
Abstract
Purpose
Oxygen therapy is often required to treat newborn infants with respiratory disorders. Prolonged exposure of neonatal rats to hyperoxia reduced alveolar septation, increased terminal air space size, and increased lung fibrosis; these conditions are very similar to those of human bronchopulmonary dysplasia. Epigenetic regulation of gene expression plays a crucial role in bronchopulmonary dysplasia development.
Method
We reared Sprague–Dawley rat pups in either room air (RA, n = 24) or an atmosphere containing 85% O2 (n = 26) from Postnatal Days 1 to 14. Methylated DNA immunoprecipitation (MeDIP) was used to analyze genome-wide DNA methylation in lung tissues of neonatal rats. Hyperoxia-exposed rats exhibited larger air spaces and thinner septa than RA-exposed rats did on Postnatal Day 14. The rats exposed to hyperoxia exhibited significantly higher mean linear intercepts than did the rats exposed to RA. We applied MeDIP next-generation sequencing for profiling changes in DNA methylation in the rat lungs exposed to hyperoxia and RA. We performed bioinformatics and pathway analyses on the raw sequencing data to identify differentially methylated candidate genes.
Results
Our in vivo model revealed that neonatal hyperoxia exposure arrested alveolarization on Postnatal Day 14. We found that the ErbB, actin cytoskeleton, and focal adhesion signaling pathways are epigenetically modulated by exposure to hyperoxia. We demonstrated that hyperoxia exposure contribute in delaying lung development through an epigenetic mechanism by disrupting the expression of genes in lungs that might be involved in alveolarization.
Conclusions
These data indicate that aberrant DNA methylation and deregulation of the actin cytoskeleton and focal adhesion pathways of lung tissues may be involved in the pathophysiology of hyperoxia-induced arrested alveolarization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Therapy involving hyperoxia is often required to treat newborn infants with respiratory disorders. However, the therapies provided to infants during lung development may have adverse long-term effects in addition to beneficial effects. Supplemental oxygen administered to newborn infants with respiratory failure can increase oxidative stress and lead to lung injury. Prolonged exposure of neonatal rats to hyperoxia reduced alveolar septation, increased terminal air space size, and increased lung fibrosis; these conditions are very similar to those of human bronchopulmonary dysplasia [1, 2]. Despite recent improvements in preventing respiratory distress syndrome in preterm infants, bronchopulmonary dysplasia remains a major cause of morbidity and mortality during the first year of life and many infants have notable respiratory problems throughout childhood, including increased airway reactivity and development of obstructive airway disease [3]. Some abnormal lung functions may persist into adulthood [4].
Epigenetics involves genetic modifications that result in changes in gene expression and function without a corresponding alteration in the DNA sequence [5]. Epigenetic mechanisms include DNA methylation, covalent histone modifications, and noncoding RNAs [6]. The epigenetic and environmental influences affecting lung development as well as injury and repair have been acknowledged as a major research area [7]. The priority for epigenetic research is understanding how epigenetics and the environment interact to influence lung development, and later susceptibility to lung injury and repair. An understanding of these mechanisms will require specialized investigations in animal models of perinatal lung injury and will lead to the clarification of potential therapies. We have found that prolonged exposure of neonatal rats to hyperoxia reduced alveolar septation, increased terminal air space size, and increased lung fibrosis [2, 8,9,10]. Animal models of epigenetic modifications secondary to neonatal hyperoxia are an invaluable tool for studying the mechanisms that determine the development of lung diseases in childhood. An understanding of the specific epigenetic mechanisms involved in the developmental origin of lung disease is vital to facilitate the identification of molecular mechanisms that enable identifying risk factors and treatments for respiratory diseases in childhood.
DNA methylation is a major epigenetic mechanism that plays essential roles in various biological processes [11, 12]. Altered DNA methylation patterns have been demonstrated in human lung epithelial-like A549 cells exposed to hyperoxia and in murine and human lungs during alveolar septation [13, 14]. However, a genome-wide analysis of DNA methylation has not been performed in the lungs of newborn rats exposed to hyperoxia. In the current study, methylated DNA immunoprecipitation (MeDIP) was used to analyze genome-wide DNA methylation in lung tissues of neonatal rats with hyperoxia-induced lung injury. Based on results of the MeDIP and arrays, gene ontology (GO) and pathway analyses were performed. These results may provide insights into the therapy and prognosis for hyperoxia-induced inhibition of alveolarization.
