
Abstract
The activity coefficient of aqueous silica was determined at 800 °C and 12 kbar by measuring the concentration of total dissolved aqueous silica (SiO2(t)) in H2O in equilibrium with four buffering mineral assemblages: quartz (Q), kyanite–corundum (KC), enstatite–forsterite (EF), and forsterite–rutile–geikielite (FRG). SiO2(t) concentrations were determined by reversed equilibration of synthetic minerals with H2O using a weight gain/weight loss technique, yielding molalities of SiO2(t) (m s) of Q, 1.413(1); KC, 0.984(43); EF, 0.276(9); FRG, 0.079(1) (1σ errors). The solubility data can be fitted well according to the homogeneous equilibrium 2 SiO2 monomers (m) = 1 Si2O4 dimer (d) assuming ideal mixing, such that the equilibrium constant (K md ) is K md =X d /X m 2, where X represents the mole fraction of SiO2(t) occurring as the subscripted species. This formulation is valid irrespective of the hydration states of the silica species because of the small silica concentration at these conditions (≤2.5 mol%). For a standard state of unit activity of the hypothetical pure monomer solution at the P and T of interest, the monomer–dimer model leads to γ s =X m /X s and
where γ s and X s are the activity coefficient and mole fraction of total silica. Our data yield \( {K_{{md}} = 155^{{ + 25}}_{{ - 39}} } \) at 800 °C and 12 kbar, in excellent agreement with results from in situ Raman spectroscopy of a quartz-saturated solution at the same P–T. In the system SiO2–H2O, a solution in equilibrium with quartz at 800 °C, 12 kbar, contains 2.5 mol% silica, of which 70% occurs in dimers, and γ s is small (0.30). Even at the low concentration of the FRG buffer (0.1 mol%), the activity coefficient is only 0.75 and the dissolved silica is substantially polymerized (25%).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Large-scale transport of silicate mineral components by aqueous fluids is an important factor in the evolution of the Earth's upper mantle and crust in such settings as subduction zones (Manning 1998), batholiths (Ferry 1994), and zones of continental reactivation (Stolz and Davies 1989). The concentration of total dissolved silica (SiO2(t)) is high in water-rich solutions over a wide range of P and T. Quantitative estimation of silica transport depends on accurate knowledge of the thermodynamic properties of solute silica. Until recently, it was thought that ideal solution of silica monomers accounts adequately for the thermodynamic behavior of SiO2(t), even in fairly concentrated solutions at high temperatures and pressures (Walther and Helgeson 1977; Berman 1988; Holland and Powell 1998), and calculations of metasomatism in deep-seated environments have employed this concept (e.g., Manning 1995; Newton and Manning 2000a). Recent experimental studies have shown, however, that this assumption is seriously in error at high P and T, even for fairly low SiO2(t) concentrations (Zhang and Frantz 2000; Zotov and Keppler 2000, 2002; Newton and Manning 2002). Zhang and Frantz (2000) challenged the conventional assumption of ideal solution of monomeric silica at high P and T based on their experimental study of the solubility of silica in H2O in the model upper mantle system MgO–SiO2–H2O. Silica activity in their experiments was buffered according to the reaction 2enstatite = forsterite + SiO2(t) (reaction EF, Table 1). They synthesized the assemblages enstatite + fluid, enstatite + forsterite + fluid, and forsterite + fluid from oxide mixtures + water of various bulk compositions in the ternary system at 10–20 kbar and 900–1,200 °C in a piston-cylinder apparatus. At constant T and P, the observed phase boundaries of the three assemblages, when projected to the subsystem SiO2–H2O, determine the total silica in solution because there is negligible solution of MgO at these conditions (Zhang and Frantz 2000). The principal conclusion of their study was that the observed SiO2 solubility of the assemblage enstatite-forsterite is two to three times smaller than the solubility to be calculated from the tabulated thermodynamic data for enstatite, forsterite, and quartz, and assuming ideal solution of aqueous silica monomers. The activity coefficient of silica would have to be as low as 0.5 for the measurements of Zhang and Frantz (2000) to be consistent with the thermodynamic data sets (e.g., Berman 1988; Holland and Powell 1998). Zhang and Frantz (2000) suggested that the formation of solute silica polymers could account for the discrepancy.
