
Abstract
Background
IgG4-related disease (IgG4RD) is a recently recognized disease entity. Differentiating IgG4RD from plasma cell type Castleman’s disease (PCD) is important but also difficult using only pathological findings. In addition, little is known about the association between these two diseases with diffuse parenchymal lung involvement.
Methods
We analyzed the serum IgG4 levels and the ratio of IgG4/IgG-positive plasmacytes in the lung and lymph node specimens of eight patients previously pathologically diagnosed of PCD with diffuse parenchymal lung involvement (DL-PCD). We also compared the clinical and laboratory findings observed in these patients.
Results
Six of the eight patients exhibited abundant IgG4-positive plasmacytes in the lung and lymph node tissues and elevated serum IgG4 levels, thereby fulfilling the diagnostic criteria of IgG4RD with DL (DL-IgG4RD) in addition to having obstructive phlebitis and massive lymphoplasmacytic infiltration with fibrosis. However, three of these six patients exhibited higher levels of serum interleukin-6 and were still diagnosed with DL-PCD. Accordingly, three of these eight patients were considered as IgG4RD with DL (DL-IgG4RD), and the other five patients were ultimately given a diagnosis of DL-PCD. These two diseases have different characteristics in terms of age, symptoms, serum levels of C-reactive protein, and IgA, complicating allergic disorders, response to corticosteroids, and prognosis.
Conclusions
This is the first report to show a high prevalence of DL-IgG4RD in DL-PCD patients, although additional large investigations are necessary. Clinical and laboratory findings are important for distinguishing between these two diseases in other organs, as previously described.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
IgG4-related disease (IgG4RD) is a recently described systemic fibroinflammatory disease that is associated with elevated circulating levels of IgG4 [1, 2]. IgG4RD is clinically characterized by the enlargement of one or more exocrine glands or extranodal tissues and elevated serum IgG4 levels. Pathologically, IgG4RD is characterized by lymphoplasmacytic infiltration with fibrosis and obliterative phlebitis, as well as an increased frequency of IgG4-positive plasmacytes [3–5]. Clinical manifestations are apparent in the pancreas, bile ducts, gallbladder, lacrimal glands, salivary glands, retroperitoneum, kidneys, prostate, and lungs [6–10].
Castleman’s disease (CD) is an uncommon lymphoproliferative disorder first reported by Castleman [11]. CD is categorized into two histological types: hyaline-vascular and plasma cell [12, 13]. Patients with the plasma cell type of CD (PCD) often manifest aggressive systemic illnesses. An overproduction of interleukin-6 (IL-6) from the affected organs is thought to be responsible for the systemic manifestations of PCD [14, 15].
The major differential diagnoses of IgG4RD with lung involvement include lung cancer, malignant lymphoma, and PCD [9]. IgG4RD and PCD are especially difficult and often impossible to distinguish based on pathological findings due to their similarities. Because PCD can express IgG4, making the differential diagnosis is not easy, even with IgG4 detection [9, 16–18]. Moreover, diffuse parenchymal lung diseases are well known to be difficult to diagnose, even in patients who undergo open lung biopsies [19, 20], and IgG4RD and PCD have been found to involve lung manifestations as a diffuse pattern. However, no reports have evaluated the existence of IgG4RD with diffuse parenchymal lung involvement (DL-IgG4RD) in patients with pathologically diagnosed PCD with diffuse parenchymal lung involvement (DL-PCD). We have suspected that many cases of pathologically diagnosed DL-PCD may include DL-IgG4RD. We therefore evaluated the IgG4 expression in the serum, lung, and lymph node tissues of patients with pathologically diagnosed DL-PCD and assessed the clinical features of these patients in the present study.
Materials and Methods
Study Subject
We examined eight patients with pathologically diagnosed DL-PCD at our university hospital and its affiliated hospitals between 1998 and 2011. One case (Patient 4) has been reported previously as a case report [21]. Written informed consent was obtained from all patients. The study complied with the Declaration of Helsinki and was approved by the Human and Animal Ethics Review Committee of the University of Occupational and Environmental Health, Japan (H23—79).
