
Abstract
Apart from the deleterious effects on the lungs, chronic obstructive pulmonary disease (COPD) should be considered a complex, systemic disease involving several organs and systems. The nature and course of systemic inflammation in COPD is important since there is a potential for anti-inflammatory therapy. The objective of the current study was to assess biomarkers of systemic inflammation in stable and exacerbation phases of COPD patients as compared to healthy controls. We also investigated the course of these biomarkers after COPD exacerbation to evaluate their usefulness for disease monitoring. Eighty-three stable patients with moderate to very severe COPD, 20 patients in exacerbation phase, and 30 subjects with normal pulmonary function were included. Serum tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and nitric oxide (NO) levels were measured once in stable COPD patients and controls and three times in the COPD exacerbation group during follow-up. TNF-α and IL-6 levels were higher than in controls in both stable and exacerbation groups. Although NO was not higher in the stable COPD group than in controls, it was higher in the exacerbation group. In follow-up after the exacerbation period, significant alteration was not detected in cytokine or NO levels compared to admission. Raised serum levels of TNF-α and IL-6 support their use as biomarkers of the systemic inflammatory response in stable COPD patients. However, the circulating biomarkers we have studied are not found to be useful either as indicators of COPD exacerbation or for monitoring recovery after exacerbation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As stated in the definition of chronic obstructive pulmonary disease (COPD), the association of an abnormal inflammatory response of the lungs to noxious particles or gases with airflow limitation indicates the critical role of the inflammatory process in the pathogenesis of COPD [1, 2]. The disease is associated with a switch from a self-limiting inflammatory response, mainly initiated by smoke inhalation, to a chronic persistent inflammatory response after prolonged exposure to cigarette smoke [3].
Today, apart from the deleterious effects on the lungs, COPD should be considered a complex, systemic disease involving the participation of several organs and systems, including musculoskeletal, cardiovascular, and endocrine systems, and metabolic changes leading to weight loss [4–12]. Recent studies have shown that COPD is associated not only with an abnormal inflammatory response of the lung but also with systemic inflammation, including systemic oxidative stress, activation of circulating inflammatory cells, and increased circulating levels of inflammatory cytokines [5, 13]. The local inflammatory process in the lungs can affect peripheral tissues either by the direct effect of released cytokines and chemokines or indirectly by activation of peripheral inflammatory cells [5].
Levels of inflammatory proteins such as C-reactive protein (CRP) and proinflammatory cytokines are increased in the systemic circulation of COPD patients [5, 13–15]. Interleukin-6 (IL-6) is produced by many different cells but the main sources are monocytes, macrophages, T and B cells, fibroblasts, epithelial cells, and smooth muscle of airways, indicative of its role in the modulation of the immune system. IL-6 plays a role in activation, proliferation, and differentiation of T cells [16]. Tumor necrosis factor-α (TNF-α) is produced from several cells, including T lymphocytes, mast cells, and cells of airway epithelium. TNF-α controls cellular migration and permeability and stimulates secretion of several cytokines [16]. IL-6 and TNF-α lead to a shift toward catabolism, resulting in muscle wasting and cachexia [3–5, 14, 15].
Reactive nitrogen species have been implicated in the pathogenesis of COPD for they have potent proinflammatory and oxidizing actions [17–19]. The formation of oxidants by phagocytic cells is fundamental in the host’s defense against infectious agents. However, in an inflammatory microenvironment, exaggerated production of nitric oxide (NO) in the presence of “oxidative stress” may produce strong oxidizing reactive nitrogen species, such as peroxynitrite, leading to nitration of most classes of biological molecules (“nitrosative stress”), ending up with cell damage and the expression of proinflammatory cytokines [17]. Stimuli such as chemokines and cytokines, including TNF-α and IL-6, or exogenous factors like infection, allergens, pollutants, and hypoxia increase NO by causing activation of inducible nitric oxide synthase (iNOS). Because the free radical NO has a short serum half-life, it cannot be directly measured and its presence must be inferred by indirect methods such as nitrite, nitrate, or nitrotyrosine [17].
The nature and course of systemic inflammation in COPD is important since there is a potential for anti-inflammatory therapy [16, 20]. Besides, biomarkers may be useful for disease monitoring. The objective of the current study was to assess systemic inflammation expressed as circulatory levels of TNF-α, IL-6, and NO in stable and exacerbation phases of COPD patients as compared to healthy controls. We also investigated the course of these biomarkers after COPD exacerbation to evaluate their potential for disease monitoring.
