
Abstract
Pulmonary arterial hypertension (PAH) is a rare form of pulmonary hypertension characterized by a progressive obliterative vasculopathy of the distal pulmonary arterial circulation that usually leads to right ventricular failure and death. Over the last 25 years, more than a dozen drugs representing five drug classes have been developed and approved for the treatment of this devastating disease. Due to the small number of patients afflicted by PAH, most health care providers have little experience with its management. To address this gap in medical knowledge, treatment guidelines have been developed by professional organizations and expert committees. Over the last few years, these guidelines have been updated to address findings from recent clinical trials and ongoing experience with these drugs. This review provides an update on the most recently published treatment guidelines for pharmacologic treatment of PAH and incorporates them into a contemporary approach to the treatment of this disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Definitions
Pulmonary hypertension (PH) refers to an elevation in mean pulmonary arterial pressure (mPAP). The pulmonary circulation is a low-pressure system, and in healthy individuals, mPAP is about 14 mmHg with a standard deviation of 3 mmHg [1]. Thus, mPAP above 20 mmHg is greater than two standard deviations above the mean and can be considered abnormally high. For this reason, many studies have defined PH as a mPAP > 20 mmHg. However, mPAP increases slightly with age such that for patients over 50 years old, two standard deviations above mPAP is approximately 23 mmHg [1]. Furthermore, precise measurement of mPAP in any patient is challenging due to the need for accurate referencing of the right heart catheter. Finally, pulmonary vascular disease typically results in severe elevation of mPAP well above two standard deviations from the mean. For these reasons 20–24 mmHg has long been considered as a borderline elevation of mPAP and PH has been defined as mPAP ≥ 25 mmHg. Recently, the 6th World Symposium on Pulmonary Hypertension (WSPH) recommended changing the definition of PH to mPAP ≥ 20 mmHg with the stipulation that pulmonary vascular resistance (PVR) be ≥ 3 Woods units [2]. The latter requirement is important because small increases in mPAP can be caused by a rise in left heart filling pressures or an increase in cardiac output without significant pulmonary vascular disease.
PH commonly occurs as the result of left-sided heart disease or chronic lung disease. Rarely, it is caused by a distinct vasculopathy of the distal pulmonary arterioles in which case it is referred to as pulmonary arterial hypertension (PAH). PH can be characterized as pre-capillary, post-capillary, or combined pre-and post-capillary, referring to the site of disease pathology existing proximal to or distal to the pulmonary capillary bed (or both). Proper determination of the type and cause of PH is crucial, because it affects the prognosis and treatment of the disease.
PH is diagnosed by a combination of clinical history and hemodynamic measurements. The hemodynamic diagnostic criteria for PH are summarized in Table 1 [3]. In addition to hemodynamic measurements, the type of PH is determined by clinical characteristics and the most likely etiology of disease. A five group classification system for PH was developed during the Second WSPH in 1998. Because the WSPH was originally sponsored by the World Health Organization (WHO), this classification system has been referred to as the WHO Group classification and was most recently updated at the 6th WSPH (Table 2). Group 1, referred to as PAH, is a rare, but severe form of PH that is defined as pre-capillary PH that is not associated with other left heart or lung disease. The word “arterial” is included in PAH to indicate a primary vasculopathy of the pulmonary arterial circulation, although increasing evidence suggests that the capillaries and post-capillary venules are frequently involved as well [4]. This disease most commonly occurs without an identifiable cause and is referred to as idiopathic PAH, but PAH has also been associated with several specific diseases including connective-tissue disease, human immunodeficiency virus (HIV), congenital left to right intra-cardiac shunts, portal hypertension, certain drug or toxin exposures, and schistosomiasis. In these cases, it can be referred to as associated PAH (APAH). Heritable PAH (HPAH) includes patients with familial PAH (PAH that occurs in two or more family members) or patients with PAH who have one of the numerous mutations described in over a dozen genes that have been associated with PAH [5].
Group 2 refers to PH that is due to left heart disease. This type of PH is distinct from the other groups in that it is caused primarily by an elevation of left heart filling pressure and is often referred to as pulmonary venous hypertension. Some patients with Group 2 PH demonstrate elevation of pulmonary arterial pressure that is greater than would be expected for the increase in pulmonary venous pressure alone and are described as having combined pre- and post-capillary PH, but are still considered WHO Group 2. These patients can be distinguished from isolated post-capillary PH hemodynamically by the presence of an elevated PVR (≥ 3 WU). The use of pulmonary vasodilators to reduce PVR in these patients may be deleterious as it can result in a further increase in left-sided filling pressures. Group 3 PH is pre-capillary PH that is due to lung disease or chronic hypoxia. Group 4 PH is pre-capillary PH that is due to chronic thromboembolic pulmonary hypertension (CTEPH) or other forms of pulmonary arterial obstruction. Lastly, Group 5 PH is PH with unclear or multifactorial etiologies.
Epidemiology
The most common types of PH are WHO Group 2 and 3, and in some studies they represent greater than 85% of all cases of elevated pulmonary artery pressure [6]. WHO Group 1 PAH is a rare disease, affecting about 30,000 people in the United States with an estimated prevalence of approximately 15 per million [7]. It is seen more commonly in women than in men by greater than a 2:1 margin and was originally described most often in the fourth or fifth decade of life. However, more recent registries suggest that most patients are diagnosed in their sixth and seventh decades of life [8]. Despite being a rare cause of PH, PAH is the most severe of the pulmonary hypertensive diseases and until recently, the only one for which specific therapy had been approved. In 2015, riociguat was approved for the treatment of Group 1 PAH and Group 4 PH. All other medications for PH are approved only for the treatment of WHO Group 1 PAH and this review is limited to the discussion of treatment guidelines for PAH. Treatment of Group 4 PH consists of removal or compression of the obstructing intravascular defects, anticoagulation to prevent recurrent pulmonary embolism, and the use of riociguat or other pulmonary vasodilators in patients who have CTEPH that is not amenable to surgical treatment or who have significant PH after surgical treatment. Treatment of Groups 2, 3, and 5 PH is directed mainly at treating the underlying heart, lung, or other disease processes.