Almost simultaneously with the work of Zhang and Frantz (2000), Zotov and Keppler (2000) identified coexisting silica monomers and dimers by in-situ Raman spectroscopy of quartz-saturated solutions in an externally heated diamond cell device. Their experiments were conducted at 500–900 °C and 5–14 kbar along isochoric P–T paths. In the higher P–T region of their investigation, they noted monomer peaks, as well as three peaks that could be attributed to silica dimers. The quantitative assessments of Zotov and Keppler (2002) indicated that the dimers could account for as much as 70% of the solute silica at their highest P–T (900 °C, 14 kbar).
Newton and Manning (2002) measured the solubility of silica in H2O in equilibrium with forsterite and enstatite at 600–900 °C, 4–14 kbar, by a precise weight change method. Their study used synthetic single crystals of enstatite and forsterite, as well as fine-grained synthetic mechanical mixtures of the two minerals and natural low-Fe enstatite and forsterite. Solubility measurements were reversed at each experimental P and T by approaching equilibrium from silica concentrations initially higher and lower than final EF solubility. As in the study of Zhang and Frantz (2000), the solubilities were much smaller than predicted assuming ideal solution of neutral monomeric silica.
Experimental verification of the partial polymerization of aqueous silica at high P and T indicates that thermodynamic models of silica in solution require revision. This can be done by comparing measured SiO2(t) concentrations in equilibrium with different buffering mineral assemblages at the same P and T. Because Mg–Si complexes and charged Si complexes can be ignored (Zhang and Frantz 2000; Newton and Manning 2002), results on enstatite + forsterite (Newton and Manning 2002) and quartz (Zotov and Keppler 2000) at the same P and T allow calibration of an activity model. Equilibrium between quartz and aqueous silica monomers (m) can be represented by
and equilibrium between quartz and aqueous silica dimers (d) by
and so on for progressively more polymerized aqueous silica species. Aqueous silica polymers are likely to be H2O-solvated species, but the extent of solvation varies with P, T, and solution composition (Walther and Helgeson 1977; Walther and Orville 1983). The problem of uncertainty in solvation configuration is avoided by defining as zero the changes in Gibbs free energy of the reactions
and
Thus, Eqs. (1) and (2) implicitly represent equilibria involving aqueous solvated silica species [Eqs. (3) and (4)] without explicit provision for H2O solvation (Walther and Helgeson 1977). This approach is useful provided that the activity of H2O is near unity. Even at the highest SiO2 concentrations encountered by Zotov and Keppler (2002), the mole fraction of H2O is immaterially different from unity (minimum of 0.96), and it can be assumed that \( {a_{{{\rm{H}}_{{\rm{2}}} {\rm{O}}}} \approx X_{{{\rm{H}}_{{\rm{2}}} {\rm{O}}}} \approx 1} \).
Homogeneous equilibrium among silica monomers and dimers can be represented by
Adopting a standard state for SiO2(t) of unit activity of the hypothetical solution of unit mole fraction of monomers and a reference state of infinite dilution, and assuming ideal solution of monomers and dimers with H2O (Newton and Manning 2002), the equilibrium constant for Eq. (5) can be written
where the subscript md denotes equilibrium involving monomers and dimers in solution [Eq. (5)] and X is the fraction of SiO2(t) occurring as the subscripted species on a molar basis. In this model, non-ideality of solute silica would result entirely from decrease of the number of monomers in solution due to Eq. (5) progressing to the right. The activity coefficient of SiO2(t), γs, is given by:
where X s is the SiO2(t) mole fraction (\( {X_{s} = n_{{{\rm{SiO}}_{{2(t)}} }} /[n_{{{\rm{SiO}}_{{2(t)}} }} + n_{{{\rm{H}}_{{\rm{2}}} {\rm{O}}}} ]} \)), and
Combination of Eqs. (6), (7), and (8) yields
Where experimental P–T ranges coincide, K md values deduced by Newton and Manning (2002) are in reasonable agreement with those found by Zotov and Keppler (2002), in spite of the very different kinds of observations and the great difference in silica concentration between quartz saturation in the Zotov and Keppler (2002) study and enstatite–forsterite saturation in the Newton and Manning (2002) study (about five times as concentrated in the former study).