Material and Methods
Subjects
One hundred three consecutive male patients who had received a diagnosis of COPD and had received continuing care at our chest clinic and 30 control subjects of the same sex and age group were admitted to the study. COPD was diagnosed and classified according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria [1]. Eighty-three of the patients had been clinically stable for at least 3 months and 20 had clinical signs of COPD exacerbation (increased dyspnea, sputum production, or sputum purulence) [21]. Stable COPD patients had been receiving optimal medical therapy, i.e., inhaled bronchodilator therapy in the form of long-acting β2 agonists and/or anticholinergic agents. Severe/very severe COPD patients (54 patients in the stable group and 18 patients in the exacerbation group) were on inhaled corticosteroids (500–1,000 μg fluticasone/day). Subjects with exacerbation were treated with antibiotics and systemic steroids for 10–14 days in addition to bronchodilatory therapy.
Exclusion criteria for both study groups included nonrespiratory infections and significant cardiac, renal, hepatic, endocrine, neurologic, or metabolic disturbance. The control group consisted of subjects with normal pulmonary function tests.
The study was approved by the institutional committee on human research and informed consent was obtained from all subjects.
Pulmonary Function Tests
Forced vital capacity (FVC) and forced expiratory volume in one second (FEV1) were measured with standard spirometric techniques according to the American Thoracic Society criteria (Minato AutoPal Spirometry, Osaka, Japan) [22]. The highest value from at least three spirometric maneuvers was used. Patients with FEV1 <50% of predicted value were considered to have severe/very severe COPD and 50% ≤ FEV1 < 80% as having moderate COPD (GOLD). An arterial blood sample was obtained while the subjects were breathing room air for at least 30 min, and was analyzed with a blood gas analyzer immediately (OMNIC, Roche, Vienna, Austria).
Assessment of Cytokines and Nitric Oxide End-products
Fasting peripheral venous blood samples were taken from the antecubital vein into plain tubes between 8:00 and 9:00 a.m. Blood was then promptly centrifuged and aliquots of serum were immediately stored at −70°C until they were analyzed for serum levels of TNF-α, IL-6, and NO.
Serum TNF-α concentration (pg/ml) was measured by a solid-phase sandwich enzyme-linked immunosorbent assay (ELISA) using an hTNF-α kit (BioSource International Inc., Camarillo, CA, USA) [23]. Serum IL-6 concentration (pg/ml) was also measured with an ELISA kit (BioSource International Inc.). NO levels (μmol/L) were determined indirectly by quantification of their oxidized products of degradation (nitrite and nitrate), using Griess reagent [24]. NO is an unstable molecule and is rapidly converted to nitrite and nitrate; therefore, serum nitrite/nitrate was used and the measured value is equivalent to NO.
Cytokine and NO levels were measured once in stable COPD patients and controls and three times in the COPD exacerbation group: on the day of admission to hospital, on the day of discharge (14th day of admission), and during recovery (4 weeks after discharge).
Statistical Analysis
Data were presented as mean ± standard deviation of the mean. Correlations between parameters were evaluated using Pearson’s rank correlation analysis. Differences between study groups were compared using Mann–Whitney U test for non-normally distributed variables. Normality was tested by the Shapiro–Wilk test. The Friedman test was used to compare baseline and follow-up cytokine and NO levels of COPD patients. Significance was determined at the 5% level.
Results
Characteristics and pulmonary function tests of the stable and exacerbation groups of COPD patients and the control subjects admitted to the study are given in Table 1. Twenty-nine (35%) of the stable COPD patients had moderate and 54 (65%) had severe/very severe COPD. There was a significant difference in pulmonary function and blood gases between the stable and exacerbation groups of COPD patients, as expected (Table 1). There was no difference in the body mass index (BMI) of COPD patients and controls (p > 0.05).
Cytokine and NO Assays
The comparison of the circulating TNF-α, IL-6, and NO levels of the stable and exacerbation-phase COPD patients and the control subjects is given in Table 2. TNF-α and IL-6 levels were higher in both the stable and the exacerbation group than in the controls. When stable and exacerbation-phase COPD patients were compared, although cytokine levels tended to be higher in the exacerbation group than in the stable group, there was no statistically significant difference; however, the NO level was higher in the exacerbation group.
Admission, discharge, and recovery serum cytokine and NO levels of the exacerbation group are given in Table 3. Although there was a transient difference between NO levels measured on admission and discharge (in 14 days), there was no significant difference in measurements between admission and recovery (in 6 weeks).