Cellular Signaling Pathways and Drug Targets for PAH
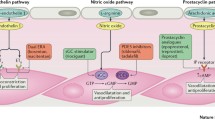
Five classes of drugs have been developed to treat PAH, and they target three major cellular signaling pathways: the endothelin pathway, the nitric oxide/cyclic guanosine monophosphate (cGMP) pathway, and the prostacyclin pathway (Fig. 1). Prostacyclin is produced by the metabolism of arachidonic acid in the vascular endothelium and stimulates production of cyclic adenosine monophosphate (cAMP), which causes relaxation of the vascular smooth muscle leading to vasodilation. Prostacyclin signaling also inhibits endothelial cell proliferation and platelet aggregation, which may lessen intravascular thrombosis [9]. Expression of prostacyclin synthase, the major enzyme responsible for prostacyclin synthesis, is decreased in pulmonary vascular endothelial cells of patients with PAH and circulating levels of prostacyclin are reduced [10, 11]. There are two classes of drugs that stimulate this pathway: (1) prostacyclin analogs, such as the synthetic prostacyclin epoprostenol or the prostacyclin derivatives treprostinil and iloprost, which have been modified for longer half-life; and (2) prostacyclin receptor agonists, such as selexipag, which binds to and activates the prostacyclin receptor (IP) but is not a prostacyclin derivative. Vascular endothelial cells also secrete endothelin-1, which causes vasoconstriction and proliferation of vascular smooth muscle cells. Pulmonary vascular expression of endothelin and circulating levels of endothelin-1 are increased in PAH [9]. The endothelin receptor antagonists (ambrisentan, bosentan, macitentan) block this pathway thereby causing pulmonary vasodilation. Nitric oxide (NO) is a potent vasodilator synthesized from l-arginine by endothelial NO synthase (eNOS). NO stimulates soluble guanylate cyclase (sGC) causing an increase in intracellular cGMP, which like cAMP, has vasodilatory and antiproliferative effects on pulmonary vascular smooth muscle [9]. The natriuretic peptides also increase cGMP in the pulmonary circulation by binding to cell surface receptors that are linked to particulate guanylyl cyclase. Deficiencies in cGMP production due to disrupted NO and natriuretic peptide signaling have also been implicated in the pathogenesis of PAH [12]. Phosphodiesterase type 5 is the major enzyme responsible for cGMP degradation in pulmonary vascular smooth muscle, and its expression is increased in patients with PAH [13]. Phosphodiesterase type-5 inhibitors (PDE5i), such as sildenafil and tadalafil, delay the metabolism of cGMP and thereby potentiate the vasodilatory effects of NO and the natriuretic peptides. Drugs that increase the activity of sGC are known as sGC simulators or sGC activators. They also cause smooth muscle relaxation and vasodilation by increasing cGMP levels, but rather than inhibiting cGMP metabolism, they increase cGMP synthesis. sGC stimulators, such as riociguat, enhance the activity of sGC in the presence of NO and can stimulate sGC activity in the absence of NO.

Adapted from [14]
The three major cellular signaling pathways targeted by PAH treatment are the endothelin pathway, the nitric oxide pathway, and the prostacyclin pathway. ERAs inhibit endothelin-1, which is increased in PAH, from binding to its receptors, thus preventing vasoconstriction and cellular proliferation. Nitric oxide, which may be decreased in PAH, stimulates soluble guanylate cyclase (sGC) to synthesize cGMP which promotes vasodilation and has antiproliferative effects. Intracellular cGMP levels can be increased by inhibiting the enzyme that degrades it—Phosphodiesterase type-5 or by increasing its synthesis via a sGC stimulator. Prostacyclin, which is decreased in PAH stimulates production of cAMP, also promotes vasodilation and has antiproliferative effects as well. Prostacyclin signaling can be increased by administration of a prostacyclin receptor agonist or by a prostacyclin derivative.
The major clinical trials that led to the approval of each drug and/or its guideline recommendations are summarized in Table 3. It should be noted that the first drug to be approved for treatment of PAH, intravenous epoprostenol, is the only drug that was found to reduce mortality [15]. That study differed from subsequent clinical trials in that patients had more advanced disease. While other drugs have not been shown to reduce mortality, they have been shown to improve exercise capacity and/or time to clinical worsening. The study design of clinical trials in PAH has evolved since the approval of epoprostenol and the initial oral agents for PAH. Earlier studies in PAH examined single outcome measures of pulmonary hemodynamics or functional capacity such as change from baseline in PVR or 6-min walking distance (6MWD) measured at a single time point after starting treatment. More recent studies have used time to clinical worsening defined by a composite of outcome events that usually include death, hospitalization for PAH, lung transplant, need for parenteral prostacyclin therapy, or evidence of disease progression demonstrated by declining 6MWD and functional status. In these more recent studies, the great majority of patients fulfill the criteria for clinical worsening by being hospitalized or meeting criteria for disease progression before mortality occurs. Due to the number of treatments available, it is no longer ethical to conduct a study that examines mortality as the primary outcome.
Clinical Presentation and Initial Work-Up
The clinical presentation of PAH is variable and can be subtle, but almost always results in shortness of breath. Most patients complain of dyspnea with exertion, but others describe fatigue and difficulty performing routine activities. As right heart failure develops, patients may report lower extremity swelling, increased abdominal girth, and loss of appetite. Exertional angina, lightheadedness, or syncope may occur when the right ventricle is no longer able to sufficiently augment cardiac output in response to exercise. Physical exam findings include signs of right ventricular failure such as jugular venous distention, right ventricular lift, peripheral edema, hepatomegaly, and ascites. Tricuspid regurgitation can manifest as a holosystolic murmur heard best at the left lower sternal border, V-wave pulsations in the jugular vein, and pulsatile liver [3]. An increased pulmonary component of the second heart sound may be appreciated. Other physical exam findings might be suggestive of associated diseases, such as clubbing in liver cirrhosis or cyanotic congenital heart disease, or telangiectasias and sclerodactyly in scleroderma. The electrocardiogram (ECG) may show right axis deviation, enlarged right atrium, or right ventricular hypertrophy or strain. Arrhythmias, especially supraventricular tachycardia (SVT), are common in patients with PH, particularly in those with decompensated disease and right ventricular failure [29]. Chest radiography is often normal, but can show enlarged right atrium, right ventricle, and/or proximal pulmonary arteries. Computed tomography (CT) may demonstrate enlargement of these structures as well, and it can also be used to evaluate the lung parenchyma, which might suggest an etiology of disease [3].