Data Collection
The clinical data were obtained from the patients’ medical records. Blood samples were collected at the time of examination for the diagnosis before starting treatment, and stored at −30 °C until the analysis, and the serum IgG4 levels were additionally measured. The blood samples were assayed for IL-6 (normal range <4.0 pg/ml), IgG (normal range 863–1,589 mg/dl), IgG4 (normal range 4–108 mg/ml), IgG4/IgG (normal range 3–6 %), IgM (normal range 35–220 mg/dl), IgE (normal range 0–170 IU/ml), IgA (normal range 110–410 mg/ml), C-reactive protein (CRP) (normal range <0.3 mg/dl), soluble interleukin-2 receptor (sIL-2R) (normal range 122–496 U/ml), Krebs von den Lungen-6 (KL-6) (normal range <500 U/ml), total protein (normal range 6.7–8.3 g/dl), lactate dehydrogenase (normal range 119–229 IU/l), total cholesterol (normal range 150–219 mg/dl), albumin (normal range 4.0–5.0 g/dl), γ-glb (normal range 11–21.1 %), and hemoglobin (normal range, male: 13.5–17.6 g/dl, female: 11.3–15.2 g/dl).
Histopathological and Immunohistochemical Examinations
The lung tissues were obtained using surgical lung biopsies in seven patients and a transbronchial lung biopsy in one patient. The lymph node tissues were obtained using mediastinal lymph node biopsies in four patients, inguinal lymph node biopsies in three patients, and a cervical lymph node biopsy in one patient. The lung tissue and lymph node specimens were fixed with 20 % formaldehyde, embedded in paraffin, and cut into 4-μm sections. The tissue sections were deparaffinized, dehydrated with graded xylene and alcohol, and incubated in 3 % hydrogen peroxide for 5 min at room temperature to eliminate the endogenous peroxidase activity. Immunostaining was performed using a BenchMark Ultra automated slide stainer (Ventana Medical Systems Inc., Tucson, AZ). Tissue sections underwent standardized heating pretreatment for antigen retrieval before the immunohistochemical procedure. Primary antibodies used were: IgG (1:6000; Dako, Glostrup, Denmark), IgG4 (1:500; The Binding Site, Birmingham, UK), CD3 (PS-1; 1:50; Novocastra, Newcastle, UK), CD5 (4C7; 1:100; Novocastra), CD10 (56C6; 1:50; Novocastra), CD20 (L26; 1:200; Novocastra), CD21 (1F8; 1:20; Dako, Carpinteria, CA), CD138 (MI15; 1:100; Dako), Bcl-2 (3.1; 1:200; Novocastra), Kappa (NCL-KAP; 1:100; Novocastra), Lambda (NCL-LAM; 1:200; Novocastra), and human herpes virus type (HHV)-8 (137B1; 1:50; Novocastra). The numbers of IgG4- and IgG-positive cells were estimated in the areas with the highest density of positive cells. Five different high-power fields (HPF) in ×400 magnification of each section were counted, and the average number of positive cells per HPF was calculated [22].
Epstein-Barr virus (EBV)-encoded small RNA (EBER) in situ hybridization was performed using Ventana XT System Benchmark in accordance with the manufacturer’s instructions, with an EBER-antisense probe (INFROM EBER probe, Ventana Medical Systems).
Evaluation of Chest Computed Tomography (CT) Images
The chest CT features of all patients were reviewed by five investigators who were blinded to the patients’ clinical status. The chest CT findings were evaluated according to the findings listed by Webb et al. [23], and mediastinal lymph node enlargement also was assessed.