Correlations
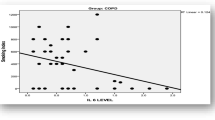
In correlation tests of study parameters, correlation was detected between TNF-α and IL-6 in the stable COPD group (r = 0.356, p = 0.001). NO was positively correlated to cigarette burden (pack-years) in stable patients (r = 0.345, p = 0.011). There was no correlation between NO, cytokines, and either severity of COPD (FEV1%) or arterial blood gases (PaO2, PaCO2).
Discussion
In the present study we investigated the circulatory levels of TNF-α, IL-6, and NO as representative markers of systemic inflammation in stable and exacerbation phases of COPD patients compared with healthy controls. We demonstrated that TNF-α and IL-6 levels were higher in both stable and exacerbation groups than in controls. The cytokine levels tended to be higher in the exacerbation group than in the stable COPD group, but there was no statistically significant difference. Although NO was not higher in the stable COPD group than in controls, it was higher in the exacerbation group. When admission and recovery measurements were compared, significant alteration was not detected in cytokine or NO levels.
We found a positive correlation between circulating NO level and cigarette burden (pack-years) of the COPD patients. Cigarette smoke, the major cause of COPD, has a high concentration of NO. Endogenous NO, released in the bronchial mucosa, plays a key role in physiologic regulation of airway functions and is implicated in airway disease. Several studies indicate that nitrosative stress may promote the progression of airflow limitation in COPD [17–19]. Recently, Ricciardolo et al. [25] investigated the markers of nitrosative stress in bronchial biopsies and bronchoalveolar lavage (BAL) of stable COPD patients. They found higher numbers of nitrotyrosine and myeloperoxidase(+) cells in the bronchial submucosa of severe COPD patients compared to mild/moderate COPD patients, smokers with normal lung function, and nonsmokers. They suggested that nitrosative stress may contribute to pathogenesis of COPD.
We demonstrated that in moderate to very severe stable COPD patients, serum TNF-α and IL-6 levels were higher than in healthy controls. IL-6 is a potent mediator of inflammation and is able to initiate catabolism by activating proteolysis [26]. IL-6 has been studied in induced sputum and exhaled breath for its role in the local inflammatory process in airways, but there are fewer studies investigating the circulatory levels. Serum IL-6 and TNF-α levels were demonstrated to be increased in both mild/moderate and severe COPD patients compared to healthy controls [27]. Schols et al. [14] searched for the role of inflammatory mediators (TNF-α and its soluble receptors IL-6 and IL-8) in metabolic derangement of COPD patients. They found increased levels of inflammatory cytokines in the serum of COPD patients with increased resting energy expenditure and decreased fat-free mass.
Inflammatory markers that increase in COPD play an important part in muscle dysfunction and exercise intolerance. The association between inflammatory markers and ventilatory limitation, muscle strength, and exercise capacity was demonstrated [28]. Recently, Yende et al [28] investigated the association of inflammatory markers C-reactive protein (CRP), TNF-α, and IL-6 with ventilatory limitation and muscle dysfunction. They reported that subjects with COPD had higher systemic levels of IL-6 and CRP compared to elderly subjects without COPD. IL-6 was found to be associated with reduced FEV1, quadriceps strength, and exercise capacity in both groups. In another study, markers of systemic inflammation were significantly elevated in all COPD patients and tended to be highest in muscle-wasted patients [29].
TNF is implicated as a mediator of cachexia in many clinical conditions, including COPD [30]. Serum TNF-α and soluble TNF receptors were reported to be elevated in stable COPD patients [31–34]. Serum TNF-α level was found to be higher in weight-losing COPD patients, whereas it was similar to that of healthy controls in weight-stable patients; increased TNF-α production was considered to be a likely cause of weight loss [32]. In the present study, serum TNF-α was found to be higher in COPD patients, but we did not evaluate its role in weight loss since the BMIs of our study population were within normal limits.
An association between severity of airflow limitation and severity of airway inflammation in smokers had been previously detected [35]. Vernooy et al. [31] searched for a relationship between local and systemic inflammation in COPD and suggested that a different method of regulating inflammation in pulmonary and systemic compartments. They reported that sputum-soluble TNF-α receptor levels were correlated to pulmonary functions. They also investigated whether systemic inflammation was related to the degree of airflow limitation and found that circulating soluble TNF-α receptors and IL-8 did not show correlation with the degree of airflow limitation. In line with their findings, we also found no correlation between markers of systemic inflammation and severity of airflow obstruction.