Although there is apparent agreement between the two experimental studies, they do not uniquely determine the activity coefficient because other models could explain the solubility data. For instance, higher polymers (trimers, etc.) may also be present (Zotov and Keppler 2000), with their abundance determined by additional equilibrium constants analogous to K md of Eq. (6). If the equilibrium constant is independent of polymer size (e.g., Masson 1965), a relation analogous to Eq. (9) for an ideal mixture of all polymers of any geometry (ap) can be derived:
(Newton and Manning 2002). Comparison of Eqs. (9) and (10) illustrates that the equilibrium constants and activity coefficients to be deduced from the solubility data differ substantially depending on the energetics and extent of polymerization.
The non-uniqueness of the derived K value means that solubility measurements are necessary on additional SiO2-buffering assemblages, in order to define the activity–concentration relations over a broad range of concentrations. Most importantly, measurements are needed for some refractory assemblage(s) that buffer SiO2 concentration in the very dilute range, where, according to Eqs. (9) and (10), the activity coefficient changes rapidly with concentration. It is necessary that the thermodynamic properties of the buffering minerals be well determined, ideally as part of a large internally consistent data set (e.g., Berman 1988; Holland and Powell 1998). This requirement proves to be a stringent one: only a few refractory silica-bearing assemblages can be found for which the known thermodynamic properties are sufficiently reliable for the purpose of defining γs–Xs relations.
The present study gives results of measurements of aqueous silica concentrations at 800 °C and 12 kbar on four SiO2-buffering assemblages that cover a wide span of concentrations. The investigated dissolution reactions and abbreviations are given in Table 1. The broad range in composition permits derivation of an isothermal–isobaric activity–concentration model for aqueous silica in the system SiO2–H2O.
Experimental methods
All experiments were conducted in a 0.75-inch (1.91-cm.)-diameter piston-cylinder apparatus with NaCl pressure media and graphite heater sleeves. The same highly polished tungsten-carbide pressure-vessel core and batch of calibrated, matched W3%Re–W25%Re thermocouples were used in all experiments. Accuracy of temperature (±3 °C) and pressure (±300 bar) measurements are discussed in Newton and Manning (2000b). Mass determinations were made with a Mettler M3 microbalance (1σ=2 μg in each weighing step).
Three different kinds of starting material were used. Most experiments used fine-grained mixtures of kyanite–corundum, enstatite–forsterite, and rutile–geikielite. These assemblages were crystallized hydrothermally from reagent-grade oxide mixtures at 900 °C and 10 kbar for 24 h. No extraneous phases were detected optically or by X-ray diffraction. Kyanite was synthesized in the same manner as in Harlov and Newton (1992; properties given therein). Synthesis of the enstatite–forsterite mix is described in Newton and Manning (2002). Crystal sizes were 5–100 μm. Both minerals of a starting mix were present in about equal weight amounts. Trigonal unit-cell constants of a o =5.052 (2) and c o =13.896 (4) for geikielite were determined by X-ray diffraction scans (CuKα, 0.5 °2θ/min) using the rutile peaks as an internal standard. The values coincide, within uncertainties, with synthetic geikielite (ASTM 6-0494). A second kind of starting material in the rutile–forsterite–geikielite system consisted of a hard, sintered compact of well-mixed rutile (70%) and forsterite (30%), which was prepared by pelletizing the mineral mix under pressure and then annealing it at 1,100 °C for 2 h. Chips of the compact weighing about 4 mg were smoothed with a diamond file, polished by rubbing with 600-mesh alundum paper and then cleaned by washing in acetone and water and drying at 350 °C. In one experiment, a natural kyanite crystal (Bahia, Brazil, 0.17 wt% TiO2, trace of FeO) was used.