Diagnosis of DL-PCD and DL-IgG4RD
Pathologically, PCD was defined as the presence of numerous lymphoid follicles with active germinal centers and interfollicular polyclonal plasmacytosis in the lymph node and lung tissue specimens, excluding other differential diagnoses based on immunohistochemical detection, as previously described [17, 18, 22, 24, 25]. The criteria of IgG4RD used in the present study were defined as fulfillment of all of the following criteria: an elevated serum IgG4 level (>135 mg/dl), pathological findings of abundant infiltration of IgG4-positive plasmacytes (>10 per HPF), and a ratio of IgG4/IgG-positive cells of more than 40 % in the tissue specimens. The pathological findings of obstructive phlebitis and prominent lymphoplasmacytic inflammation with fibrosis were also considered for the diagnosis of IgG4RD as described previously [1, 9, 10]. However, hyper IL-6 syndrome, which was characterized by high levels of serum IL-6 and inflammation, was not included within the IgG4RD diagnosis, even if the diagnostic criteria for IgG4RD were fulfilled, as previously described [1, 18, 22].
Statistical Analysis
Comparisons between groups (DL-PCD vs. DL-IgG4RD) were made using the Mann–Whitney U test with the SPSS software program (version 19.0). P values < 0.05 were considered to be statistically significant. The values are expressed as the mean ± standard deviation (SD) when appropriate.
Results
Diagnosis of DL-PCD and DL-IgG4RD
The histopathological findings of the biopsied lung and lymph node specimens and the laboratory findings are shown in the Fig. 1 and Table 1. The pathological lung and lymph node findings of all eight patients were compatible with a diagnosis of PCD. In addition, there were no HHV-8- and EBER-positive cells, and the immunoglobulin light chain reactivity of plasma cells showed a polyclonal pattern in all tissue specimens. IgG4-positive plasmacytes accounted for more than 40 % of the IgG-positive plasmacytes in the lung and lymph node tissue specimens in six patients (Patients 1–6) and less than 40 % of the IgG-positive plasmacytes in the lung and lymph node tissue specimens in the other two patients (Patients 7, 8). Massive lymphoplasmacytic inflammation with fibrosis within the peribronchial and perivascular connective tissues was observed in the lung specimens of all eight patients, and obstructive phlebitis was observed in the lung tissue specimens of seven patients (Patients 1–6, 8). Eosinophilic infiltration and granulomas were rarely seen in the lung specimens of all of the eight patients. The serum IgG4 levels were elevated in all seven analyzed patients (Patients 1–7). As such, six of eight subjects (75 %) fulfilled the pathological criteria for DL-IgG4RD (Patients 1–6). However, three of these six patients (Patients 4–6) were still considered to have DL-PCD, because they exhibited higher serum IL-6 levels. Accordingly, three of the eight patients (37.5 %) fulfilled the diagnostic criteria and were considered as DL-IgG4RD (Patients 1–3) and five of the eight patients (Patients 4–8) were considered as DL-PCD.

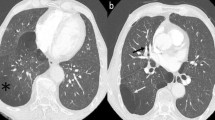
Radiological and histopathological findings diagnosed as plasma cell-type Castleman’s disease with diffuse parenchymal lung involvement with abundant IgG4-positive plasmacytes (patient 4). Chest computed tomography (CT) scans obtained on admission demonstrated mediastinal lymphadenopathies, interlobular septal thickening with peribronchovascular consolidation, and patchy ground-glass and nodular attenuations in the bilateral lungs (a, b). Hematoxylin-eosin (H-E) staining of the lung specimens obtained with surgical lung biopsies showed peribronchovascular inflammatory cell infiltration and follicular hyperplasia with fibrosis (c), plasmacyte and lymphocyte infiltration to the bronchioles (d), and obstructive phlebitis (e). Immunohistochemically, the infiltrating plasmacytes were highly positive for IgG4 (IgG4/IgG = 56 %) in the lung tissue (f). H-E staining of the mediastinal lymph nodes showed hyperplastic germinal centers and hypercellular interfollicular spaces (g), plasma cell infiltration (h), and mildly atrophic concentric germinal centers containing hyalinized blood vessels (i). Immunohistochemically, the infiltrating plasma cells were also highly positive for IgG4 (IgG4/IgG = 50.5 %) in the lymph node tissue. Original magnification: ×100 (c, g), ×400 (d, e, f, h, i, j)