Methods
Animals
The study was performed in accordance with guidelines provided and approved by the Animal Care Use Committee of Taipei Medical University. Time-dated pregnant Sprague–Dawley rats (Bio-LASCO Taiwan Co. Ltd., Taipei, Taiwan) were housed in individual cages with free access to laboratory food and water ad libitum, subjected to a 12:12-h light–dark cycle, and allowed to deliver vaginally at term (22 days).
Exposure to Hyperoxia
Within 12 h of birth, the litters were pooled and randomly redistributed to the mothers, and the pups were then randomly assigned to RA (n = 24) or hyperoxia (n = 26) treatment. The pups in the O2 treatment group were reared in an atmosphere containing 85% O2 from Postnatal Days 1 to 14. The pups in the RA control group were reared in normal RA for 14 days. To prevent oxygen toxicity in the nursing mothers, they were rotated between the O2 treatment and RA control litters every 24 h. An oxygen-rich atmosphere was maintained in a transparent 40 × 50 × 60-cm3 plexiglass chamber (NexBiOxy, Hsinchu, Taiwan) receiving O2 continuously at 4 L/min. Oxygen levels were monitored using a ProOx P110 monitor (BioSpherix, Redfield, NY, USA). Carbon dioxide was removed from the chamber by washout and absorption with calcium hydroxide. The animals were sacrificed using an intraperitoneal injection of pentobarbital sodium and exsanguinated through aortic transaction on Postnatal Day 14. The heart and lungs were removed en bloc.
Lung Histology
A single lobe of the right lung tissue was harvested and fixed in 4% paraformaldehyde. Lung tissue sections 5 μm in thickness were stained with hematoxylin and eosin, examined using light microscopy, and assessed for lung morphometry. The mean linear intercept (MLI), an indicator of mean alveolar diameter, was assessed in ten nonoverlapping fields. The number of cross sections of an alveolar wall with half of the ruler was counted. Each alveolar wall was counted as two crossings. Two numbers per field were obtained and the average was used in the equation: MLI = (0.57/average intercepts) × 1000 (μm). The coefficient 0.57 is calculated from the length of the ruler and the coefficient of shrinkage = 0.612 [15]. Five portions of every section were randomly selected and captured at ×100 magnification by a digital camera, and then analyzed the septal thickness by the computerized image analysis system (Image-Pro Plus 5.1 for Windows; Media Cybernetics, Inc., Bethesda, MD, USA) [16]. For radial alveolar count (RAC), a perpendicular line was placed from the junction of epithelium of conducting and respiratory bronchiole to the nearest connective septum or pleura, and the air space traversed by the line were counted [17].
DNA Preparation and Methylated DNA Immunoprecipitation
Total genomic DNA was extracted using the Genomic DNA Miniprep Kit (Axygen, CA, USA) according to the manufacturer’s instructions. The DNA quality was evaluated using agarose gel electrophoresis and a BioPhotometer Plus spectrophotometer (Eppendorf, Germany). Subsequently, lung tissues (35 mg) were sonicated to produce DNA fragments ranging from 100 to 500 bp. After end repairing, phosphorylating, and A-tailing the DNA fragments by using the Paired-End DNA Sample Prep Kit (Illumina, CA, USA), the DNA was ligated to an Illumina sequencing primer adaptor. Subsequently, the fragments were used for MeDIP enrichment by employing the Magnetic Methylated DNA Immunoprecipitation kit (Diagenod, Belgium) according to the manufacturer’s recommendations and the qualifying DNA was used for PCR amplification. Then, bands between 220 and 320 bp were excised from the gel and purified with the QIAquick Gel Extraction Kit (Qiagen, Germany). The products were quantified with the Quant-iT™ dsDNA HS Assay Kit (Invitrogen, MA, USA) using an Agilent 2100 analyzer (Agilent Technologies, CA, USA). Following qPCR qualification, DNA libraries were sequenced on the Illumina HiSeq 2000 (Illumina) to generate paired-end 50-bp reads by the Beijing Genomics Institute (BGI, China).
Bioinformatics Analysis
For bioinformatics analysis, adapters and reads for which more than 10% was affected by N contamination and more than half of the bases had quality scores lower than 20 were excluded from the clean data pool. The rest of the reads were mapped to the reference genome, and the depths of reads with unique alignments were calculated in each 10-kb window. Furthermore, peak distributions in the CpG islands, defined as short DNA regions of genome containing a high frequency of CG dinucleotides, were measured to differentiate methylated regions, followed by the Χ 2 and false discovery rate statistics attributed to GO biological function and the KEGG signaling pathway.