Transthoracic echocardiography has become the most frequently used method of assessing pulmonary arterial pressure and right heart function. In addition, it provides important information regarding left heart function, which is necessary to exclude Group 2 PH caused by reduced left ventricular systolic function, diastolic dysfunction, or valvular heart disease. Pulmonary function tests (PFTs) are needed to exclude chronic lung disease. In patients with PAH, PFTs often show normal spirometry and lung volumes, but a reduced diffusion capacity of carbon monoxide (DLCO). Overnight oximetry and/or polysomnography are helpful to exclude nocturnal oxygen desaturation and evaluate for obstructive or central sleep apnea. Ventilation perfusion imaging (VQ scan) should be performed to exclude CTEPH and is more sensitive for this diagnosis than CT pulmonary angiogram. Exclusion of CTEPH is needed even in the absence of a clinical history of venous thromboembolism, as nearly half of all cases have no recollection of pulmonary embolism. When the diagnosis of PAH is suspected based on clinical presentation and/or non-invasive testing, the diagnosis needs to be confirmed by right heart catheterization (RHC). This procedure is the only reliable method of accurately assessing pulmonary arterial pressure, left heart filling pressure, and cardiac output. It is also helpful in detecting left to right intra-cardiac shunts and is needed to assess acute pulmonary vasoreactivity. The importance of proper diagnosis in PAH is critical considering the gravity of the prognosis and the need for expensive and often cumbersome lifelong therapy. In addition to obtaining cardiopulmonary hemodynamics, additional measures are usually made to assess overall disease severity. These measures include 6MWD, vital signs, dyspnea score, and determination of functional class using the WHO modification of the New York Heart Association functional classification for heart failure. The functional class (FC) scoring system categorizes patients by exercise limitation, with FC I representing asymptomatic patients and FC IV representing those who have symptoms with any activity or at rest. FC II and III are distinguished by whether the patient gets short of breath with ordinary or with less than ordinary activity. For example, a patient who is only symptomatic while performing a strenuous, but ordinary activity, such as mowing the lawn, would be considered FC II, whereas a patient who is symptomatic with less strenuous activities, such as walking to the mailbox or washing the dishes, would be considered FC III (Table 4). Figure 2 depicts a flow chart for initial work-up of PH.

Algorithm for initial work-up of PAH. If PH is clinically suspected, obtain echocardiogram to evaluate for left heart disease. If there are no signs of left heart disease, obtain electrocardiogram, pulmonary function tests, chest imaging (chest x-ray and/or CT scan and VQ scan), and polysomnography. Assess patient for risk factors for PAH such as connective tissue disease or exposures to drugs/toxins. Lung disease and left heart disease should be treated aggressively, and patients should be followed closely for clinical response. If no treatable causes of PH are identified, right heart catheterization should be performed. Furthermore, patients with Groups 2 and 3 disease who have signs of worsening PH despite optimal treatment of heart and lung disease should undergo right heart catheterization to evaluate for PH out of proportion to underlying disease which might benefit from PAH therapy. If the diagnosis of pre-capillary PH is confirmed by right heart catheterization, response to acute vasodilator therapy should be evaluated, and–if positive–calcium channel blockers can be considered. If there is no acute vasodilator response, treatment for PAH can be initiated based on the treatment algorithm displayed in Fig. 3. PH pulmonary hypertension, ECG electrocardiogram, PFTs pulmonary function tests, PoPH portopulmonary hypertension, CTD-PAH connective tissue disease-associated pulmonary arterial hypertension, RHC right heart catheterization
Treatment Guidelines
Numerous review articles have discussed treatment options for pulmonary hypertension, but few have developed comprehensive guidelines for the management of PAH. As the number of medications for this rare disease increased over the first decade of this century, several professional organizations developed and updated evidence-based guidelines for clinical practice. They include the joint committees of the European Society of Cardiology and the European Respiratory Society (ESC/ERS), the WSPH, and the American College of Chest Physicians (ACCP). Each of these guidelines is discussed below, but all share common features as follows.
General Measures and Supportive Care
All three guidelines recommend that patients with a diagnosis of PAH should receive routine preventative care including pneumococcal and influenza vaccinations [3, 30]. Women should be counseled to avoid pregnancy, not only because pregnancy is associated with an increased maternal mortality, but because some PAH medications can cause fetal defects. Prior to the advent of PAH-specific medications, maternal mortality rates were about 30% and fetal mortality > 10% [31]. Although recent reports have described more successful delivery of woman with PAH [32], broad-based studies that assess the overall mortality rate of pregnancy have not been done in the age of modern PAH treatment.
To improve exercise tolerance, patients are encouraged to participate in supervised exercise programs [3, 30]. Non-essential surgery should be avoided due to increased risk of peri-operative complications [30]. If possible, the use of epidural anesthesia instead of general anesthesia is recommended, as the latter can reduce right ventricular contractility and cause hemodynamic instability. Due to the rarity of PAH, most care providers are expected to have limited experience in diagnosis and treatment of PAH, and for this reason, all three guidelines suggest consideration of patient referral to a center with expertise in PAH to aid in patient care. Supportive care, such as supplemental oxygen to maintain oxygen saturation greater than 90% and diuretics as needed to prevent volume overload and excess fluid retention, is also recommended [3, 30, 33]. Patients are cautioned against air travel, because hypoxia caused by reduced cabin pressure can cause acute worsening of PH. Patients who choose to fly should undergo altitude simulation testing to determine if supplemental oxygen is necessary to maintain an arterial oxygen tension ≥ 60 mmHg or oxygen saturation of > 90% [3, 30]. Evidence that anticoagulation is helpful in PAH is limited. Most recent guidelines do not recommend anticoagulation for patients with associated PAH, but for those with IPAH, heritable PAH, or drug-induced PAH, it is suggested that patients be considered for anticoagulation on a case by case basis [1, 8].