Clinical and Laboratory Findings
We compared the clinical and laboratory findings of the three patients with DL-IgG4RD and the five patients with DL-PCD. The clinical and laboratory findings in the DL-PCD and DL-IgG4RD groups are summarized in Tables 2 and 3. Three patients with DL-IgG4RD had an average age of 71.0 ± 12.3 years and consisted of two males and one female. Five patients with DL-PCD demonstrated an average age of 51.0 ± 9.8 years and consisted of two males and three females. The average age of the patients with DL-IgG4RD was higher than that of the patients with DL-PCD (P = 0.025). Seven patients (Patients 2–8) had respiratory symptoms, such as exertional dyspnea, coughing, and/or general malaise, and one patient with DL-IgG4RD (Patient 1) had no clinical symptoms. All patients with DL-PCD had systemic or strong symptoms such as fever, general malaise, and dyspnea. In contrast, the patients with DL-IgG4RD did not exhibit such systemic or strong symptoms (100 vs. 0 %). Serum antibodies against the human immunodeficiency virus (HIV) were negative in all eight patients. Two cases of DL-IgG4RD (Patients 2, 3) involved comorbidities of IgG4RD, such as autoimmune pancreatitis and IgG4-related membranous nephropathy (67 vs. 0 %), whereas neither of the patients with DL-PCD had any comorbidities. Complicating allergic disorders, including allergic conjunctivitis, bronchial asthma, and allergic dermatitis, were more common in the DL-IgG4RD group and not observed in the DL-PCD group (100 vs. 0 %). The levels of serum IL-6 were significantly higher in the DL-PCD group than in the DL-IgG4RD group (5.0 ± 1.6 vs. 23.3 ± 8.1 pg/ml, P = 0.025). The serum IgG levels were elevated in all patients with DL-IgG4RD and DL-PCD (3,736 ± 1,620 vs. 3,946 ± 2,109 mg/dl, P = 0.88). The serum IgG4 levels and the IgG4/IgG ratios also were elevated in the patients with DL-IgG4RD and all four patients with DL-PCD (1,813 ± 1,635 vs. 677 ± 407 mg/dl, P = 0.29; 43.1 ± 21 vs. 18.9 ± 15.1 %, P = 0.16). Although the levels of serum IgM and IgE were not different between the two groups, the levels of serum IgA were normal in all patients with DL-IgG4RD and elevated in four of the five patients with DL-PCD (209 ± 120 vs. 698 ± 284 mg/dl, P = 0.025). The serum CRP levels were significantly higher in the patients with DL-PCD (0.46 ± 0.05 vs. 7.7 ± 4.1 mg/ml, P = 0.025). However, the levels of serum soluble IL-2 receptor, KL-6, total protein, albumin, γ-globulin, lactate dehydrogenase, total cholesterol, and hemoglobin were not different between the two groups.
Chest CT Findings
The chest CT findings are shown in the Fig. 1 and Table 4. Mediastinal lymph node enlargement, thickening of the bronchovascular bundles, and the presence of centrilobular nodules were observed in all eight patients. Interlobular septal thickening were observed in all three patients with DL-IgG4RD and in four of five patients with DL-PCD. Ground glass opacities were observed in one of the three patients with DL-IgG4RD and in all five patients with DL-PCD. These findings were similar between the DL-IgG4RD and DL-PCD patients.
Responses to Treatment and Prognoses
The responses to treatment and prognoses of the patients are shown in Table 5. The patients were followed up for a median 59.2 months (range: 15–135 months). During the observation period, one of the three patients with DL-IgG4RD (Patient 1) was observed using a wait-and-see policy without receiving any treatment because the patient’s symptoms were minor and no radiological progression was observed. The other two patients with DL-IgG4RD (Patients 2, 3) and all five patients with DL-PCD received initial therapy with corticosteroids (Patients 4–8). The two patients with DL-IgG4RD (Patients 2, 3) and two of the five patients with DL-PCD (Patients 5, 6) received corticosteroid treatment alone, achieving good responses in clinical, laboratory, and radiological findings. The other three patients with DL-PCD showed either no response to corticosteroids or repetitive worsening of the symptoms during the tapering of corticosteroids. As a result, either immunosuppressive agent (cyclosporine in Patients 4, 7) or monoclonal anti-IL-6 antibody (tocilizumab in Patient 8) was added to the treatment regimen in such cases. During the observation period, one of the five patients with DL-PCD died after developing respiratory failure, 25 months after disease presentation (Patient 8).