Statistical Analysis
Data were expressed as means ± SDs. The Student t test and one-way ANOVA were used appropriately for comparison between groups. The differences were considered significant at P < 0.05.
Results
Six female rats birthed 60 pups, and 24 and 36 pups were randomly distributed to the RA and hyperoxia groups, respectively. Ten rats exposed to hyperoxia died.
Body and Lung Weight and Lung-to-Body Weight Ratio
Table 1 presents the effect of hyperoxia on body weight, lung weight, and the lung-to-body weight ratio on Postnatal Day 14. The hyperoxia-exposed rats exhibited significantly lower body and lung weights than did the RA-exposed rats on Postnatal Day 14. The lung-to-body weight ratios of the hyperoxia-exposed rats were significantly higher than were those of the RA-exposed rats on Postnatal Day 14.
Lung Histology
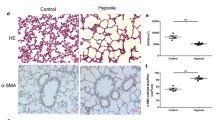
The lungs of the hyperoxia-exposed rats exhibited larger air spaces and thinner septa than did those of the RA-exposed rats on Postnatal Day 14 (Fig. 1a). The rats exposed to hyperoxia exhibited significantly higher MLI and lower RAC and septal thickness than did the rats exposed to RA (Fig. 1b).

a Representative lung sections and b mean linear intercept, radial alveolar count, and septal thickness from lungs of room air- (n = 24) and hyperoxia-exposed (n = 26) rats on Postnatal Day 14 (***P < 0.001)
Hyperoxia Caused Widespread Changes in DNA Methylation
Widespread changes in gene-specific DNA methylation were apparent in both groups (RA and hyperoxia), as depicted in the heat map (Fig. 2a). These results identified genes that exhibited an increase or a decrease in methylation in the rats exposed to hyperoxia compared with those exposed to RA. Approximately 100% of the methylated genes were at the intersection of these two groups (Fig. 2b). Furthermore, most genes exhibited a similar proportion of decreased (86.5%) and increased (84.5%) methylation on UTR region in hyperoxia group (Fig. 2c). To confirm the previous analysis, a histogram of methylation peaks with associated P values is illustrated in Fig. 3a. We found that the graph of the log P value indicated a normal distribution; that is, less than 5% of the thousands of peaks that were identified as methylated were expected to be false positives. To investigate the relation between DNA methylation changes and the genes, six gene-related groups (upstream2k, 5′UTR, CDS, intron, 3′UTR, and downstream2k) were formed. The data indicated that hyperoxia methylation samples expressed a greater number of downregulated signals, particularly in the 5′UTR (Fig. 3b).

Heat map of differential DNA methylation and the number of genes showing increased and decreased methylation. a Heat map of differential DNA methylation enrichment peaks of genes, b Venn diagram of the number of methylated genes in the lung following room air and hyperoxia treatment, and c Venn diagram of the number of decreased and increased methylated genes in the lung following hyperoxia treatment

Differential methylation analysis revealed epigenetic signatures. a Histogram of log value from the methylation read analysis. b Methylation status according to gene-related annotation. Gene-related groups: upstream2k, 5′UTR, CDS, intron, 3′UTR, and downstream2k
Elemental Distribution
Hyperoxia-induced DNA methylation alterations were initially observed from the elements. A total of 29,917,928 (RA group) and 28,359,665 (hyperoxia group) candidate methylated reads were distributed in the different gene elements (Fig. 4). The results indicated the number and proportion of reads on each element in the RA group: 1,691,169 on CDS (4.47%), 134,229 on CpG islands (0.35%), 654,444 on downstream 2k (1.73%), 1,235,374 on the 5′UTR (3.27%), 13,478,095 on the gene body (35.64%), 11,832,777 on introns (31.29%), 404,842 on the 3′UTR (1.07%), and 486,998 on upstream2k (1.29%). In the hyperoxia group, the following numbers and proportions were determined: 1,509,551 on CDS (4.15%), 122,764 on CpG islands (0.34%), 627,058 on downstream 2k (1.72%), 1,181,973 on the 5′UTR (3.25%), 12,754,526 on the gene body (35.03%), 11,286,653 on introns (31.00%), 378,576 on the 3′UTR (1.04%), and 498,564 on upstream 2k (1.37%).