Pulmonary Vasodilator Responders
All three guidelines suggest that patients with IPAH, heritable PAH, or drug-induced PAH undergo pulmonary vasodilator challenge during RHC [30]. A positive pulmonary vasodilator response is defined as a reduction in mPAP by ≥ 10 mmHg to a value of ≤ 40 mmHg, with unchanged or improved cardiac output in response to administration of a rapidly active pulmonary vasodilator, such as inhaled nitric oxide or intravenous epoprostenol. A positive vasodilator response is observed in about 10% of patients with idiopathic or heritable PAH [3]. These patients often respond favorably to treatment with high-dose calcium channel blockers, such as nifedipine or amlodipine, and have a better overall prognosis [1, 8]. However, caution should be used in patients with advanced disease evidenced by reduced cardiac output, hypotension, or syncope, as calcium channel blockers have negative inotropic effects and cause systemic vasodilation leading to hypotension. Patients who have a positive vasodilator response and are treated with calcium channel blockers should be followed closely for response to treatment, because up to half of these patients demonstrate disease progression requiring additional therapy [34]. Patients with PAH associated with connective tissue disease rarely demonstrate or maintain an acute vasodilatory response [35] and some guidelines suggest that vasodilator testing in these patients may be unnecessary [1, 8]. Acute vasodilator testing is not recommended for patients in whom pulmonary veno-occlusive disease (PVOD), pulmonary capillary hemangiomatosis (PCH), or other causes of elevated pulmonary capillary or pulmonary venous pressure are suspected, because pulmonary vasodilators may cause acute pulmonary edema in these conditions [36, 37].
Initial Treatment
All current treatment guidelines recommend that patients with PAH who do not demonstrate an acute pulmonary vasodilator response should be treated with PAH-specific therapy based on severity of symptoms or risk of clinical deterioration [3, 30, 33]. The general approach to therapy entails determining severity of disease and initiating oral therapy in less severe PAH and continuous intravenous infusion of prostacyclin in patients with severe PAH.
The European Society of Cardiology/European Respiratory Society Guidelines
The ESC/ERS released official guidelines for the diagnosis and treatment of pulmonary hypertension in 2004 and updated them in 2009 and in 2015 [3]. The most recent document represents the most comprehensive of the three recently updated treatment guidelines and includes over 35 tables, 48 pages of text, and 456 references. A key feature of the ESC/ERS guidelines is that initial treatment is determined by risk stratification using an assessment of numerous clinical factors to determine the probability of 1-year mortality (Table 5). Risk of death is divided into low, intermediate, and high-risk categories defined as estimates of 1-year mortality of < 5%, 5–10%, or > 10%, respectively. Although each of the variables used to calculate mortality risk have been associated with prognosis, the threshold values that divide patients into each of the risk categories were assigned somewhat arbitrarily by expert opinion, and their ability to accurately predict 1-year mortality has not been formally studied. Furthermore, many patients have risk variables in different columns (for example, BNP and 6MWD meet criteria for high risk, but functional class and right atrial pressure meet criteria for intermediate risk) making it difficult to determine the overall risk. The purpose of risk stratification is not to accurately determine 1-year survival, but rather to develop an overall assessment of disease severity. Oral medications are then felt to be appropriate for patients who are at low or intermediate risk. As patients demonstrate more high-risk factors, greater consideration should be given for using continuous prostacyclin infusion.
Monotherapy
Oral monotherapy is recommended for patients who are at low or intermediate risk. Each drug is scored on class of recommendation as follows: I—recommended, IIa—should be considered, IIb—may be considered, or III—not recommended. Treatment recommendations are also graded on level of evidence as follows: A—multiple large randomized clinical trials or meta-analyses, B—single randomized clinical trial or large non-randomized trials, or C—combination of expert opinion and small studies, retrospective studies, or registries. By this scoring system, therapy can be initiated in low and intermediate-risk patients with an ERA, such as ambrisentan (I, A), bosentan (I, A), or macitentan (I, B); a phosphodiesterase type -5 inhibitor (PDE5i), such as sildenafil (I, A), tadalafil (I, B), or vardenafil (IIb, B); the guanylate cyclase stimulator (GCS) riociguat (I, B); or the oral prostacyclin receptor agonist (PRA), selexipag (I, B). Low or intermediate-risk patients in WHO functional class III PAH could alternatively be treated with oral or parenteral prostacyclin analogs, such as intravenous epoprostenol (I, A), inhaled iloprost (I, B), intravenous iloprost (IIa, C), subcutaneous or inhaled treprostinil (I, B), or intravenous treprostinil (IIa, C).
The ESC/ERS guidelines recommend that patients in the high-risk category be treated with intravenous prostacyclin in combination with a PDE5i or ERA. The preferred prostacyclin therapy for functional class IV PAH is intravenous epoprostenol (I, A), but other prostacyclin analogs such as inhaled or intravenous iloprost; or subcutaneous, inhaled, intravenous, or oral treprostinil may be considered, albeit at a lower level of recommendation (IIb, C). Oral agents such as ERAs, PDE5is, or GCS are not generally recommended unless patients are unwilling or unable to be treated with prostacyclin infusion therapy (IIb, C).
Combination Therapy
As an alternative to monotherapy, patients at low to intermediate risk can be treated with up-front combination oral therapy, typically with ambrisentan and tadalafil (I, B), but combinations of other ERAs and PDE5i can be used (IIa, C). Combination therapy that includes a parenteral prostacyclin can also be used to treat patients at intermediate risk and should be used to treat patients at high risk. Specifically intravenous epoprostenol plus bosentan and/or sildenafil (IIa, C) should be used in high-risk patients. Other combinations of ERA, PDE5i, and parenteral prostacyclins can also be considered to treat WHO functional class III or IV PAH (IIb, C). Although parenteral prostacyclins are the mainstay of treatment in functional class IV disease, combination ambrisentan, and tadalafil (or another combination of ERA and PDE5i) can also be considered (IIb, C). It should be noted that at the time of publication of the ESC/ERS guidelines, data on up-front combination therapy in low and intermediate-risk patients had just become available, and its use was not uniformly adapted. As a result these guidelines left the choice between monotherapy or combination therapy to the provider.
Regardless of the initial choice of therapy, the 2015 ECS/ERS guidelines recommend that patients be followed closely and re-evaluated after 3–6 months of treatment. The goal of treatment is to maintain or achieve a low (I, C) or intermediate (IIa, C)-risk status. Patients who fail to do so are felt to have had an inadequate response to therapy and should be considered for additional therapy. These therapies are added sequentially such that those on monotherapy who do not achieve low-risk status are treated with dual therapy and if low-risk status is not reached 3–6 months later, triple therapy is initiated. Triple therapy may include three oral agents or two oral agents with an inhaled or parenteral prostacyclin. Those who progress to high-risk status despite triple therapy that includes a parenteral prostacyclin are referred for evaluation for lung transplantation.
