
Abstract
Xeroderma pigmentosum is a rare autosomal recessive disease characterized by hypersensitivity to UV light which is due to alterations of the nucleotide excision repair pathway. Eight genes (XPA to XPG and XPV) are responsible for the disease. Among them, the XPC gene is known to be the most mutated in Mediterranean patients. The aim of this study was to determine the frequency of the most common XPC mutation and describe the clinical features of Moroccan patients with xeroderma pigmentosum. Twenty four patients belonging to 21 unrelated Moroccan families and 58 healthy subjects were investigated. After clinical examination, the screening for the c.1643_1644delTG (p.Val548AlafsX25) mutation in the XPC gene was performed by PCR and automated sequencing of exon 9 in all patients and controls. The molecular analysis showed that among the 24 patients, 17 were homozygous for the c.1643_1644delTG mutation and all their tested parents were heterozygous, whereas the others (7 patients) did not carry the mutation. The frequency of this mutation was estimated to be 76.19 % (16/21 families). None of the 58 healthy individuals carried this mutation. In addition, clinical investigation showed that the majority of the patients bearing this mutation have the same clinical features. Our results revealed that the p.Val548AlafsX25 mutation is the major cause (76.19 %) of xeroderma pigmentosum in Moroccan families. This would have an important impact on improving management of patients and their relatives.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Xeroderma pigmentosum (XP) is a rare, autosomal, recessive syndrome characterized by a sun-hypersensitivity associated with skin abnormalities (poikiloderma, skin atrophy, telangiectasia, actinic keratoses, angiomas, and keratoacanthomas) and cancers [3, 27]. Patients may also develop ocular abnormalities (photophobia, conjunctivitis, keratitis, ectropion, and entropion) and/or neurological disorders (mental deterioration, sensorineural deafness, hyporeflexia, spasticity and ataxia) [13]. The incidence of XP was about 1/1,000,000 births in the USA and Europe [4], 1:20,000–100,000 in Japan [10, 18] and 1:10,000–30,000 in North Africa [8, 12, 19, 29]. XP is caused by genetic defects in the nucleotide excision repair (NER) pathway involved in the repair of bulky DNA damage, such as UV-induced DNA lesions [24]. Two NER sub-pathways have been described. The first, transcription-coupled repair (TCR), is a fast system specialized in the repair of DNA lesions located on actively transcribed genes. The second, global genome repair (GGR), is a slower mechanism for repairing lesions located in the rest of the genome [22]. Defects in NER are associated with hypermutability and UV-induced cancer predisposition in humans and in animal models [9]. Seven complementation groups, from XP-A to XP-G, leading to defective DNA NER and their corresponding genes have been characterized. An additional form, XP variant (XP-V), has clinical XP leading to defective DNA post-replication repair [3, 14]. Among the seven complementation groups, the XPC, XPA and XPD are the most frequent groups in Europe, North Africa, Japan and the USA. These groups are responsible for about 90 % of XP patients worldwide [24].
The XPC protein is responsible for the recognition of target lesions and recruitment of the incision complex in the first step of the NER during GGR but not during TCR [1, 16, 25, 28]. To date, more than 36 pathogenic variants have been reported in the XPC gene and the c.1643_1644delTG is known to be the most common mutation in Mediterranean XP patients [2, 21, 23]. This frameshift mutation causes the absence of XPC protein [20]. It has been found that the DNA repair level induced by this mutation was reduced to 10–20 % of that of proficient normal cells [6, 23]. Here, we performed screening for the most common XPC mutation and described the clinical features of XP Moroccan patients.