The main cause for the presence of systemic inflammatory response in COPD still remains to be elucidated. Systemic hypoxia is suggested to be a good candidate, for tissue hypoxia markedly induces local expression of inflammatory cytokines, including TNF-α and IL-6 [34, 36]. Takabate et al. [34] studied the relationship between hypoxemia and the activity of the TNF-α system. They found an inverse correlation between PaO2 and circulating TNF-α and soluble TNF receptor levels in weight-losing COPD patients but not in normally nourished ones. We did not find a correlation between hypoxemia and TNF-α or IL-6 in stable COPD patients, but it should be noted that most of our patients did not have severe hypoxemia.
The definition of exacerbation in COPD remains controversial and the criteria remain subjective [37]. This creates difficulty, both in clinical practice and research, in reliably distinguishing exacerbations from day-to-day symptom variation. Therefore, there is a need for a “biomarker” for objective confirmation of exacerbation. Markers of airway neutrophilic inflammation (myeloperoxidase, IL-8, IL-6, and TNF-α) in induced sputum were shown to be increased at the time of acute exacerbation and declined 1 month later [38, 39]. Malo et al. [40] investigated whether inflammatory markers (CRP, IL-8, IL-6, and TNF-α) increased in serum during COPD exacerbation or were modified by intravenous corticosteroid treatment. Their results showed that there was evidence of systemic inflammation during COPD exacerbation but no change was detected during recovery (up to 2 months). Likewise, in the present study there was no significant alteration in TNF-α and IL-6 levels on follow-up (up to 6 weeks), and they remained higher than controls.
Increased numbers of inflammatory cells and elevated levels of various mediators have been reported in biopsy, BAL, and induced sputum samples of patients with COPD exacerbation, indicating that oxidative stress is further increased during COPD exacerbation [38, 39]. iNOS expression is induced under inflammatory and oxidant conditions, and nitrotyrosine formation is increased in the presence of reactive oxygen species. Tsoumakidou et al. [41] demonstrated that markers of airway inflammation, including nitrotyrosine and iNOS in induced sputum of patients with severe exacerbation of COPD, were increased compared with the stable state. Therefore, there may be a clinical benefit from inhibiting iNOS expression and nitrotyrosine formation during exacerbation, suggesting a potential role for NO modulators.
The measurements of NO reflect the degree of airway inflammation in asthma and can be used to guide anti-inflammatory therapy; however, changes in exhaled NO in COPD are less impressive and more difficult to interpret. Although exhaled NO has been shown to increase during exacerbations and remains elevated for at least a week, the level of exhaled NO has not been associated with the cause of exacerbation or response to treatment [42]. There is yet limited information on the course of serum NO levels during exacerbation and the recovery phase. In our study NO levels of the exacerbation group were higher than those of the controls. However, we did not have information on the baseline parameters of patients with exacerbation. Their baseline pulmonary function parameters might be lower than those of the stable group and NO might be found to be higher since they had more advanced disease than those in the stable group. Although there was a transient difference between NO levels measured on admission and discharge, there was no significant difference in measurements between admission and recovery. This transient decrease might be the effect of intensive anti-inflammatory therapy given during hospitalization, since corticosteroids could reduce the systemic inflammatory state induced by COPD [43]. Increased exhaled NO levels during exacerbation in hospitalized patients were reported to be normalized several months after discharge [18]. Our follow-up time of 6 weeks might be insufficient to evaluate the potential of TNF-α, IL-6, and NO for monitoring recovery from exacerbation.
Recently, Pinto-Plata et al. [44] investigated the relationship of systemic inflammatory markers to symptoms and physiologic changes during exacerbation. They demonstrated that hospitalized patients with COPD exacerbation experienced significant changes in systemic cytokine levels that correlated with symptoms and lung function; and they concluded that a COPD exacerbation represented not only a worsening of airflow obstruction but also increased systemic demand in a host with limited ventilatory reserve.
In summary, raised serum levels of TNF-α and IL-6 support their use as biomarkers of the systemic inflammatory response in stable COPD patients. However, the circulating biomarkers we have studied are not found to be useful either as indicators of COPD exacerbation or for monitoring recovery after exacerbation. Further studies with longer follow-up times are needed since knowledge of the course of the biomarkers of systemic inflammation during stable and exacerbation phases of COPD will form the basis for designing clinical trials on anti-inflammatory therapies to decrease morbidity in COPD.