The Sixth World Symposium on Pulmonary Hypertension
The Sixth World Symposium on Pulmonary Hypertension (WSPH) was held in 2018. Thirteen task forces, each focusing on a different topic developed recommendations that were published as a series of manuscripts in the European Respiratory Journal in 2019 [2, 4, 38,39,40,41,42,43,44,45,46,47,48]. The most notable difference between the 2018 WSPH and the 2015 ESC/ERS treatment guidelines was the recommendation for a reduced role for monotherapy in patients at low or intermediate risk. Specifically, these guidelines recommend up-front use of combination therapy with an ERA and PDE5i in low and intermediate-risk patients unless patients have multiple risk factors for left ventricular diastolic dysfunction, high probability of PVOD, or PAH that was not studied in the AMBITION trial, such as PAH associated with portal hypertension or very mild PAH. For patients in functional class IV or who are in the high-risk category, it is recommended that treatment be initiated with intravenous epoprostenol in combination with a PDE5i. Monotherapy can be considered, as it has been shown to improve exercise capacity, hemodynamics, and outcomes compared with no treatment, but only in situations in which parenteral prostacyclin and combination therapy are not available or are not agreeable to the patient. The recommendation of up-front initial combination therapy is due to the findings of the AMBITION trial which was published just after the release of the 2015 ECS/ESR guidelines. In that study, 500 patients were randomized 2:1:1 to receive a combination of tadalafil and ambrisentan, ambrisentan alone, or tadalafil alone. Those assigned to the combination arm had a lower rate of clinical failure events defined as death, hospitalization for PAH, disease progression, or unsatisfactory 6-month clinical response than patients randomized to tadalafil or ambrisentan alone [28]. As with the 2015 ECS/ERS guidelines, the 2019 WSPH guidelines agree that a “multiparametric risk stratification approach” using “clinical, exercise, right ventricular function and hemodynamic parameters” should be used to assign patients to a low, intermediate, or high-risk category that drives both initial and follow-up treatment choices. However, other prognostic tools aside from the 2015 ECS/ESR risk stratification table (Table 5) may be used to assign risk category and readers are referred to risk assessment scores derived from the REVEAL, COMPERA, Swedish PAH, and French PH Network registries.
The American College of Chest Physicians Guidelines
The ACCP first published guidelines for the management of PAH in the journal CHEST in 2004 [49]. The CHEST guidelines were updated in 2007, 2014, and 2019 [30]. For these guidelines, a systematic literature search was conducted to address the question of what is the comparative effectiveness and safety of monotherapy or combination therapy for PAH. The quality of evidence from clinical trials that met the search criteria was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach [50]. Graded recommendations and ungraded consensus-based statements were developed and voted on using a modified Delphi technique to achieve consensus. Similar to the 2015 ESC/ERS guidelines, the 2019 CHEST guidelines recommend the use of oral therapies for patients with mild or moderate disease and intravenous epoprostenol for patients with severe disease. However, the CHEST guidelines rely more on patient functional class than on a comprehensive risk assessment to determine disease severity. As such, oral therapies are recommended for patients in functional class II or III and intravenous prostacyclin for patients in functional class IV. The 2019 CHEST guidelines also agree with the 2019 WSPH guidelines that up-front combination therapy is preferred over monotherapy for treatment-naïve patients in functional class II and III. The 2019 CHEST guidelines are summarized as follows:
WHO Functional Class I
The CHEST guidelines do not recommend initiating therapy in patents with WHO functional class I disease due to the lack of any clinical trials that evaluated PAH therapy in this group, but recommends that these patients should be monitored regularly for progression to functional class II. This is not a recommendation against the early treatment of PAH, but rather the inability to make a recommendation regarding treatment in a group of patients who have not been formally studied.
WHO Functional Class II
The CHEST guidelines recommend initiating combination oral therapy with an ERA and a PDE5i, specifically ambrisentan and tadalafil, to improve 6MWD. Patients who cannot tolerate combination therapy can be started on monotherapy with an ERA (ambrisentan to improve 6MWD; or bosentan or macitentan to delay time to clinical worsening), a PDE5i (tadalafil or sildenafil to improve 6MWD); or GCS (riociguat to improve 6MWD, improve functional class, and delay time to clinical worsening). The CHEST guidelines recommend against inhaled or parenteral prostanoids as initial therapy in WHO functional class II disease.
WHO Functional Class III
First-line initial therapy for patients with WHO functional class III PAH is combination therapy with an ERA and a PDE5i, specifically ambrisentan and tadalafil to improve 6MWD. Patients who cannot tolerate combination therapy should be treated with monotherapy with an ERA (bosentan to improve 6MWD and decrease PH-related hospitalizations; ambrisentan to improve 6MWD; or macitentan to improve functional class and delay time to clinical worsening), a PDE5i (sildenafil to improve 6MWD and functional class; or tadalafil to improve 6MWD, improve functional class and delay time to clinical worsening); or GCS (riociguat to improve 6MWD, improve functional class, and delay time to clinical worsening). Patients in functional class III who have evidence of rapid progression or other high-risk signs can be initiated on parenteral prostanoids, specifically intravenous epoprostenol, intravenous treprostinil, or subcutaneous treprostinil, all of which have been shown to improve 6MWD.
WHO Functional Class IV
Patients with WHO functional class IV disease should be treated with intravenous epoprostenol, which has been shown to improve 6MWD, improve functional class and improve survival, or intravenous or subcutaneous treprostinil which have been shown to improve 6MWD. Those who are not good candidates for parenteral therapy can be treated with combination therapy with an ERA and a PDE5i and considered for triple therapy by adding inhaled or oral prostacyclin derivatives or a PRA. Patients may be started on a PDE5i in addition to parenteral prostanoids, but the CHEST guidelines do not recommend the combination of bosentan and intravenous epoprostenol due to lack of demonstrate efficacy [30].