The distribution of reads in different gene elements. The x axis indicates different gene elements and the y axis indicates the proportion of reads in a specific gene element. Reads are concentrated in introns because the introns are considerably longer than the other elements
GO Annotation and Pathway Analysis
The GO project provides a controlled vocabulary to describe attributes of genes and gene products in any organism (http://www.geneontology.org). Ontology covers three domains: biological processes, cellular components, and molecular functions (Fig. 5). In the present study, GO ontology was used to perform GO term analysis of the 1,235,374 reads in the RA group and 1,181,973 reads in hyperoxia group on the 5′UTR region. Results indicated that the candidate reads were associated with 216 biological processes, 246 cellular components, and 227 molecular functions. Genetic studies have largely focused on candidate reads on the 5′UTR involved in development, intracellular parts, and enzyme binding. Therefore, all genes associated with the three GO terms were selected (Table 2). GO analysis of all candidate genes revealed that 54 genes were involved in development, 199 genes in intracellular parts, and 41 genes in enzyme binding. Next, four methylated genes from the GO term results that were epigenetically modified by hyperoxia were selected (Table 3). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (http://www.genome.jp/kegg) was used to perform pathway analysis of these candidate genes. Three signaling pathways were downregulated by hyperoxia, namely ErbB, regulation of the actin cytoskeleton, and the focal adhesion signaling pathway (Table 4).

GO classification for differential genes. The horizontal axis indicates GO items (biological process, cellular component, and molecular function), the left vertical axis indicates the proportion of genes involved, and the right vertical axis indicates the exact number of genes
Genes that Exhibited Changes in Methylation Following Hyperoxia Treatment
The genes that exhibited an increase (Supplementary file 1: Table S1A) or a decrease (Supplementary file 2: Table S1B) in methylation in the rat lung following hyperoxia treatment were assessed using peak analysis.
Discussion
Our in vivo model revealed that neonatal hyperoxia exposure arrested alveolarization, as confirmed by a higher MLI and lower RAC on Postnatal Day 14. In the present study, we used MeDIP next-generation sequencing on lung tissue DNA from RA- and hyperoxia-exposed rats to investigate hyperoxia-induced arrested alveolarization. Using bioinformatics analysis, we identified specific genes that are epigenetically reprogrammed by hyperoxia and associated with arrested alveolarization. These results indicate that aberrant DNA methylation of lung tissues may be involved in the pathophysiology of hyperoxia-induced arrested alveolarization.
Our study demonstrated that rats reared in hyperoxic conditions exhibited significantly lower body and lung weights and higher lung-to-body weight ratios on Postnatal Day 14 than those reared in RA did. Rats reared in hyperoxic conditions exhibited a mortality rate of 27.8%. Newborn rat pups exposed to hyperoxia exhibited pulmonary inflammation and edema [18]. These results suggest that hyperoxia induced rat lung injury and that the cause of death was hyperoxia-induced lung injury.
The degree of alveolarization was evaluated by MLI and RAC methods. MLI is the average distance between alveolar walls. RAC is the average number of alveoli transected by a perpendicular line drawn from the center of a respiratory bronchiole to the nearest septal division. The MLI defined by Campbell and Tompkieff is the most common morphometric measure for assessing alveolarization in animal models [19]. In this study, we assessed alveolarization using the MLI because the value decreases as the lung undergoes secondary septation and the parenchymal complexity increases and is highly inversely correlated with functional assessment of gas exchange [20].
According to our KEGG pathway analysis, focal adhesion was one of the downregulated pathways in this genome-wide analysis of DNA methylation. Decreased cell adhesion and phosphorylation of focal adhesion kinase was observed in alveolar type II cells isolated from rats exposed to hyperoxia and high tidal volume mechanical ventilation [21]. Hyperoxia treatment reduced phosphorylation of focal adhesion kinase in mouse lung epithelial type II cells [22, 23]. Focal adhesion kinase is a crucial regulator protein for focal adhesion, which has been implicated in numerous cellular events, including cell spreading, migration, and proliferation [24, 25]. Thus, the focal adhesion signaling pathway may have an essential role in the development of hyperoxia-induced arrested alveolarization.
