
Abstract
Vascular endothelial growth factor (VEGF) plays important roles in the regulation of angiogenesis and inflammation in both pathological and physiological wound repair. Several strategies have been developed for keloid therapy; however, a universally effective treatment has not been explored to date. In this study, three potential short interfering RNA (siRNA) sequences for the VEGF gene were cloned into expression plasmids and transfected into keloid fibroblasts. PGC-VEGF shRNA 2 (short hairpin RNA 2) plasmid significantly inhibited VEGF expression determined by real-time polymerase chain reaction (PCR), enzyme-linked immunosorbent assay (ELISA), and fibroblasts growth was also significantly by (methyl thiazolyl tetrazolium) MTT assay and apoptosis detection, whereas the control transfection showed no obviously effects. Plasminogen activator inhibitor-1 (PAI-1) expression in pGC-VEGF shRNA2 group was also obviously downregulated when compared to the pGC-VEGF shRNA negative control and mock group. These results suggest that modulation of VEGF production by vector-based RNAi in keloid fibroblasts may be a therapeutic potential strategy for keloid.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Keloids, which are pathological scars defined as benign cutaneous tumors extending beyond wound margins, are distinguished by substantial deposition of collagen in the dermis, resulting in an imbalanced production and aggregation of extracellular matrix [4, 13, 28]. It usually develops in response to trauma of the skin, from either controlled injuries (e.g., earlobe piercing or surgery) or pathological events (e.g., acne, chickenpox, burn, or inflammation) [25]. But the basic mechanism involved in keloid development is still unknown.
VEGF (Vascular endothelial growth factor), one of the most widely studied secreted factors involved in angiogenesis, has been implicated as crucial to normal and pathological wound healing [11]. As an angiogenic peptide composed a variety of isoforms, VEGF is also a vascular permeability factor that promotes neo-vascularization and cell growth [19, 3]. The most abundant of the five VEGF isoforms, VEGF-A, is typically utilized in biological angiogenic studies [7, 23]. Recently, Gira et al. [8] indicated that VEGF production is abundant in the underlying dermis of keloids. In vitro studies have indicated that VEGF is expressed at higher levels in keloid-derived fibroblasts than in normal skin fibroblasts [28, 8, 24]. Also, VEGF stimulates expression of PAI-1 which is highly expression in keloid tissues and cultured fibroblasts compared with normal bordering skin, at both mRNA and protein levels, in a dose- and time-dependent manner in keloid fibroblasts. This findings support the concept that an altered balance of activator and inhibitor activities in the plasminogen systerm, in particular, an overexpression of PAI-1, may partly contribute to keloid formation and tissue fibrosis [28]. Therefore, VEGF likely plays a significant role in keloid formation by changing the extracellular matrix, suggesting that modulating VEGF would be as a potential drug target for keloid reduction therapy.
RNAi is a new technique for specific gene inhibition. RNAi is a post-transcriptional gene-silencing mechanism that can be initiated by double-stranded RNA (dsRNA) homologous in sequence to the target gene. A vector-based approach can achieve suppression of gene expression in mammalian cells. The expression vectors employ RNA polymerase III promoters from either the U6 small nuclear RNA or the H1 ribonuclease (RNase) gene to transcribe short hairpin RNA (shRNA) [1, 13, 16, 17, 25]. Taken together, inhibition of VEGF to interfere with keloid neovascularization may be one of more important strategy in anti-keloid formation. Therefore, to strive for a new strategy which could inhibit, or at least in part, expression of VEGF in keloid fibroblasts, in this study, we show here for the first time to apply RNAi technology in keloid fibroblasts in vitro.