Discussion
In the present study, six of eight patients (75 %) with pathologically diagnosed DL-PCD exhibited IgG4/IgG ratios of more than 40 % of plasmacytes in the lung and lymph node tissues in addition to high serum IgG4 levels. Obstructive phlebitis and massive lymphoplasmacytic inflammation with fibrosis also were observed in the lung tissue specimens of these six patients. However, three of these six patients were still categorized as DL-PCD due to higher serum IL-6 levels in the laboratory findings. Accordingly, three of the eight patients (37.5 %) fulfilled the diagnostic criteria of IgG4RD and the other five patients were still considered as DL-PCD. Even though the sample size of this study was small, these two disease entities were clearly different with respect to age, symptoms, serum levels of CRP and IgA, complicating allergic disorders, response to corticosteroids, and prognosis.
This is the first report to show a high prevalence of DL-IgG4RD among patients with pathologically diagnosed DL-PCD. PCD and IgG4RD often are difficult or occasionally impossible to distinguish each other based only on pathological findings due to their similarities. In addition, it has been reported that PCD patients express IgG4 [17, 18, 26]. Although there are no reports showing differences between IgG4RD and DL-PCD patients with diffuse lung diseases, a few reports do show differences in other organs. Sato et al. [18] reported that six cases of multicentric CD involved the presence of abundant IgG4-positive cells in tissue specimens. Kojima et al. [17] showed that three of four patients (75 %) with PCD of the retroperitoneum should be diagnosed with IgG4RD and concluded that the majority of PCD cases arising in the retroperitoneum might actually be IgG4RD. These reports and our results suggest that many cases of pathologically diagnosed DL-PCD might involve increased expressions of IgG4 and that some of these cases can be categorized as DL-IgG4RD based on the combination of clinical and laboratory findings.
PCD has been reported to be associated with strong systemic inflammation related to IL-6 production from hypertrophic lymph nodes [14, 15]. In contrast, IgG4RD is associated with relatively mild inflammatory reactions and is also considered to be an allergic disorder. Masaki et al. [4] reported that 30 of 64 cases (47 %) of IgG4RD were complicated by allergic disorders. Zen et al. [27] demonstrated that the expressions of T-helper 2 cytokines, such as IL-4, -5, and -13, induced from localized lesions, complicating allergic disorders in patients with IgG4RD. Therefore, patients with PCD usually exhibit strong systemic symptoms and inflammation in the laboratory findings, whereas patients with IgG4RD show relatively mild symptoms and inflammation. In the present study, age, symptoms, the serum CRP levels, the serum IgA levels, and complicated allergic disorders were significantly different in the DL-IgG4RD and DL-PCD groups. These results are similar to those of the report by Sato and colleagues [22], suggesting that our diagnosis is appropriate and that considering clinical and laboratory findings to distinguish these two diseases is very important. In contrast, in this study, there were no differences in the serum IgG, IgM, total protein, albumin, total cholesterol, or hemoglobin levels between the groups. These results are different from those of Sato and colleagues [22], and further examination is needed.
In the present study, the chest CT findings were similar in the patients with both DL-IgG4RD and DL-PCD, as described previously. For example, Inoue et al. [28] and Johkoh et al. [29] reported that the chest CT findings of patients with IgG4RD and CD with lung involvement are characterized by lymph node enlargement, thickening of the bronchovascular bundles, and the presence of centrilobular nodules. In our study, all eight patients exhibited these three radiologic findings. These findings are usually seen in patients with lymphoproliferative disorders and are not specific to DL-IgG4RD and DL-PCD. Collectively, the previous reports and our study suggest that it is difficult to distinguish these two diseases based only on chest CT findings.