Another downregulated pathway in this genome-wide analysis of DNA methylation was the actin cytoskeleton pathway. A cytoskeleton is a system of intracellular filaments and tubules that extend throughout the cytoplasm, from the nucleus to the plasma membrane. The cytoskeleton is crucial for cell shape, division, and function [26, 27]. Rao et al. reported that oxidative stress may induce tyrosine phosphorylation and dissociate the occludin–zonula occludens-1 complexes from the cytoskeleton [28]. Hyperoxia caused remodeling of the cytoskeleton through the reorganization of actin and microtubules in primary rat type II alveolar epithelial cells and a mouse alveolar epithelial cell line [29]. Our in vivo model indicated that hyperoxia disrupted the actin cytoskeleton pathway. Future studies will investigate the effects of hyperoxia on the cytoskeleton components in rat lungs.
The ErbB signaling pathway was modified in this genome-wide analysis of DNA methylation. However, the role of the molecules belonging to this category in the alveolarization processes has not been studied. In light of our results, further studies on this pathway would be of interest.
In total, four methylated genes are associated with hyperoxia-induced inhibition of alveolarization, namely Crk, Fyn, growth factor receptor-bound protein 2 (Grb2), and β1 integrin (Itgβ1). Grb2 is an adaptor protein involved in signal transduction and cell communication [30]. Integrins are a family of transmembrane heterodimeric glycoproteins that link the cytoskeleton to the extracellular environment and act as adhesion receptors, signaling receptors, and mechanoreceptors to regulate cell growth, migration, and differentiation [31]. A recent study on lung ontogeny reported that Itgβ1 is required for lung branching morphogenesis and alveolarization [32]. These results suggest that the aberrant methylation of these genes has a functional role in hyperoxia-induced inhibition of alveolarization.
There are several limitations in this study that must be addressed. First, analyzing the DNA methylation level at each CpG site through MeDIP sequencing is difficult. Second, our study was conducted on entire lung tissue, consisting of several cell types. This might have resulted in the contamination of results because of the inclusion of cells that do not directly contribute to alveolarization. Further studies involving laser capture microdissection and analysis of specific gene methylation changes in each identified network by evaluating protein levels are warranted.
Conclusions
This microarray analysis performed in a model of hyperoxia-induced arrested alveolarization indicated the involvement of several pathways. Our results indicate that deregulation of the actin cytoskeleton and focal adhesion pathways may be crucial components of the mechanism inducing arrested alveolarization observed in hyperoxia. Our findings provide new insights into the potential mechanisms by which hyperoxia arrests alveolarization in newborn rats, and these insights may facilitate the development of novel preventive and therapeutic options for human bronchopulmonary dysplasia.
Abbreviations
- ErbB:
-
Avian erythroblastosis oncogene B
- GO:
-
Gene ontology
- Grb2:
-
Growth factor receptor-bound protein 2
- Itgβ1:
-
Β1 integrin
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MeDIP:
-
Methylated DNA immunoprecipitation
- MLI:
-
Mean linear intercept
- RA:
-
Room air
- RAC:
-
Radial alveolar count
References
Manji JS, O’Kelly CJ, Leung WI et al (2001) Timing of hyperoxic exposure during alveolarization influences damage mediated by leukotrienes. Am J Physiol Cell Mol Physiol 281:L799–L806
Chen CM, Wang LF, Chou HC et al (2007) Up-regulation of connective tissue growth factor in hyperoxia-induced lung fibrosis. Pediatr Res 62:128–133
Lemons JA, Bauer CR, Oh W et al (2001) Very low birth weight outcomes of the National Institute of Child Health and Human Development neonatal research network, January 1995 through December 1996. NICHD Neonatal Research Network. Pediatrics 107:E1
Northway WH Jr, Moss RB, Carlisle KB et al (1990) Late pulmonary sequelae of bronchopulmonary dysplasia. N Engl J Med 323:1793–1799
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Goldberg AD, Allis CD, Bernstein E (2007) Epigenetics: a landscape takes shape. Cell 128:635–638
