemical composition and consequences of the evolution of the French Massif Central precambrian crust. Contrib Mineral Petrol 62:283–300
Loomis TP (1978) Multicomponent diffusion in garnet: II. Comparison of models with natural data. Am J Sci 278:1119–1137
Loomis TP (1983) Compositional zoning of crystals: a record of growth and reaction history. In: Saxena SK (ed) Kinetics and equilibrium in mineral reactions (Advances in physical geochemistry, vol 3). Springer, New York, pp 1–60
Loomis TP, Ganguly J, Elphick SC (1985) Experimental determination of cation diffusivities in aluminosilicate garnets II. Multicomponent simulation and tracer diffusion coefficients. Contrib Mineral Petrol 90:45–51
Lorimer GW, Cliff G (1976) Analytical electron microscopy of minerals. In: Wenk HR (ed) Electron microscopy in mineralogy. Springer, Berlin, pp 506–519
Meissner E, Sharp TG, Chakraborty S (1998) Quantitative measurement of short compositional profiles using analytical transmission electron microscopy. Am Mineral 83:546–552
Morniroli JP, Cordier P, van Cappellen E, Zuo JM, Spence J (1997) Application of the convergent beam imaging (CBIM) technique to the analysis of crystal defects. Microsc Microanal Microstruct 8:187–202
Patiño Douce A, Johnston AD (1991) Phase equilibria and melt productivity in the pelitic system: implications for the origin of peraluminous granitoids and aluminous granulites. Contrib Mineral Petrol 107:202–218
Perchuk LL, Lavrent’eva IV (1983) Experimental investigation of exchange equilibria in the system cordierite-garnet-biotite. Adv Phys Geochem 3:199–239
Perchuk A, Philippot P (2000) Geospeedometry and timescales of high-pressure metamorphism. Int Geol Rev 42:207–223
Philippot P, Blichert-Toft J, Perchuk AL, Costa S, Gerasimov VY (2001) Lu-Hf and Ar-Ar geochronology confirms extreme rate of subduction zone metamorphism deduced from geospeedometry. Tectonophysics 342:23–38
Schwandt CS, Cygan RT, Westrich HR (1995) Mg self-diffusion in pyrope garnet. Am Mineral 80:483–490
Schwandt CS, Cygan RT, Westrich HR (1996) Ca self-diffusion in grossular garnet. Am Mineral 81:448–451
Shannon RD (1976) Revised effective ionic radii and systematic study of interatomic distances in halides and chalcogenides. Acta Crystallogr A 32:751–767
Tanaka M, Saito R, Ueno K, Harada Y (1980) Large-angle convergent-beam electron-diffraction. J Electron Microsc 29:408–412
Tracy RJ (1982) Compositional zoning and inclusions in metamorphic minerals. In: Ferry JM (ed) Characterization of metamorphism through mineral equilibria (Reviews in mineralogy, vol 10) Mineral. Soc Am Washington, DC, pp 355–397
van Cappellen E, Doukhan JC (1994) Quantitative transmission X-ray microanalysis of ionic compounds. Ultramicroscopy 53:343–349
Vielzeuf D, Clemens JD (1992) The fluid-absent melting of phlogopite + quartz: Experiments and models. Am Mineral 77:1206–1222
Vielzeuf D, Holloway JR (1988) Experimental determination of the fluid-absent melting relations in the pelitic system. Contrib Mineral Petrol 98:257–276
Vielzeuf D, Champenois M, Valley JW, Brunet F, Devidal JL (2005a) SIMS analyses of oxygen isotopes: matrix effects in Fe-Mg-Ca garnets. Chem Geol 223:208–226
Vielzeuf D, Lupulescu A, Perchuk A, Laporte D, Baronnet A, Addad A (2005b)—Uphill diffusion and zero flux planes in garnets: an experimental and ATEM study. TMS Lett (The Minerals, Metals and Materials Society) 2:89–90
Vielzeuf D, Veschambre M, Brunet F (2005c) Oxygen isotope heterogeneities and diffusion profile in composite metamorphic-magmatic garnets from the Pyrenees. Am Mineral 90:463–472
Yardley BWD (1977) An empirical study of diffusion in garnet. Am Mineral 62:793–800
Acknowledgements
This work was supported by Centre National de la Recherche Scientifique—Institut National des Sciences de l’Univers through grants DyETI 2002 and DyETI 2005 to D.Vielzeuf. We thank J.-L. Devidal and F. Faure for their assistance with the electron microprobe and SEM at LMV, and C. Vanni, W. Saikali, and C. Dominici for their help with the FIB at CP2M. The preparation of the polished garnet seeds benefited from techniques developed by J. L. Paquette (LMV). We performed the ATEM analyses at the French Earth Science TEM facility (Marseille and Lille). We thank A. Addad for his supervision during the analytical sessions at Lille, and P. Cordier for the LACBED images published in this paper. K. Benzerara is gratefully acknowledged for performing a preliminary series of Ca, Fe, and Mg mapping of a FIB foil (Dif1) by Scanning Transmission X-ray Microscopy on a Synchrotron source at Berkeley. A portion of this work was done while D.V. was at Caltech for a year as part of a CNRS/Caltech exchange; E. M. Stolper is gratefully acknowledged for his financial support. A.L. Perchuk acknowledges Université Blaise Pascal for providing a 4-month Visiting Professor position at LMV—Clermont-Ferrand, and the Russian Foundation for Basic Research for financial support (project 06-05-65204). This paper benefited from suggestions by B. Devouard, discussions with P. Asimow, and formal reviews by C.S. Schwandt and S. Chakraborty.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T.L. Grove.
Rights and permissions
About this article
Cite this article
Vielzeuf, D., Baronnet, A., Perchuk, A.L. et al. Calcium diffusivity in alumino-silicate garnets: an experimental and ATEM study. Contrib Mineral Petrol 154, 153–170 (2007). https://doi.org/10.1007/s00410-007-0184-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-007-0184-x