Mineral mixes or sintered chips were contained in 2–3-mm-long segments of Pt tubing, welded at one end and lightly crimped at the other. The capsules were usually perforated with between 6 and 20 100–200-μm holes to allow access of the fluid without loss of particulate matter (in two experiments on the geikielite–rutile mix, there was visible evidence after the run of escaped crystals, with attendant anomalous weight changes; these runs were discounted). The perforated inner capsules were placed into 2.5-mm OD Pt tubes with a weighed amount of H2O and, in most cases, a fragment of a natural quartz crystal to produce a predetermined initial SiO2 fluid content, and sealed by arc-welding.
Experiments were terminated by turning off the apparatus power. Temperatures fell below 200 °C in about 12 s. Retrieved outer capsules were cleaned of salt by soaking in H2O. Weights of retrieved capsules demonstrated that no leakage occurred during runs. H2O contents were redetermined by puncturing and drying at 115 °C for 15 min, followed by 320 °C for 15 min. The weight changes corresponded to the original H2O contents within 0.3% with no systematic discrepancies relative to the initial H2O values. This showed that welding-related weight losses, typically about 1 mg, included negligible H2O.
If the initial SiO2 concentration was less than the equilibrium amount, the inner capsules lost weight; if there was initial SiO2 oversaturation, the capsules gained weight through growth of crystals in the inner capsule. Thus, the silica concentrations for all assemblages approached equilibrium from both the undersaturated and oversaturated directions (i.e., were reversed). Experiments have shown that quartz dissolution at these conditions is very fast compared with reaction of the fluid with the experimental charges (Newton and Manning 2002).
Uncertainty in each weighing step was propagated to final solubility determinations. Resulting precision estimates are reported parenthetically as 1σ in the final digit(s) unless otherwise noted. Reported errors do not include the uncertainty in accuracy of P–T of experiments.
Unless otherwise noted, the thermodynamic data of Holland and Powell (1998) were used in all calculations.
Results
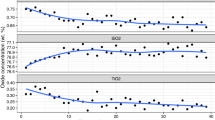
Experimental details are given in Table 2 and shown in Fig. 1, which demonstrates the time independence and reversibility of the SiO2 molality for each of the assemblages (Q, KC, EF, and FRG, Table 1). The solubility of MgO is below detection (<4 μg, 2σ) at these conditions (Newton and Manning 2002). Additional experiments show that the solubilities of Al2O3 and MgO + TiO2 make only slight contributions to the weight changes (Table 3). SiO2(t) molalities in KC and FRG experiments were adjusted accordingly (Table 2).

Silica concentration vs. experiment duration. 2σ errors in solubility measurements are smaller than the symbol size. Note break in scale. Upward- and downward-pointing triangles denote experiments in which equilibrium was approached from initial under- and oversaturation, respectively. Lines and associated numerical values are the mean solubility (Q, FRG) or the midpoint in the range of permissible solubility (KC, EF). See Table 1 for abbreviations
Weight losses of the inner capsules in runs GE-6 and GE-8 (Table 2) are inferred to result from the spontaneous nucleation and growth of geikielite, with loss of SiO2 into the fluid, according to reaction FRG. This interpretation was verified by scanning electron microscopy of the charge of experiment #GE-8. Figure 2A shows that euhedral, equant, and tabular geikielite crystals, up to 80 μm across, are perched on the surface of the sintered rutile–forsterite pellet. Similarly, forsterite was identified in the charges of runs on geikielite–rutile mixes (+ quartz), as shown in Fig. 2B. The forsterite is interpreted to have nucleated and grown according to reaction FRG (Table 1) in reverse.

Backscattered-electron micrographs of experimental run products, 800 °C, 12 kbar. A Euhedral geikielite neoblasts on a fine-grained, sintered rutile + forsterite compact, grown by reaction with initially pure H2O (experiment GE-8, 98 h). B Euhedral, twinned forsterite crystals (fo) in a rutile–geikielite aggregate, grown by reaction with silica oversaturated solution. Geikielite crystals are equant and solution-rounded; rutile is prismatic and euhedral (experiment GE-7, 114 h). C Euhedral corundum platelets occupying solution cavities in kyanite crystal, grown by reaction with initially pure H2O (experiment KY-2, 94 h). D Doubly terminated quartz crystal, grown in initially pure H2O from a ground ellipsoid. Faceting developed despite net threefold dissolution of the crystal (experiment QZB-81, 23 h)
The data for reaction FRG are especially important because they define the most dilute SiO2 concentration point of the present experiments and, hence, represent the closest approach to ideal solutions. The measurements ranging from 89 to 114 h seem to be independent of duration, H2O volume, or direction of approach to equilibrium (Fig. 1, Table 2). Therefore, we treated them on a statistical basis; the average of the six solubility determinations corresponding to reaction FRG gives a total silica molality (m s ) of 0.079 when adjusted for a MgO + TiO2 solubility of 0.022 wt% (Table 3). 1σ in the parent population of FRG experiments is 0.005 molal, yielding a standard error in the mean of 0.002 mol. The latter value was used in all calculations.