Castro M, Ramirez MI, Gern JE et al (2009) Strategic plan for pediatric respiratory diseases research: an NHLBI working group report. Proc Am Thorac Soc 6:1–10
Chou HC, Lang YD, Wang LF et al (2012) Angiotensin II type 1 receptor antagonist attenuates lung fibrosis in hyperoxia-exposed newborn rats. J Pharmacol Exp Ther 340:169–175
Jiang JS, Lang YD, Chou HC et al (2012) Activation of the renin-angiotensin system in hyperoxia-induced lung fibrosis in neonatal rats. Neonatology 101:47–54
Su CL, Chou HC, Huang LT et al (2014) Combined effects of maternal inflammation and neonatal hyperoxia on lung fibrosis and RAGE expression in newborn rats. Pediatr Res 75:273–280
Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13:484–492
Day JJ, Sweatt JD (2011) Epigenetic mechanisms in cognition. Neuron 70:813–829
Panayiotidis MI, Rancourt RC, Allen CB et al (2004) Hyperoxia induced DNA damage causes decreased DNA methylation in human lung epithelial like A549 cells. Antioxid Redox Signal 6:129–136
Cuna A, Halloran B, Faye-Petersen O et al (2015) Alterations in gene expression and DNA methylation during murine and human lung alveolar septation. Am J Respir Cell Mol Biol 53:60–73
Roubliova XI, Biard JM, Ophalvens L, Gallot D, Jani JC, Verbeken EK, Van De Ven CP, Tibboel D, Deprest JA (2007) Morphology of the developing fetal lung—the rabbit experimental model. In: Méndez-Vilas A, Díaz J (eds) Modern research and educational Topics in microscopy. Formatex, Badajoz, pp 417–425
Gao C, Li R, Huan J, Li W (2011) Caveolin-1 siRNA increases the pulmonary microvascular and alveolar epithelial permeability in rats. J Trauma 70:210–219
Cooney TP, Thurlbeck WM (1982) The radial alveolar count method of Emery and Mithal: a reappraisal 2–intrauterine and early postnatal lung growth. Thorax 37:580–583
Almario B, Wu S, Peng J et al (2012) Pentoxifylline and prevention of hyperoxia-induced lung-injury in neonatal rats. Pediatr Res 71:583–589
Campbell H, Tomkeieff SI (1952) Calculation of the internal surface of a lung. Nature 170:116–117
Ahlfeld SK, Gao Y, Conway SJ et al (2015) Relationship of structural to functional impairment during alveolar-capillary membrane development. Am J Pathol 185:913–919
Desai LP, Sinclair SE, Chapman KE et al (2007) High tidal volume mechanical ventilation with hyperoxia alters alveolar type II cell adhesion. Am J Physiol Lung Cell Mol Physiol 293:L769–L778
Wilhelm KR, Roan E, Ghosh MC et al (2014) Hyperoxia increases the elastic modulus of alveolar epithelial cells through Rho kinase. FEBS J 281:957–969
Zhang L, Yuan LJ, Zhao S et al (2015) The role of placenta growth factor in the hyperoxia-induced acute lung injury in an animal model. Cell Biochem Funct 33:44–49
Infusino GA, Jacobson JR (2012) Endothelial FAK as a therapeutic target in disease. Microvasc Res 83:89–96
Belvitch P, Dudek SM (2012) Role of FAK in S1P-regulated endothelial permeability. Microvasc Res 3:22–30
Fletcher DA, Mullins RD (2010) Cell mechanics and the cytoskeleton. Nature 463:485–492
Wickstead B, Gull K (2011) The evolution of the cytoskeleton. J Cell Biol 194:513–525
Rao RK, Basuroy S, Rao VU et al (2002) Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-betacatenin complexes from the cytoskeleton by oxidative stress. Biochem J 368:471–481
Roan E, Wilhelm K, Bada A et al (2012) Hyperoxia alters the mechanical properties of alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 302:L1235–L1241
Lowenstein EJ, Daly RJ, Batzer AG et al (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70:431–442
Roth-Kleiner M, Post M (2005) Similarities and dissimilarities of branching and septation during lung development. Pediatr Pulmonol 40:113–114
Plosa EJ, Young LR, Gulleman PM et al (2014) Epithelial β1 integrin is required for lung branching morphogenesis and alveolarization. Development 141:4751–4762
Funding
This work was supported by the Ministry of Science and Technology, Taiwan under Grant (MOST 103-2314-B-038-019-MY3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
The study was performed in accordance with guidelines provided and approved by the Animal Care Use Committee of Taipei Medical University (LAC-2013-0159).
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, CM., Liu, YC., Chen, YJ. et al. Genome-Wide Analysis of DNA Methylation in Hyperoxia-Exposed Newborn Rat Lung. Lung 195, 661–669 (2017). https://doi.org/10.1007/s00408-017-0036-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-017-0036-z