Materials and methods
Keloid fibroblasts cultures
Seven patients (two men and five women with age range of 13–42 years as a mean age of 26.5 years) had received no previous treatment for keloid before surgical excision. Prior written informed consent was obtained from the patients and the study received the approval of the ethics board at XiJing Hospital. The study was conducted according to The Helsinki Declaration of 1975 (revised 1983). The epidermis and any remaining fat was excised from the excised tissue, which was then cut into small (1 mm3) pieces, and placed as explants in 25 cm2 tissue culture flasks (Corning Coster Corpration, Cambridge). Growth medium consisted of: Dulbecco’s Modified Eagle’s Medium (DMEM), L-glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin (all Gibco, NY, USA), and fetal calf serum (Hyclone, USA). Fibroblasts were grown in the moist atmosphere of a CO2 incubator (5% CO2, 95% air) at 37°C. Growth medium was changed every 3–4 days. When keloid fibroblasts outgrowth had become well established, the skin explants were dislodged prior to subculture of the monolayer. The fibroblasts were grown to confluence and then to a maximum passage of three prior to further experiments.
Generation of vascular endothelial growth factor short hairpin RNA expression plasmids
The following criteria were used to identify target for siRNA from the VEGF cDNA coding sequence: (a) start with an AA dinucleotide, (b) 21 nucleotides in length, (c) G/C content of <50%, and (d) no sequence homology to other coding sequences on BLAST search. Using these criteria, three pairs of cDNA oligonucleotides targeting human VEGF mRNA at different locations were choosen (Table 1) [5]. One negative control siRNA containing a scrambled sequence with the same nucleotide composition was also selected. Sense and antisense primer containing the sense siRNA sequence, 9 bp loop sequence, antisense siRNA sequence, and RNA polymerase III terminator sequence were created with BamHI and HindIII restriction sites on the 5′ and 3′ ends, respectively. These primers were annealed and inserted into pGCsi-U6 (Genechem, Inc., Shanghai, China) downstream of the U6 RNA polymerase III promoter following the manufacturer’s instructions. The resulting plasmids containing siRNA sequences VR1, VR2, and VR3 and negative control sequence were named pGC-VEGF shRNA1, pGC-VEGF shRNA2, pGC-VEGF shRNA3, and pGC-VEGF shRNANC, respectively.
Experimental protocol and transfection with the shRNA expression vector
Negative control keloid fibroblasts were treated with pGC-VEGF shRNANC using liposome GenePORTER 2 (Gene Therapy System, Inc., San Diego, CA) (50 nM), where specific control keloid fibroblasts were treated with pGC-VEGF shRNAs (50 nM). In parallel wells, keloid fibroblasts were transfected with transfection reagent alone (Mock). According to the manufacture’s instructions, keloid fibroblasts were plated in six-well plates at 1 × 106 cells/well, grown for 24 h, then transfected with shRNA expression vectors. Diluted 10 μl of the GenePORTER2 reagent with serum-free medium; 10 μl of 50 nM expression vector with DNA diluent, then added the diluted DNA to the diluted GenePORTER2 reagent, and incubated for 5–10 min to form lipid/DNA complexes(lipoplexes), Post 24 h transfection, cells were replaced fresh growth media. Forty-eight hours after transient transfection, the extracellular medium was collected with removal of cell sediment for determination of secreted VEGF protein. For transient transfection with neomycin selection, the day after following transfection, new growth media with neomycin 400 μg/ml was added, and the extracelluar medium was collected after 7 days.
ELISA assay of VEGF level in culture media
Conditioned media was collected from keloid fibroblasts and centrifuged at 3,000 rpm and 4°C for 10 min to remove cell debris and then stored at −80°C. The concentration of VEGF in culture supernatant was measured using a commercial VEGF enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s instructions (Jingmei, Shenzhen, China). ELISA plates were read using the Sunrise Reader (Tecan Trading AG, Switzerland) and lower-and-upper detection limits of the ELISA were 7 and 2,000 pg/ml. Data reflects the mean of samples done in triplicates.