Results for reaction EF were given by Newton and Manning (2002). Two determinations at 800 °C, 12 kbar, approached equilibrium from initially under- and oversaturated conditions (Table 2). Because the initially undersaturated experiment has lower final SiO2 solubility than the initially oversaturated experiment, the measurements bracket the true m s to between 0.267(2) and 0.285(2). With only two constraints, it must be assumed that there is uniform probability of the true value anywhere within the bracket limits.
The KC experiments also approached equilibrium from initial super- and undersaturation. Figure 2C shows hexagonal corundum crystals formed by dissolution of the natural kyanite in initially pure H2O, according to reaction KC. Kyanite growth occurred in experiment KY-4, which had excess SiO2 (Table 2). As with EF results, the three KC experiments bracket m s to between 0.941(2) and 1.027(2), with equal probability of the actual value anywhere between these constraints.
The two quartz solubility measurements yielded nearly identical molalities (Table 2). The residual quartz was highly faceted (Fig. 2D), even though there were crystal weight losses of three- and eightfold in the two experiments. When considered with the very rapid rate of dissolution at these conditions (Newton and Manning 2002), the recrystallization strongly suggests the attainment of solution equilibrium. This permits averaging the two values to give m s =1.413(1), which is in good agreement with the prediction of Manning (1994) of 1.50(2).
Discussion
Combination of silica solubility measured at quartz saturation with that at any other reaction B of the form
at the same P and T, fixes the activity of the thermodynamic component SiO2. Provided that no other components contribute significantly to the weight changes in our experiments, measured SiO2(t) concentration at any two mineral buffers constrain the ratio of activity coefficients, \( {\Gamma } \). Accordingly, the quartz buffer (Q) can be combined with any buffer B to give
where \( {\Delta G^{ \circ }_{{B - Q}} } \) is the difference between the standard apparent standard molar Gibbs free energies of buffers B and Q, which is readily derived from standard thermodynamic data sets for minerals.
Reliable values of \( {\Delta G^{ \circ }_{{B - Q}} } \) at 800 °C and 12 kbar of KC-Q and EF-Q are provided by the Berman (1988, 1992 update) and Holland and Powell (1998) data sets, which are derived from many experimental phase-equilibrium data involving the minerals enstatite, forsterite, kyanite, corundum, and quartz. Thermodynamic data for rutile are similarly well constrained, but the tabulated Gibbs free energy of formation of geikielite is poorly constrained by the reconnaissance experiments of Haselton et al. (1978) on the equilibrium
Derived thermodynamic properties of geikielite differ considerably between the two data sets. However, the recent experimental data of Ferry et al. (2002) on this equilibrium make possible a much improved estimate of \( {\Delta G^{ \circ }_{{FRG - Q}} } \). We used the midpoint of bracketed values of \( {\Delta _{f} H^{ \circ }_{{geikielite}} } \) based on Ferry et al. (2002) to compute \( {\Delta G^{ \circ }_{{FRG - Q}} } \).