Optimal treatment strategies for IgG4RD and CD have not been established, because both diseases are relatively rare and studies of large numbers of patients have not yet been conducted. However, patients with IgG4RD generally exhibit good responses to corticosteroid therapy [1, 2, 9, 10]. On the other hand, many patients with PCD do not exhibit good responses to corticosteroid treatment alone (steroid responsiveness: 30–70 %) [30–32]. Therefore, patients with PCD occasionally receive systemic chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone) [31], or monoclonal anti-IL-6 antibodies [33, 34]. In this study, all of the patients with DL-IgG4RD exhibited favorable outcomes with or without corticosteroid treatment, whereas three of five patients with DL-PCD required additive treatment in addition to corticosteroids. These results suggest that differentiating between these diseases is important to predict the response to corticosteroids and the prognosis, as previously described in patients with IgG4RD affecting other organs.
There were several limitations to this study. First, no international consensus has been reached yet with regard to the precise diagnostic criteria for IgG4-related lung disease. Therefore, we employed the latest comprehensive diagnostic criteria for IgG4RD proposed from Japan [1]. Hyper IL-6 syndrome and IgG4RD often are similar in pathological findings, but the clinical and laboratory findings are very different [22]. Thus, Hyper IL-6 syndrome is excluded from IgG4-related syndrome in the proposed criteria from Japan, although the definition of Hyper IL-6 syndrome is not clearly stated. In the present study, the patients with DL-IgG4RD and DL-PCD exhibited different clinical aspects in terms of age, symptoms, serum levels of CRP, and IgA, complicating allergic disorders, response to corticosteroids, and prognosis. Therefore, we believe that the diagnostic criteria and the concept of DL-IgG4RD used in this study are appropriate. Second, the sample size of this study was small. We examined only eight cases because DL-IgG4RD and DL-PCD are both relatively rare diseases. Accordingly, additional examinations with larger numbers of patients with these diseases are necessary to confirm our results. Third, the understanding of IgG4-related lung disease is still evolving, and further detailed knowledge of this disease would modify the diagnostic criteria for IgG4-related lung disease in the future.
In conclusion, this is the first report to show that relatively many cases of pathologically diagnosed DL-PCD involve an expression of IgG4, and some of them can be categorized as DL-IgG4RD. Clinical and laboratory findings are important to distinguish these two diseases in patients with diffuse lung diseases, as described previously in other organs.
References
Umehara H, Okazaki K, Masaki Y et al (2012) IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol 22:1–14
Stone JH, Zen Y, Deshpande V (2012) IgG4-related disease. N Engl J Med 366:539–551
Cheuk W, Yuen HK, Chu SY et al (2008) Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol 32:671–681
Masaki Y, Dong L, Kurose N et al (2009) Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis 68:1310–1315
Smyrk TC (2011) Pathological features of IgG4-related sclerosing disease. Curr Opin Rheumatol 23:74–79
Zen Y, Nakanuma Y (2010) IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol 34:1812–1819
Fujinaga Y, Kadoya M, Kawa S et al (2010) Characteristic findings in images of extra-pancreatic lesions associated with autoimmune pancreatitis. Eur J Radiol 76:228–238
Matsui S, Taki H, Shinoda K et al (2012) Respiratory involvement in IgG4-related Mikulicz’s disease. Mod Rheumatol 22:31–39
Matsui S, Hebisawa A, Sakai F et al (2013) Immunoglobulin G4-related lung disease: clinicoradiological and pathological features. Respirology 18:480–487
Ryu JH, Sekiguchi H, Yi ES (2012) Pulmonary manifestations of immunoglobulin G4-related sclerosing disease. Eur Respir J 39:180–186