Total RNA extraction and Real-Time quantitative RT-PCR
Total RNA from the keloid fibroblasts was prepared using Trizol® Reagent (Invitrogen) according to the manufacturer’s instructions. cDNA synthesis was conducted according to the RNA PCR kit protocol (TAKARA). Reverse transcription was performed using 5 μg of total RNA in 10 μl of the following solution: 2 μl MgCl2, 1 μl 10× RT buffer, 1 μl dNTP, 0.25 μl RNase inhibitor, 0.5 μl AMV reverse transcriptase, 0.5 μl oligo dT primer and 3.75 μl RNase free ddH2O. The reaction was incubated at 30°C for 10 min, 42°C for 50 min, 95°C for 5 min, and 4°C for 5 min. Primers for PCR were the following: VEGF forward 5′TCTTGGGTGCATTGGAGCCTC3′ and VEGF reverse 5′ AGCTCATCTCTCCTATGTGC3′ and PAI-1 forward 5′TGCTGGTGAATGCCCTCTACT3′ and PAI-1 reverse 5′ CGGTCATTCCCAGGTTCTACT3′. The GAPDH forward primer was 5′ GCACCGTCAAGGCTGAGAAC3′ and the GAPDH reverse primer was 5′TGGTGAAGACGCCAGTGGA3′. The housekeeping gene GAPDH was employed as a reference. Real-time PCR was performed using the Chromo 4™ Four-Color Real-Time System (Bio-Rad Laboratories, Inc., CA, USA) and was carried out with SYBR Premix Ex Taq (Perfect Real Time) kit (TAKARA) in a 25-μl volume of the following solution: 12.5 μl SYBR Premix Ex Taq, 0.5 μl PCR forward primer, 0.5 μl PCR reverse primer, 2.0 μl template, 2 μl 25 mMMgCl2, 7.5 μl ddH2O, cycle conditions for VEGF and PAI-1 PCR were 10 min at 95°C followed by forty cycles of at 94°C, 30 s at 60°C, and 20 s at 72°C and GAPDH PCR 10 min at 95°C followed by twenty five cycles of at 94°C, 30 s at 56°C, and 20 s at 72°C. During each extension step, SYBR green fluorescence was monitored and provided the real-time quantitative measurements of the fluorescence. Quantitation was carried out using an external standard curve. PCR results were analyzed using Opticon Monitor Analysis 2.0 software (Bio-Rad Laboratories, Inc., CA, USA).
Western blot analysis for PAI-1
The conditioned media were collected for determination of PAI-1 protein. Equal amounts of protein sampled from conditioned medium were subjected to electrophoresis on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels and electro blotted onto polyvinylidene difluoride membranes (Hybond-P, Amersham Pharmacia Biosciences). After blocking with Tris-buffered saline (TBS)–5% skim milk, the membranes rinsed and then incubated with primary antibodies anti-human PAI-1 (Abcam, Inc., Cambridge, UK) diluted 1:500 in PBS containing 0.05% Tween 20 for overnight. Membranes were subsequently washed again as described above, incubated with a horseradish peroxides (HRP)-conjugated goat anti-mouse IgG (1:5,000; Pierce) for 1 h at room temperature, and visualized with enhanced chemiluminescence (ECL) detection. BandScan 5.0 software was used for gray scale scan to evaluate the relative value of protein expression.
The cell growth following transfection was evaluated by MTT assay
Cells at a concentration of 2.0 × 104 per well were seeded in the 96-well plate and transfected with effective plasmid by using liposome GenePORTER 2. A total of 20 μM 3-(4,5 dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL) was added into each well and incubated at 37°C for 4 h. The media were carefully aspirated, and 200 μL of dimethyl sulfoxide (DMSO) was added to each well and pipetted up and down to dissolve crystals. Then, the cells continued incubating at 37°C for 5 min before being measured for absorbance (A) at 550 nm on a Sunrise Reader (Tecan Trading AG, Switzerland).
Apoptosis detection
The in situ cell death detection kit (TUNEL, Roche Diagnostics GmbH, Mannheim, Germany) was used to detect apoptosis and to quantify DNA strand breaks in keloid fibroblasts. The cell monolayers were grown directly in tissue culture flasks, starved for 24 h in fetal calf serum without fetal calf serum and then incubated in shRNANC or shRNA2 (50 nM) for 72 h. The flasks were then fixed in buffered paraformaldehyde, permeabilised with Triton-x, and labeled with TUNEL reaction mixture according to the manufacture’s instruction. Samples were analysed using a Leitz Diaplan microscope (Leica, Milan, Italy) equipped with epifluorescence. A negative control (obtained by incubating a slide with labeled solution without terminal transferase) and a positive control (obtained by treating a slide with Dnase I solution) were included in each assay run.