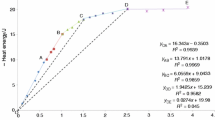
Our solubility constraints, values of \( {\Delta G^{ \circ }_{{B - Q}} } \), and \( {\log \Gamma _{{B - Q}} } \) calculated using Eq. (12) are listed in Table 4. The \( {\log \Gamma _{{B - Q}} } \) constraints (Fig. 3) can be fitted very well with the monomer-dimer model [Eq. (9)]. A \( {\log K_{{md}} } \) value of 2.19 satisfies all of the bracketing data using the Holland and Powell (1998) data set (Fig. 3); a similar value of 2.14 is obtained using Berman (1988, updated 1992). The results of Ferry et al. (2002) permit a range of \( \Delta _f H_{geikielite}^ \circ , \) which, in turn, allows a range in the equilibrium constant. The maximum allowable range is that defined by the EF brackets, which translates to \( {\Delta _{f} H^{ \circ }_{{geikielite}} } \)=–1,568.17 to −1,567.74 kJ/mol, consistent with Holland and Powell (1998), and yields \( {\log K_{{md}} } \)=2.06 to 2.25 (Fig. 3). The same calculations using Berman (1988, updated 1992) yield \( {\Delta _{f} H^{ \circ }_{{geikielite}} } \)=–1,570.31 to −1,569.89 kJ/mol and \( {{\rm{l}}ogK_{{md}} } \)=1.94 to 2.12.

\( {{\rm{Log}}\Gamma _{{B - Q}} } \) vs. mole fraction of total silica in solution at 800 °C, 12 kbar. Values are from Table 4 using thermodynamic data from Holland and Powell (1998). Experimental results on KC and EF experiments plotted with upward and downward pointing triangles. Actual solubility is any value between these limits. Results of FRG and Q experiments shown with filled circles, corresponding to the mean of all experimental results. Variation in \( {{\rm{l}}og\Gamma _{{B - Q}} } \) with X s also shown for monomer-dimer model log K =2.19. Range in permissible values of log K is 2.06–2.25 (see text), yielding \( {{\rm{l}}og\Gamma _{{B - Q}} - X_{s} } \) variation shown in light solid lines. The all-polymer model (dashed line) fails to fit all the experiments simultaneously. Abbreviations as in Fig. 1
As noted above, our model does not account explicitly for H2O solvation. It can be demonstrated that this does not introduce significant error by recasting the monomer and dimer species as H4SiO4 and H6Si2O7. Homogeneous equilibrium between these species involves liberation of 1 mol of H2O for every mol of dimers produced because 2H4SiO4 = H6Si2O7 + H2O. The log K for this equilibrium explicitly involves \( {a_{{{\rm{H}}_{{\rm{2}}} {\rm{O}}}} } \). Assuming ideal mixing of silica and H2O, it is only ~0.01 less than \( {\log K_{{md}} } \). Differences in the H2O solvation configuration between monomers and dimers would have to be very large to change \( {\log K_{{md}} } \) significantly.
Using the all-polymer model [Eq. (10)], no single log K ap value that is consistent with all solubility constraints could be found. This is a result of the less pronounced dependence of K ap on \( {\Gamma } \) at low X s . A value of \( {\log K_{{ap}} } \)=1.39 is shown for illustration in Fig. 3.
Our \( {\log K_{{md}} } \) value at 800 °C and 12 kbar is the same, within the uncertainties, as that found at the same P–T conditions by Zotov and Keppler (2002) by in situ Raman spectroscopy of quartz-saturated solutions (\( {\log K_{{md}} } \)=2.07±0.07). The close agreement between the present determination of K md and that of Zotov and Keppler (2002), arrived at by very different techniques, must be in part fortuitous, because the latter reconstruction involved extrapolating the Fournier and Potter (1982) empirical quartz solubility equation, which gives m s =1.511 at 800 °C and 12 kbar. Nevertheless, the agreement would appear to be a solid justification of the use of the monomer-dimer model, embodied in Eq. (9), in the general P–T range 5–15 kbar and 600–900 °C.
Using \( {\log K_{{md}} } \)=2.19, Eq. (9) can be rearranged to give the activity coefficient-mole fraction relation of aqueous silica at 800 °C and 12 kbar (Fig. 4). Strong concentration-dependent non-ideality, most pronounced for small concentrations, is evident. For concentrations as dilute as 0.1 mol%, the activity coefficient is markedly different from unity. It is to be emphasized that the construction of Fig. 4 does not depend strongly on the validity of the monomer–dimer model. Equations (6), (7), (8), and (9) serve as a convenient and rational basis for integrating the data, but the activity–concentration relations of Fig. 4 are based mainly on the measured quantities expressed in the brackets of Fig. 3, namely the solubilities and Gibbs free energies for the simple minerals involved, which are among the best determined and most reliable in the thermodynamic data sets.