Determining Response to Therapy
All three sets of guidelines recommend evaluating response to treatment after 3–6 months. ESC/ERS recommends risk-stratifying patients as low, intermediate, or high risk as described above. ESC/ERS and WSPH recommend that if low-risk status has been achieved (or maintained), current therapy should be continued. Patients who are intermediate or high risk after 3–6 months of therapy should be initiated on an additional agent. If monotherapy was used, options for combination therapy include macitentan + sildenafil, riociguat + bosentan, selexipag + ERA, or selexipag + PDE5i. The use of riociguat + PDE5i is not recommended because in the only clinical trial performed this combination did not improve efficacy and was associated with greater adverse effects [51]. If patients do not respond to combination therapy, triple therapy, such as selexipag + ERA + PDE5i can be used or parenteral prostacyclins can be considered. Referral to transplant center should also be considered in this population [3, 45].
CHEST guidelines primarily use WHO functional class to determine response to treatment. For patients who continue to be WHO functional class III or IV despite therapy with ERA and PDE5i, inhaled treprostinil or inhaled iloprost can be added. Treatment for WHO functional class IV PAH on stable doses of intravenous epoprostenol can be optimized by either up-titrating epoprostenol or adding sildenafil. Patients with WHO functional class III or IV disease who are symptomatic despite two classes of PAH drugs, should have a third agent added. Other combinations endorsed by CHEST include adding riociguat to bosentan, ambrisentan, or inhaled prostanoids; adding macitentan to PDE5i or inhaled prostanoids; and adding tadalafil to ambrisentan [30]. Figure 3 depicts a flow chart of how to initiate therapy and adjust treatment regimens based on response to initial therapy.

Algorithm for initiating and adjusting PAH therapy. Assess functional class and mortality risk. WHO functional class I does not require therapy, but patients should be monitored closely for progression to functional class II. Guidelines recommend that initial therapy for WHO functional class II/III is combination oral therapy with ERA and PDE5i. Alternative therapies include monotherapy with an oral agent or intravenous prostacyclin. Recommended therapy for WHO functional class IV is intravenous prostacyclin alone or in combination with PDE5i or ERA. Alternative approaches for patients who do not want or cannot tolerate recommended therapy include combination oral therapy. Response to treatment should be monitored closely. Those who remain WHO functional class III/IV or are intermediate/high risk should have additional therapies added. A second oral agent can be added to oral monotherapy; a third oral agent or inhaled/parenteral prostacyclin can be added to combination oral therapy; combination inhaled/parenteral prostacyclin and oral therapy can be supplemented with a second oral agent; inhaled/subcutaneous prostacyclin can be changed to intravenous prostacyclin; or the dose of intravenous prostacyclin can be increased. Patients who are refractory to maximal treatment should be considered for lung transplant. Black arrows designate recommended therapies. Blue arrows represent alternative therapies when recommended therapies cannot be instituted. WHO world health organization, ERA endothelin receptor antagonist, PDE5i phosphodiesterase-5-inhibitor, SQ subcutaneous, IV intravenous
Knowledge Gaps and Future Studies
The treatment algorithms in the above guidelines have been driven in part by the results of the AMBITION study that demonstrated a significant reduction in clinical failure events, greater increase in 6MWD, and greater reduction in proBNP levels in patients who received up-front combination therapy with a PDE5i and an ERA compared to those who were treated with either agent alone. However, it remains unclear if the beneficial effect of combination therapy is due to additive or synergistic effects of these two drug classes or if using two drugs increases the likelihood of finding one that the patient is able to respond to. Furthermore, it is not known if other combinations of oral therapies such as sGC stimulator and a PRA or an oral prostacyclin would be more or less effective than the PDE5i + ERA combination. At the time that the AMBITION study was initiated, PDE5i and ERAs were the only oral agents approved for treatment of PAH. Since that time, three new classes of orally active agents have been approved (riociguat, selexipag, and oral treprostinil), providing numerous other potential combination therapies that have yet to be tested. Finally, it is possible that the benefits of dual versus monotherapy could be exceeded by up-front triple combination therapy. One ongoing phase III trial (TRITON) is presently evaluating such an approach by examining the efficacy of up-front treatment with macitentan + tadalafil + selexipag versus macitentan + tadalafil + placebo in treatment-naïve PAH patients (Clinicaltrials.gov NCT02558231). Study completion is expected in the year 2020.
Present guidelines also suggest that patients who fail to respond to up-front combination therapy be treated with additional therapy resulting in an increased likelihood that most patients will eventually be on three drugs. An alternative approach that is just beginning to be tested is to switch therapies in patients who have an unsatisfactory response to their initial treatment. In a recently completed, prospective, open-labelled study, 61 patients treated with PDE5i alone or in combination with an ERA who failed to achieve a satisfactory response defined as functional class III with a cardiac index < 3.0 L/min/m2 and PVR > 400 dyne.cm.sec.4 were switched from their PDE5i to riociguat [52]. Significant improvements in 6MWD and NT-proBNP levels were seen at the completion of the 24-week study, and just over half of the patients had an improvement in WHO functional class. A larger prospective, randomized, placebo-controlled study (REPLACE) has recently completed enrollment and results of this study should help to determine if this strategy can be successful (Clinicaltrials.gov NCT02891850). Due to the lack of data, neither triple combination therapy nor switching established therapy has been discussed in the most recently updated guidelines, and neither approach can be recommended until further studies are completed. However, for those patients who are unwilling or unable to follow current treatment guidelines, these alternatives present additional options (Fig. 4). It should be emphasized, however, that neither sequential add-on therapy nor switching drugs from one class to another should delay the initiation of parenteral prostacyclin therapy in patients who progress to a high-risk status.