Materials and methods
Subjects
Our study included 24 XP cases (16 females and 8 males; sex ratio (M/F) = 0.5) with different clinical presentations belonging to 21 unrelated Moroccan families, recruited between July 2010 and June 2012. The age of patients ranged from 1 to 28 years with a mean of 10.48 years. All patients were diagnosed and treated at the department of Dermatology in Ibn Rochd University Hospital of Casablanca, Morocco. Five patients were familial cases with at least one related affected by XP and 19 were sporadic cases (no familial history of XP). The clinical data were collected from medical records in a detailed questionnaire and informed consent was obtained from each patient or his parents. On the other hand, we also carried out genetic investigation of 58 unrelated Moroccan healthy subjects without any history of XP. Patients and controls originated from different regions of the country. Whole blood samples were collected from controls; patients and their relatives in standard EDTA collection tubes. This study was approved by the local ethical committee.
Molecular investigation
Genomic DNA was extracted from whole blood by the salting-out method [17]. Screening of most common mutation, c.1643_1644delTG (p.Val548AlafsX25), was performed by PCR amplification and sequencing of the complete exon 9 of XPC gene having a length of 609 bp using the following primers—XPC-9F: 5′-CCAGGGTGTCTTATAAAGAGG-3′ and XPC-9R: 5′-CAAGGCCTTACCTCCAAG-3′ [2]. PCR was performed in final volume of 15 μl with 30–50 ng of DNA, 6 pmol of each primer, 200 μM of dNTPs, 3 mM of MgCl2, 1× PCR buffer, 0.75 U of GoTaq polymerase (Promega, Madison, USA). The PCR conditions included initial denaturation at 95 °C for 5 min, 35 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 35 s, extension at 72 °C for 40 s, and final elongation at 72 °C for 7 min. PCR products clean-up was performed with exonuclease I and shrimp alkaline phosphatase enzymes prior to sequencing. Sequencing reactions were carried out using the BigDye Terminator v 1.1 Standard Kit (Applied Biosystems, Foster City, CA, USA) in accordance with the manufacturer’s recommendations. Sequences were determined with an ABI 3130 Genetic Analyzer and compared with the reference sequence of XPC gene (Genbank cDNA sequences, NM_004628). All patient and healthy subjects were tested for this mutation.
Results
In this study, we describe a series of 24 XP patients from 21 unrelated families. The clinical data of XP patients are summarized in Table 1. Consanguinity was registered in 76.19 % (16/21) of families. Among them, 81.25 % were of first degree and 18.75 % of second degree. XP symptoms have begun at a mean age of 21.78 months (range 3–84 months). Skin photosensitivity, poikilodermia and xeroderma were present in all patients. However, skin atrophy and telangiectasia were present in 79.17 % (19/24) and 83.33 % (20/24) of cases, respectively. Skin tumors occurred in 62.5 % (15/24) of patients, with BCC in 41.67 % (10/24), SCC in 54.17 % (13/24) and melanoma in 4.17 % (1/24). Ophthalmological abnormalities were noted in 87.5 % (21/24) of cases: photophobia in 87.5 %, conjunctivitis in 16.67 %, keratitis in 37.5 %, ectropion in 8.33 %, corneal opacity in 4.17 %, and ocular tumors in 12.5 %. However, neurological symptoms have not been observed in any of our patients. In addition, three patients had other clinical symptoms, one patient had lymphoma, another had sarcoma and last one had intracerebral hemorrhage.
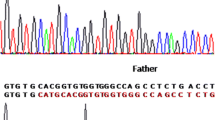
Mutation analysis revealed that 17/24 XP patients are homozygous for the most common deletion of two bases TG in exon 9 at position c.1643_1644 (NM_004628) (Fig. 1). However, 7/24 patients were negative for this mutation. Additional investigation showed that all tested parents and some healthy relatives of XP patients were heterozygous for this mutation. Thereby, the frequency of this XPC alteration was estimated to be 76.19 % (16/21) in XP Moroccan families. The majority of patients with the p.Val548AlafsX25 mutation had the same clinical characteristics. Indeed, skin photosensitivity and xeroderma were present in all these patients, poikilodermia and skin atrophy in 15/17, telangiectasia in 14/17, skin tumors in 11/17 and ophthalmological disorders in 14/17 subjects; with absence of neurological symptoms. Moreover, molecular results showed the presence of two SNPs: c.1362T>C (rs3731128) in XP18 and XP19; c.1496C>T (rs2228000) in XP12.