Variation in activity coefficient of total silica with mole fraction of total silica, 800 °C, 12 kbar, based on the monomer-dimer model [Eq. (9)] and \( {{\rm{l}}ogK_{{md}} } \)=2.19 (bold line). Light, solid lines show the range in possible values consistent with \( {\Delta _{f} H^{ \circ }_{{geikielite}} } \) from Ferry et al. (2002) and the EF buffer (see text)
The activity coefficient relations shown in Fig. 4 represent the fraction of total silica present at any concentration that is in the form of monomers, as per Eq. (7). Although the derived fraction of monomers is mainly model-independent, there may well be among the polymerized population a smaller quantity of trimers and even more highly polymerized species (Newton and Manning 2002). The existence of a second critical end point in the system SiO2–H2O (Kennedy et al. 1962) implies that higher polymeric species will be present in increasingly greater quantities at higher silica concentrations and at temperatures and pressures beyond those of this study.
The present analysis gives an activity coefficient at quartz saturation at 800 °C and 12 kbar of 0.301. This yields an apparent standard molar Gibbs free energy of reaction of buffer Q (\( {\Delta G^{ \circ }_{Q} } \)) of 43.69 kJ/mol at this P and T. Using the maximum range in \( {\Delta _{f} H^{ \circ }_{{geikielite}} } \) defined by the EF brackets limits \( {\gamma ^{Q}_{S} } \) between 0.283 and 0.339 and \( {\Delta G^{ \circ }_{Q} } \) between 42.64 and 44.24 kJ/mol at 800 °C, 12 kbar.
Geological applications
The present work shows that solubility and metasomatic phase relations involving SiO2–H2O fluids under deep crust/upper mantle P–T conditions can be calculated from tabulated thermodynamic data with the aid of activity–concentration relations based on a relatively small number of measured solubility data. The most rigorous calculations are possible for refractory systems where SiO2 is the most soluble component, as in the ultramafic upper mantle. The solubility of enstatite–forsterite as predicted by the monomer–dimer model is adequate to calibrate activity–concentration relations over an extended P–T region appropriate to deep-Earth conditions. Solubility measurements in addition to those presented by Newton and Manning (2002) are desirable for this purpose, as are, importantly, additional measurements of quartz solubility. Accurate control of quartz solubility may be most important in the >900 °C temperature range, where the deviations of existing experimental data from the Manning (1994) solubility equation are greatest.
Other factors that may enhance the solubility of silica of natural assemblages are alkalis and acid/base relations. It is possible that silica may be present as feldspar-like solute species. The system albite (NaAlSi3O8)–H2O becomes supercritical in an elevated P–T range (Shen and Keppler 1997), and extremely silicate-rich solutions are in equilibrium with albite, which are virtually H2O-rich melts with SiO2 near 50 wt%. Alkaline solutions emitted by deep-seated magmas may leach SiO2 to a greater degree than possible by pure water, as in the fenite aureoles of carbonatite complexes. The most direct application of the present data may be in some subduction zones, where great silica transport is recorded in the form of quartz vein complexes, and where other geochemical criteria suggest that the fluid transporting medium was dominantly H2O (e.g., Bebout and Barton 1989). The non-ideality of aqueous silica at elevated P–T leads to larger changes in solute transporting power with silica concentration. Compared to an assumed ideal solution of monomers, a polymerized solution requires lower integrated fluid fluxes to transport silica.