Strategies in the treatment of PAH. Guideline-recommended therapy depends on mortality risk. Combination PDE5i + ERA is the recommended treatment for low-risk patients in FC II/III. Current guidelines do not recommend monotherapy but it can be considered with any of the approved oral therapies for PAH in low-risk patients who cannot obtain or tolerate combination treatment. Inhaled/oral prostacyclin or selexipag should be considered for patients who are treated with PD5i + ERA who progress from FC II to FC III or from low to intermediate risk, or in patients in FC III or intermediate risk who do not improve. Intravenous epoprostenol should be used in patients who are FC IV or high risk and oral therapy alone or in combination should be considered in these patients. An alternative approach for intermediate-risk patients who do not improve on treatment with PD5i or PD5i + ERA is to switch PD5i to riociguat in and then add prostacyclin therapy if there is no improvement. RAP, BNP, 6MWD, and WHO FC provide only approximate measures of 1-year mortality risk. See text for more detailed methods of risk assessment. Treatments that are recommended by current guidelines are in black font. Currently available options to recommended treatments are shown in blue. RAP right atrial pressure, BNP brain natriuretic peptide, 6MWD 6-min walk distance, WHOFC world health organization functional class, PDE5i phosphodiesterase-5-inhibtor, ERA endothelin receptor antagonist
Adverse Effects and Drug Costs
Most of the major side effects associated with the use of PAH-specific medications are related to their vasodilating effects outside of the pulmonary circulation. Although considerable overlap of adverse effects exists between drugs, some symptoms appear unique to particular drug classes. PDE5is commonly cause headache and have also been associated with esophageal reflux and muscle pains. Rarely, PDE5is have been associated with visual or auditory disturbances. Due to the possible association between PDE5i and non-arteritic anterior ischemic optic neuropathy (NAION), PDE5i should be stopped immediately in patients who report change in vision until they can be evaluated by an ophthalmologist. The use of ERAs has been associated with worsening peripheral edema and sinus congestion or rhinorrhea. These agents may also cause a mild anemia. Elevation of liver transaminases is common in bosentan and has been associated with hepatitis, but rarely occurs in ambrisentan or macitentan. The side effect profile of prostacyclins differs from the other agents because this class of drug is usually titrated to maximum tolerable effect. Prostacyclins are well known for causing facial flushing, erythema, diarrhea, and jaw pain when chewing. Some patients will also complain of musculoskeletal pain especially in their lower extremities or on the soles of their feet when standing. Other less specific complaints include fatigue, GI upset, and lightheadedness and are seen with all drug classes. All PAH-specific medications can lower systemic blood pressure. Although this effect is generally mild, in some cases, it can necessitate dose reduction or discontinuation of the patient’s anti-hypertensive medications. The combination of PDE5i and nitrate therapy can cause more severe systemic hypotension and is contraindicated. The frequency and severity of side effects from PAH medications vary considerably between patients, but in some cases may make it difficult to follow recommended treatment guidelines. Side effect profiles should be considered when designing a specific regimen for a patient. The large number of drugs available for treatment of PAH provides the patient and clinician with a variety of options for individualizing patient care.
Although PAH is a rare disease, medical treatment can contribute significantly to health care costs. In general, PDE5i are the least expensive class of PAH drugs, whereas the retail prices of ERAs, GCS, PRA, and inhaled or oral prostanoids are estimated to cost considerably more. Under current pricing conditions, combination therapy with two or three oral agents can exceed hundreds of thousands of dollars a year in the US. Generic PDE5i and ERAs are available, but their cost savings have generally been minimal. Cost estimates for parenteral prostacyclins and oral treprostinil are difficult to estimate because they depend on the dose, which varies considerably between patients. However, for many patients, the cost of parenteral therapy may be substantially less than combination therapy with oral agents. Health care providers will need to be cognizant of the overall cost of prescribed therapies in the present-day health care system.
Summary and Conclusions
PAH is a rare disease that is progressive and incurable. The general approach to management includes proper classification of disease, determination of disease severity, initiating treatment, and frequent assessments of response to therapy. Proper diagnosis is imperative as patients with PAH have a poor prognosis and require long-term therapy. Over a dozen drugs are currently approved for the treatment of PAH providing both patient and clinician numerous options for what was once considered a disease with no treatment. Several comprehensive treatment guidelines have recently been updated. In general, these guidelines suggest that PAH patients be assessed for disease severity and likelihood of 1-year mortality using a combination of symptoms and clinical findings and that those who are at low or intermediate risk be treated initially with up-front combination therapy comprised of an ERA and a PDE5i. Patients at high risk of death within the following year and/or in WHO functional class IV should be treated with continuous intravenous infusion of epoprostenol alone or in combination with an oral PDE5i or a PDE5i and an ERA. All patients should be assessed at regular intervals for response to therapy. Patients who do not attain, low or intermediate-risk status, should be considered for additional therapy. More recently developed medications such as GCS, PRA, and oral treprostinil provide patients with a number of options to expand therapy, but should not delay the initiation of parenteral prostacyclins in high-risk patients. Consideration should be given for referral to a center experienced in diagnosis and treatment of PAH at the time of diagnosis due to the rarity of this disease and the limited experience that most practitioners may have in its management. Patients who remain at high risk despite medical therapy should be considered for lung transplant evaluation.