Electropherograms showing sequencing results: a mutation c.1643-1644delTG at a homozygous state, b wide type sequence and c mutation c.1643-1644delTG at a heterozygous state
On the other hand, screening for the c.1643_1644delTG mutation in 58 healthy subjects showed the absence of carriers in the general population.
Discussion
XP is a highly heterogeneous disease at both clinical and genetic levels. Our study focused on the screening of c.1643_1644delTG (p.Val548AlafsX25) mutation of XPC gene and described clinical features in Moroccan XP patients. We found this mutation at a high frequency in XP patients with about 76 % of screened families. Similar findings were reported by previous studies. In fact, approximately 74 % of XP probands were homozygous for the common XPC Val548AlafsX25 mutation in a series of 66 Maghrebi patients [23]. In addition, this mutation was found in the majority of examined XP patients from Tunisia, Algeria, Egypt and Italy [2, 6, 7, 11, 15]. Moreover, the Val548AlafsX25 mutation was found in about 87 and 96 % of XPC patients from North Africa Maghrebian patients, respectively [23]. This common mutation leads to a premature termination codon and absence of normal XPC protein [20]. Indeed, it affects interaction capacities between XPC protein and DNA, RAD23B, CETN2 and TFIIH molecules, which are necessary for DNA damage recognition, excision, gap-filling synthesis and ligation during GG-NER [5]. Consequently, DNA repair level induced by this mutation has been found decreased to 10–20 % in normal fibroblast cells [6, 23]. Like other Mediterranean patients [2, 23], we found that the great majority of our patients with Val548AlafsX25 mutation have the same clinical characteristics: skin photosensitivity and lesions in all patients, the tumors and ophthamological disorders were in 11 and 14/17 subjects, respectively. Unlike our patients who showed absence of neurological symptoms, 2/49 patients from Maghreb showed mental retardation, microcephaly, deafness, spasticity and ataxia [23].
On the other hand, the origin of the high frequency of the common Val548AlafsX25 mutation has been investigated in North African patients. Haplotype analysis of microsatellites markers surrounding XPC gene suggested the presence of a common founder effect specific to the Mediterranean region with a common haplotype comprising specific alleles [2, 23]. The age of occurrence of this mutation was estimated to be 1,250 years ago which approximately corresponds to the beginning of European invasion by muslims from Arabia (the “Saracens”) [23]. In our study, the high frequency of homozygous patients for this mutation could mainly be explained by the high rate of consanguinity reaching 75 % of mutated families, but not due to carrier frequency in general population, as we do not detect this mutation in 58 healthy Moroccan unrelated subjects and that the heterozygous carriers frequency was estimated to be about 1:500 worldwide [26].
In conclusion, our findings showed the predominance of the c.1643_1644delTG (p.Val548AlafsX25) mutation in Moroccan patients with xeroderma pigmentosum associated with a similar clinical profile in the majority of cases. These results would help to establish an early diagnosis to improve the management of XP patients and offer the possibility to perform genetic counseling for XP relatives in our country.