References
Bebout GE, Barton MD (1989) Fluid flow and metasomatism in a subduction zone hydrothermal system: Catalina Schist terrane, California. Geology 17:976–980
Berman RG (1988) Internally consistent thermodynamic data for minerals in the system Na2O–K2O–CaO–MgO–FeO–Fe2O3–Al2O3–SiO2–TiO2–H2O–CO2. J Petrol 29:445–522
Ferry JM (1994) A historical review of metamorphic fluid flow. J Geophys Res 99:15487–15498
Ferry JM, Newton RC, Manning CE (2002) Experimental determination of the equilibria: rutile + magnesite = geikielite + CO2 and zircon + 2 magnesite = baddeleyite + 2CO2. Am Mineral 87:1342–1350
Fournier RO, Potter RW II (1982) An equation correlating the solubility of quartz in water from 25° to 900 °C at pressures up to 10,000 bars. Geochim Cosmochim Acta 46:1969–1973
Harlov DE, Newton RC (1992) Experimental determination of the reaction 2magnetite +2kyanite + 4quartz = 2almandine + O2 at high pressure on the magnetite-hematite buffer. Am Mineral 77:558–564
Haselton HT, Sharp WR, Newton RC (1978) CO2 fugacity at high temperatures and pressures from experimental decarbonation reactions. Geophys Res Lett 5:753–756
Holland TJB, Powell R (1998) An internally consistent thermodynamic data set for phases of petrologic interest. J Metamorph Geol 16:309–343
Kennedy GC, Wasserburg GJ, Heard HC, Newton RC (1962) The upper three-phase region in the system SiO2–H2O. Am J Sci 260:501–521
Manning CE (1994) The solubility of quartz in the lower crust and upper mantle. Geochim Cosmochim Acta 58:4831–4839
Manning CE (1995) Phase-equilibrium controls on SiO2 metasomatism by aqueous fluids in subduction zones: reaction at constant pressure and temperature. Int Geol Rev 37:1074–1093
Manning CE (1998) Fluid composition at the blueschist–eclogite transition in the model system Na2O–MgO–Al2O3–SiO2–H2O–HCl. Swiss Bull Mineral Petrol 78:225–242
Masson CR (1965) An approach to the problem of ionic distribution in liquid silicates. Proc R Soc Lond A287:201–221
Newton R C, Manning CE (2000a) Metasomatic phase relations in the system CaO–MgO–SiO2–H2O–NaCl at high temperatures and pressures. Int Geol Rev 42:152–162
Newton RC, Manning CE (2000b) Quartz solubility in concentrated aqueous NaCl solutions at deep crust–upper mantle metamorphic conditions: 2–15 kbar and 500–900 °C. Geochim Cosmochim Acta 64:2993–3005
Newton R C, Manning CE (2002) Solubility of enstatite + forsterite in H2O at deep crust/upper mantle conditions: 4 to 15 kbar and 700 to 900 °C. Geochim Cosmochim Acta 66:4165–4176
Shen AH, Keppler H (1997) Direct observation of complete miscibility in the albite–H2O system. Nature 385:710–712
Stolz AJ, Davies GR (1989) Metasomatized lower crustal and upper mantle xenoliths from north Queensland: chemical and isotopic evidence bearing on the composition and source of the fluid phase. Geochim Cosmochim Acta 53:649–660
Walther JV, Helgeson HC (1977) Calculation of the thermodynamic properties of aqueous silica and the solubility of quartz and its polymorphs at high pressures and temperatures. Am J Sci 277:1315–1351
Walther J, Orville PM (1983) The extraction-quench technique for determination of the thermodynamic properties of solute complexes: application to quartz solubility in fluid mixtures. Am Mineral 68:731–741
Zhang Y-G, Frantz JD (2000) Enstatite–forsterite–water equilibria at elevated temperatures and pressures. Am Mineral 85:918–925
Zotov N, Keppler H (2000) In-situ Raman spectra of dissolved silica species in aqueous fluids to 900 °C and 14 kbar. Am Mineral 85:600–603
Zotov N, Keppler H (2002) Silica speciation in aqueous fluids at high pressures and high temperatures. Chem Geol 184:71–82
Acknowledgements
This research was funded by NSF EAR-990583. We thank John Walther and Andy Shen for helpful reviews of the manuscript, and George Jarzebinski for assistance with SEM petrography.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial responsibility: T.L. Grove
Rights and permissions
About this article
Cite this article
Newton, R.C., Manning, C.E. Activity coefficient and polymerization of aqueous silica at 800 °C, 12 kbar, from solubility measurements on SiO2-buffering mineral assemblages. Contrib Mineral Petrol 146, 135–143 (2003). https://doi.org/10.1007/s00410-003-0483-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-003-0483-9