References
Kovacs G, Berghold A, Scheidl S, Olschewski H (2009) Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 34(4):888–894
Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M et al (2019) Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. https://doi.org/10.1183/13993003.01913-2018
Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A et al (2016) 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37(1):67–119
Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR et al (2019) Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. https://doi.org/10.1183/13993003.01887-2018
Southgate L, Machado RD, Gräf S, Morrell NW (2020) Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol 17(2):85–95. https://doi.org/10.1038/s41569-019-0242-xEpub 2019 Aug 12
Strange G, Playford D, Stewart S, Deague JA, Nelson H, Kent A et al (2012) Pulmonary hypertension: prevalence and mortality in the Armadale echocardiography cohort. Heart 98(24):1805–1811
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V et al (2006) Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 173(9):1023–1308
Association PH (2019) Preliminary data: demographic characteristics of participants. Pulmonary Hypertension Association. http://phassociation.org/phar/preliminary-data/demographics. Accessed 29 Oct 2019
Humbert M, Sitbon O, Simonneau G (2004) Treatment of pulmonary arterial hypertension. N Engl J Med 351(14):1425–1436
Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM et al (1992) An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 327(2):70–75
Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L et al (1999) Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 159(6):1925–1932
Klinger JR, Abman SH, Gladwin MT (2013) Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med 188(6):639–646
Wharton J, Strange JW, Moller GM, Growcott EJ, Ren X, Franklyn AP et al (2005) Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med 172(1):105–113
Humbert M, Lau EM, Montani D, Jaïs X, Sitbon O, Simonneau G (2014) Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 130(24):2189-2208. https://doi.org/10.1161/CIRCULATIONAHA.114.006974
Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB et al (1996) A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 334(5):296–301
Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC et al (2002) Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 165(6):800–804
Jing ZC, Parikh K, Pulido T, Jerjes-Sanchez C, White RJ, Allen R et al (2013) Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation 127(5):624–633
McLaughlin VV, Benza RL, Rubin LJ, Channick RN, Voswinckel R, Tapson VF et al (2010) Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 55(18):1915–1922
Olschewski H, Simonneau G, Galiè N, Higenbottam T, Naeije R, Rubin LJ et al (2002) Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 347(5):322–329
Galie N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA et al (2008) Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 117(23):3010–3019
Rubin LJ, Badesch DB, Barst RJ, Galiè N, Black CM, Keogh A et al (2002) Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 346(12):896–903
Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA et al (2013) Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 369(9):809–818
Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D et al (2005) Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 353(20):2148–2157
Simonneau G, Rubin LJ, Galie N, Barst RJ, Fleming TR, Frost AE et al (2008) Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med 149(8):521–530
Galie N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z et al (2009) Tadalafil therapy for pulmonary arterial hypertension. Circulation 119(22):2894–2903
Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC et al (2013) Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369(4):330–340
Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N et al (2015) Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 373(26):2522–2533
Galie N, Barbera JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV et al (2015) Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 373(9):834–844
Tongers J, Schwerdtfeger B, Klein G, Kempf T, Schaefer A, Knapp JM et al (2007) Incidence and clinical relevance of supraventricular tachyarrhythmias in pulmonary hypertension. Am Heart J 153(1):127–132
Klinger JR, Elliott CG, Levine DJ, Bossone E, Duvall L, Fagan K et al (2019) Therapy for pulmonary arterial hypertension in adults: update of the CHEST guideline and expert panel report. Chest 155(3):565–586
Weiss BM, Zemp L, Seifert B, Hess OM (1998) Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol 31(7):1650–1657
Lim K, Chang SA, Oh SY, Lee JH, Song J, Kang IS et al (2019) Pulmonary arterial hypertension and pregnancy: single center experience in current era of targeted therapy. Korean Circ J 49(6):545–554
Galie N, McLaughlin VV, Rubin LJ, Simonneau G (2019) An overview of the 6th world symposium on pulmonary hypertension. Eur Respir J. https://doi.org/10.1183/13993003.02148-2018
Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S et al (2005) Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 111(23):3105–3111
Montani D, Savale L, Natali D, Jais X, Herve P, Garcia G et al (2010) Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur Heart J 31(15):1898–1907
Palmer SM, Robinson LJ, Wang A, Gossage JR, Bashore T, Tapson VF (1998) Massive pulmonary edema and death after prostacyclin infusion in a patient with pulmonary veno-occlusive disease. Chest 113(1):237–240
Preston IR, Klinger JR, Houtchens J, Nelson D, Mehta S, Hill NS (2002) Pulmonary edema caused by inhaled nitric oxide therapy in two patients with pulmonary hypertension associated with the CREST syndrome. Chest 121(2):656–659
Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F et al (2019) Genetics and genomics of pulmonary arterial hypertension. Eur Respir J. https://doi.org/10.1183/13993003.01899-2018
Vonk Noordegraaf A, Chin KM, Haddad F, Hassoun PM, Hemnes AR, Hopkins SR et al (2019) Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J. https://doi.org/10.1183/13993003.01900-2018
Sitbon O, Gomberg-Maitland M, Granton J, Lewis MI, Mathai SC, Rainisio M et al (2019) Clinical trial design and new therapies for pulmonary arterial hypertension. Eur Respir J. https://doi.org/10.1183/13993003.01908-2018
Frost A, Badesch D, Gibbs JSR, Gopalan D, Khanna D, Manes A et al (2019) Diagnosis of pulmonary hypertension. Eur Respir J. https://doi.org/10.1183/13993003.01904-2018
Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S et al (2019) Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J. https://doi.org/10.1183/13993003.01916-2018
McGoon MD, Ferrari P, Armstrong I, Denis M, Howard LS, Lowe G et al (2019) The importance of patient perspectives in pulmonary hypertension. Eur Respir J. https://doi.org/10.1183/13993003.01919-2018
Kim NH, Delcroix M, Jais X, Madani MM, Matsubara H, Mayer E et al (2019) Chronic thromboembolic pulmonary hypertension. Eur Respir J. https://doi.org/10.1183/13993003.01915-2018
Galie N, Channick RN, Frantz RP, Grunig E, Jing ZC, Moiseeva O et al (2019) Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. https://doi.org/10.1183/13993003.01889-2018
Hoeper MM, Benza RL, Corris P, de Perrot M, Fadel E, Keogh AM et al (2019) Intensive care, right ventricular support and lung transplantation in patients with pulmonary hypertension. Eur Respir J. https://doi.org/10.1183/13993003.01906-2018
Vachiery JL, Tedford RJ, Rosenkranz S, Palazzini M, Lang I, Guazzi M et al (2019) Pulmonary hypertension due to left heart disease. Eur Respir J. https://doi.org/10.1183/13993003.01897-2018
Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H et al (2019) Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. https://doi.org/10.1183/13993003.01914-2018
Badesch DB, Abman SH, Ahearn GS, Barst RJ, McCrory DC, Simonneau G et al (2004) Medical therapy for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 126(1 Suppl):35s–62s
Balshem H, Helfand M, Schunemann HJ, Oxman AD, Kunz R, Brozek J et al (2011) GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol 64(4):401–406
Galie N, Muller K, Scalise AV, Grunig E (2015) PATENT PLUS: a blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. Eur Respir J 45(5):1314–1322
Hoeper MM, Simonneau G, Corris PA, Ghofrani HA, Klinger JR, Langleben D et al (2017) RESPITE: switching to riociguat in pulmonary arterial hypertension patients with inadequate response to phosphodiesterase-5 inhibitors. Eur Respir J. https://doi.org/10.1183/13993003.02425-2016
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Klinger's institution receives grant funding for pulmonary hypertension studies from United Therapeutics and Actelion, and he serves as a non-paid committee member on a pulmonary hypertension clinical trial for Bayer.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vazquez, Z.G.S., Klinger, J.R. Guidelines for the Treatment of Pulmonary Arterial Hypertension. Lung 198, 581–596 (2020). https://doi.org/10.1007/s00408-020-00375-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-020-00375-w