References
Araki M, Masutani C, Takemura M, Uchida A, Sugasawa K et al (2001) Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem 276:18665–18672
Ben Rekaya M, Messaoud O, Talmoudi F, Nouira S, Ouragini H et al (2009) High frequency of the V548A fs X572 XPC mutation in Tunisia: implication for molecular diagnosis. J Hum Genet 54:426–429
Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ (1998) Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW (eds) The genetic basis of human cancer. McGraw-Hill, New York, pp 245–274
Bootsma D, Kraemer KH, Cleaver JJ (2001) Hoeijmakers nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. In: Sly W, Valle D, Scriver AB (eds) The metabolic basis of inherited disease. McGraw-Hill Book Co, New York, pp 245–274
Bunick CG, Miller MR, Fuller BE, Fanning E, Chazin WJ (2006) Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry 45:14965–14979
Chavanne F, Broughton BC, Pietra D, Nardo T, Browitt A et al (2000) Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein, and transcript levels. Cancer Res 60:1974–1982
Daya-Grosjean L, James MR, Drougard C, Sarasin A (1987) An immortalized xeroderma pigmentosum, group C, cell line which replicates SV40 shuttle vectors. Mutat Res 183:185–196
Fazaa B, Zghal M, Bailly C, Zeglaoui F, Goucha S et al (2001) Melanoma in xeroderma pigmentosum: 12 cases. Ann Dermatol Venereol 128:503–506
Hanawalt PC (2002) Subpathways of nucleotide excision repair and their regulation. Oncogene 21:8949–8956
Hirai Y, Kodama Y, Moriwaki S, Noda A, Cullings HM et al (2006) Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1 % of the Japanese population. Mutat Res 601:171–178
Khan SG, Oh KS, Shahlavi T, Ueda T, Busch DB et al (2006) Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis 27:84–94
Khatri ML, Bemghazil M, Shafi M, Machina A (1999) Xeroderma pigmentosum in Libya. Int J Dermatol 38:520–524
Kraemer KH, Lee MM, Scotto J (1987) Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 123:241–250
Lehmann AR (2003) DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85:1101–1111
Li L, Bales ES, Peterson CA, Legerski RJ (1993) Characterization of molecular defects in xeroderma pigmentosum group C. Nat Genet 5:413–417
Maillard O, Solyom S, Naegeli H (2007) An aromatic sensor with aversion to damaged strands confers versatility to DNA repair. PLoS Biol 5:e79
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Moriwaki S, Kraemer KH (2001) Xeroderma pigmentosum—bridging a gap between clinic and laboratory. Photodermatol Photoimmunol Photomed 17:47–54
Moussaid L, Benchikhi H, Boukind EH, Sqalli S, Mouaki N et al (2004) Cutaneous tumors during xeroderma pigmentosum in Morocco: study of 120 patients. Ann Dermatol Venereol 131:29–33
Rezvani HR, Ged C, Bouadjar B, de Verneuil H, Taieb A (2008) Catalase overexpression reduces UVB-induced apoptosis in a human xeroderma pigmentosum reconstructed epidermis. Cancer Gene Ther 15:241–251
Rivera-Begeman A, McDaniel LD, Schultz RA, Friedberg EC (2007) A novel XPC pathogenic variant detected in archival material from a patient diagnosed with xeroderma pigmentosum: a case report and review of the genetic variants reported in XPC. DNA Repair (Amst) 6:100–114
Sarasin A, Stary A (2007) New insights for understanding the transcription-coupled repair pathway. DNA Repair (Amst) 6:265–269
Soufir N, Ged C, Bourillon A, Austerlitz F, Chemin C et al (2010) A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa. J Invest Dermatol 130:1537–1542
Stary A, Sarasin A (2002) The genetics of the hereditary xeroderma pigmentosum syndrome. Biochimie 84:49–60
Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ et al (1998) xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell 2:223–232
Thoms K-M, Kuschal C, Emmert S (2007) Lessons learned from DNA repair defective syndromes. Exp Dermatol 16(6):532–544
Van Steeg H, Kraemer KH (1999) xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol Med Today 5:86–94
Venema J, van Hoffen A, Karcagi V, Natarajan AT, van Zeeland AA et al (1991) xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes. Mol Cell Biol 11:4128–4134
Zghal M, El-Fekih N, Fazaa B, Fredj M, Zhioua R et al (2005) Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 49 Tunisian cases. Tunis Med 83:760–763
Acknowledgments
We thank all families for their cooperation. We also thank the team of dermatology of Ibn Rochd Hospital (Casablanca, Morocco) for their collaboration and effective participation in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Senhaji, M.A., Abidi, O., Nadifi, S. et al. c.1643_1644delTG XPC mutation is more frequent in Moroccan patients with xeroderma pigmentosum. Arch Dermatol Res 305, 53–57 (2013). https://doi.org/10.1007/s00403-012-1299-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00403-012-1299-0