References
NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) (updated 2005) Global strategy for the diagnosis, management and prevention of chronic obstructive lung disease. Available at http://www.goldcopd.com/
Celli BR, MacNee W, ATS/ERS Task Force (2004) Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 23:932–946
Oudijk JD, Lammers WJ, Koenderman L (2003) Systemic inflammation in chronic obstructive pulmonary disease. Eur Respir J 22(Suppl 46):5s–13s
Wouters EF, Creutzberg EC, Schols AM (2002) Systemic effects in COPD. Chest 121(Suppl):127s–130s
Agusti AGN, Noguera A, Sauleda J, Sala E, Pons J, Busquets X (2003) Systemic effects of chronic obstructive pulmonary disease. Eur Respir J 21:347–360
Karadag F, Cildag O, Yurekli Y, Gurgey O (2003) Should COPD patients be routinely evaluated for bone mineral density? J Bone Miner Metab 21:243–247
Karadag F, Karul AB, Cildag O, Altun C, Gurgey O (2004) Determinants of BMI in patients with COPD. Respirology 9:70–75
Karadag F, Cildag O, Altınısık M, Kozacı LD, Kıter G, Altun C (2004) Trace elements as a component of oxidative stress in chronic obstructive pulmonary disease. Respirology 9:33–37
Karadag F, Polatli M, Ozcan H, Cildag O (2004) Role of arterial blood gas abnormalities on oedema formation in chronic obstructive pulmonary disease. Respirology 9:481–484
Karakas S, Karadag F, Karul AB, Gurgey O, Gurel S, Güney E, Cildag O (2005) Circulating leptin and body composition in chronic obstructive pulmonary disease. Int J Clin Pract 59:1167–1170
Karadag F, Ozcan H, Karul AB, Ceylan E, Cildag O (2007) Correlates of erectile dysfunction in moderate-to-severe chronic obstructive pulmonary disease. Respirology 112:248–253
Karadag F, Ozcan H, Karul AB, Yilmaz M, Cildag O (2007) Correlates of non-thyroidal illness syndrome in chronic obstructive pulmonary disease. Respir Med 101:1439–1446
Gan WQ, Man SF, Senthilselvan A, Sin DD (2004) Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax 59:574–580
Schols AM, Buurman WA, Staal van den Brekel AJ, Dentener MA, Wouters EF (1996) Evidence for a relation between metabolic derangements and increased levels of inflammatory mediators in a subgroup of patients with chronic obstructive pulmonary disease. Thorax 51:819–824
Eid AA, Jonescu AA, Nixon LS, Lewis-Jenkins V, Matthews SB, Griffiths TL, Shale DJ (2001) Inflammatory response and body composition in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 164:1414–1418
Chung KF (2001) Cytokines in chronic obstructive pulmonary disease. Eur Respir J 18(Suppl 34):50s–59s
Ricciardolo FL, Di Stefano A, Sabatini F, Folkerts G (2006) Reactive nitrogen species in the respiratory tract. Eur J Pharmacol 533:240–252
Agusti AG, Villaverde JM, Togores B, Bosch M (1999) Serial measurements of exhaled nitric oxide during exacerbations of chronic obstructive pulmonary disease. Eur Respir J 4:523–528
Kharitonov SA, Barnes PJ (2003) Nitric oxide, nitrotyrosine, and nitric oxide modulators in asthma and chronic obstructive pulmonary disease. Curr Allergy Asthma Rep 3:121–129
Barnes PJ, Stockley RA (2005) COPD: current therapeutic interventions and future approaches. Eur Respir J 25:1084–1106
Anthonisen NR, Connett JE, Enright PL, Manfreda J, Lung Health Study Research Group (2002) Hospitalizations and mortality in the Lung Health Study. Am J Respir Crit Care Med 166:333–339
ATS/ERS Task Force (2005) Standardization of lung function testing. Eur Respir J 26:319–338
Aukrust P, Müller F, Lien E, Nordoy I, Liabakk N, Kvale D, Espevik T, Stig S (1999) Tumor necrosis factor (tnf) system levels in human immunodeficiency virus-infected patients during highly active antiretroviral therapy: persistent tnf activation is associated with virologic and immunologic treatment failure. J Infec Dis 179:74–82
Navarro-Gonzalvez JA, Garcia-Benayas C, Arenas J (1998) Semiautomated measurement of nitrate in biological fluids. Clin Chem 44:679–681
Ricciardolo FL, Caramori G, Ito K, Capelli A, Brun P, Abatangelo G, Papi A, Chung KF, Adcock I, Barnes PJ, Donner CF, Rossi A, Di Stefano A (2005) Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J Allergy Clin Immunol 116:1028–1035