Castleman B, Iverson L, Menendez VP (1956) Localized mediastinal lymph node hyperplasia resembling thymoma. Cancer 9:822–830
Frizzera G (1988) Castleman’s disease and related disorders. Semin Diagn Pathol 5:346–364
Weisenburger DD, Nathwani BN, Winberg CD, Rappaport H (1985) Multicentric angiofollicular lymph node hyperplasia: a clinicopathologic study of 16 cases. Hum Pathol 16:162–172
Yoshizaki K, Matsuda T, Nishimoto N et al (1989) Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood 74:1360–1367
Nishimoto N (2005) Clinical studies in patients with Castleman’s disease, Crohn’s disease, and rheumatoid arthritis in Japan. Clin Rev Allergy Immunol 28:221–230
Ishida F, Kitano K, Kobayashi H, Saito H, Kiyosawa K (1997) Elevated IgG4 levels in a case with multicentric Castleman’s disease. Br J Haematol 99:981–982
Kojima M, Nakamura N, Motoori T et al (2010) Castleman’s disease of the retroperitoneum: with special reference to IgG4-related disorder. J Clin Exp Hematop 50:39–44
Sato Y, Kojima M, Takata K et al (2010) Multicentric Castleman’s disease with abundant IgG4-positive cells: a clinical and pathological analysis of six cases. J Clin Pathol 63:1084–1089
Shrestha B, Sekiguchi H, Colby TV et al (2009) Distinctive pulmonary histopathology with increased IgG4-positive plasma cells in patients with autoimmune pancreatitis: report of 6 and 12 cases with similar histopathology. Am J Surg Pathol 33:1450–1462
Strehl JD, Hartmann A, Agaimy (2011) A Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol 64:237–243
Ogoshi T, Yatera K, Nagata S et al (2011) A case of multicentric Castleman disease with massive infiltration of plasmacytes presenting IgG4. JJRS 49:437–442
Sato Y, Kojima M, Takata K et al (2009) Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman’s disease. Mod Pathol 22:589–599
Webb WR, Muller NL, Naidich DP (2001) Illustrated glossary of high-resolution computed tomography term. High-resolution CT of the lung, 3rd edn. Lippincott Williams & Wilkins, Philadelphia
Flendrig JA, Schillings PHM (1969) Benign giant lymphoma: the clinical signs and symptoms. Folia Med Neerl 12:119
Keller AR, Hochholzer L, Castleman B (1972) Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 29:670–683
Jo JH, Park YS, Jeon YK, Nam SJ, Huh J (2011) Comparison of plasma cell type of Castleman’s disease and IgG4-related sclerosing disease: a histopathological and immunohistochemical study. Pathobiology 78:227–232
Zen Y, Fujii T, Harada K et al (2007) Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology 45:1538–1546
Inoue D, Zen Y, Abo H et al (2009) Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology 251:260–270
Johkoh T, Muller NL, Ichikado K et al (1998) Intrathoracic multicentric Castleman disease: CT findings in 12 patients. Radiology 209:477–481
Frizzera G, Peterson BA, Bayrd ED et al (1985) A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease: clinical findings and clinicopathologic correlations in 15 patients. J Clin Oncol 3:1202–1216
Herrada J, Cabanillas F, Rice L, Manning J, Pugh W (1998) The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med 128:657–662
Bowne WB, Lewis JJ, Filippa DA et al (1999) The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer 85:706–717
Beck JT, Hsu SM, Wijdenes J et al (1994) Brief report: alleviation of systemic manifestations of Castleman’s disease by monoclonal anti-interleukin-6 antibody. N Engl J Med 330:602–605
van Rhee F, Fayad L, Voorhees P et al (2010) Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol 28:3701–3708
Conflicts of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ogoshi, T., Kido, T., Yatera, K. et al. Assessment of Pathologically Diagnosed Patients with Castleman’s Disease associated with Diffuse Parenchymal Lung Involvement Using the Diagnostic Criteria for IgG4-Related Disease. Lung 191, 575–583 (2013). https://doi.org/10.1007/s00408-013-9497-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-013-9497-x