Ebisui C, Tsujinaka T, Morimote T, Kan K, Iijima S, Yano M, Kominami E, Tanaka K, Monden M (1995) Interleukin-6 induces proteolysis by activating intracellular proteases in C2C12 myotubes. Clin Sci 89:431–439
Yasuda N, Gotoh K, Minatoguchi S, Asano K, Nishigaki K, Nomura M, Ohno A, Watanabe M, Sano H, Kumada H, Sawa T, Fujiwara H (1998) An increase of soluble Fas, an inhibitor of apoptosis, associated with progression of COPD. Respir Med 92:993–999
Yende S, Waterer GW, Tolley EA, Newman AB, Bauer DC, Taaffe DR, Jensen R, Crapo R, Rubin S, Nevitt M, Simonsick EM, Satterfield S, Harris T, Kritchevsky SB (2006) Inflammatory markers are associated with ventilatory limitation and muscle dysfunction in obstructive lung disease in well functioning elderly subjects. Thorax 61:10–16
Van Helvoort HA, Heijdra YF, Thijs HM, Vina J, Wanten GJ, Dekhuijzen PN (2006) Exercise-induced systemic effects in muscle-wasted patients with COPD. Med Sci Sports Exerc 38:1543–1552
Tracey KJ, Cerami A (1990) Metabolic responses to cachectin/TNF. A brief review. Ann NY Acad Sci 587:325–331
Vernooy JH, Kucukaycan M, Jacobs JA, Chavannes NH, Buurman WA, Dentener MA, Wouters EF (2002) Local and systemic inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 166:1218–1224
Di Francia M, Barbier D, Mege JL, Orehek J (1994) Tumor necrosis factor-alpha levels and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 150:1453–1455
De Godoy I, Donahoe M, Calhoun WJ, Mancino J, Rogers RM (1996) Elevated TNF-α production by peripheral blood monocytes of weight-losing COPD patients. Am J Respir Crit Care Med 153:633–637
Takabatake N, Nakamura H, Abe S, Inoue S, Hino T, Saito H, Yuki H, Kato S, Tomoike H (2000) The relationship between chronic hypoxemia and activation of tumor necrosis factor-α system in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 161:1179–1184
Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C et al (1998) Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 158:1277–1285
Yan SF, Tritto I, Pinsky D, Liao H, Huang J, Fuller G, Brett J, May L, Stern D (1995) Induction of interleukin 6 (IL-6) by hypoxia in vascular cells. Central role of the binding site for nuclear factor-IL-6. J Biol Chem 270:11463–11471
Rodriguez-Roisin R (2000) Toward a consensus definition for COPD exacerbations. Chest 117(Suppl 2):398s–3401s
Aaron SD, Angel JB, Lunau M, Wright K, Fex C, Le Saux N, Dales RE (2001) Granulocyte airway markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 163:349–355
Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA (2000) Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax 55:114–120
Malo O, Sauleda J, Busquets X, Miralles C, Agusti A, Noguera A (2002) Systemic inflammation during exacerbations of chronic obstructive pulmonary disease. Arch Bronconeumol 38:172–176
Tsoumakidou M, Tzanakis N, Chrysofakis G, Siafakas NM (2005) Nitrosative stress, heme oxygenase-1 expression and airway inflammation during severe exacerbations of COPD. Chest 127:1911–1918
Vestbo J (2006) Clinical assessment, staging, and epidemiology of chronic obstructive pulmonary disease exacerbations. Proc Am Thorac Soc 3:252–256
Sin DD, Lacy P, York E, Man SF (2004) Effects of fluticasone on systemic markers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 170:760–765
Pinto-Plata VM, Livnat G, Girish M, Cabral H, Masdin P, Linacre P, Dew R, Kenney L, Celli BR (2007) Systemic cytokines, clinical and physiological changes in patients hospitalized for exacerbation of COPD. Chest 131:37–43
Acknowledgment
The study was funded by Adnan Menderes University Research Foundation (Project No. TPF-04027).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Karadag, F., Karul, A.B., Cildag, O. et al. Biomarkers of Systemic Inflammation in Stable and Exacerbation Phases of COPD. Lung 186, 403–409 (2008). https://doi.org/10.1007/s00408-008-9106-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-008-9106-6