Statistical analysis
Each experiment was performed independently a minimum of three times. Results are expressed as mean ± SEM. Statistical comparisons between groups were evaluated using ANOVA, by using a statistical software package (SPSS 10.0, Chicago, IL, USA). A value of P ( 0.05 was accepted as significant.
Results
Three shRNA-expressing plasmids (shRNA1, shRNA2, shRNA3) were constructed using the pGCsi-U6 vector to target human VEGF and, as a negative control, shRNANC was also constructed. To examine the efficacy of these plasmids in silencing VEGF expression, plasmids were transiently transfected into keloid fibroblasts along with the negative control plasmid shRNANC. Total RNA was isolated 48 h post transfection and the house keeping gene GAPDH mRNA levels were measured using Real-Time RT-PCR. ShRNA plasmids showed variable efficacy in decreasing VEGF mRNA, with the most effective shRNA, shRNA2, and decreasing VEGF mRNA by 78% (Fig. 1a). ShRNA1, shRNA3 decreased VEGF by 28, 51%, respectively, as compared to the negative control plasmid transfected fibroblasts. These results were in agreement with previous research [5]. Because these were transient transfection, a significant proportion of VEGF mRNA may have been from untransfected cells. To reduce VEGF mRNA level by these untransfected cells, transiently transfected keloid fibroblasts were exposed for 7 days to selection with neomycin, and VEGF mRNA levels were then measured. Again, the shRNA2 was the most effective shRNA, decreasing VEGF mRNA level by 90% (Fig. 1b). The other shRNAs only decreased VEGF mRNA by 31–62%.

Effects of VEGF shRNA on the expression of VEGF by keloid fibroblasts. shRNANC or shRNA1, 2, and 3 (50 nM) were introduced into keloid fibroblasts, respectively, at subconfluence using GenePORTER. In parallel wells, fibroblast cells were transfected with transfection reagent alone (Mock). Forty-eight hours after transfection, RNA was extracted. VEGF mRNA levels were determined by real-time RT-PCR (a) and transfetcion with selection (b). Media was replaced at 24 h after transfection, and conditioned media was collected at 48 h for measurement of VEGF (c) and transfection with selection (d) using ELISA. Results are mean ± SEM and are standardized as a percentage of control. *P < 0.05 vs. negative control (shRNANC). **P < 0.01 vs. negative control (shRNANC). ***P < 0.001 vs. negative control (shRNANC)
Next, we performed an ELISA using a commercial VEGF enzyme-linked immunosorbent assay (ELISA) kit to quantify the amount of VEGF165 protein present in the conditioned media. Similar to above experiment protocol, when keloid fibroblasts were transfected with shRNA expression plasmids and selected with neomycin, the VEGF165 concentration in the conditioned media was markedly reduced (Fig. 1c, d). These results were in agreement with the amount of VEGF165 mRNA present, as analyzed by real-time RT-PCR experiments. These data indicated that VEGF expression can be strongly inhibited both on mRNA and protein levels by vector-based RNAi. Among these shRNA plasmids, shRNA2 was the most effective, which was applied in the subsequent experiments.
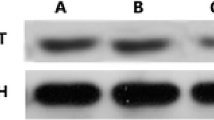
To test whether VEGF is involved in the regulation of the plasminogen system in keloid fibroblasts, we treated keloid fibroblasts with shRNA2 (50 nM) and assayed PAI-1 mRNA in the keloid fibroblasts by quantitive Real-Time RT-PCR. As shown in Fig. 2a, the PAI-1 mRNA level in shRNA transfected keloid fibroblasts was markedly reduced by 48%. On the contrary, no obvious changes in PAI-1 mRNA level were observed between the negative control group and mock group. Next, we studied the effect of shRNA on PAI-1 protein expression in keloid fibroblasts by western blot. The results showed that treatment of keloid fibroblasts with shRNA2 also significantly induced. This reduction was further confirmed by western blot (Fig. 2b, c). Consistent with findings from mRNA analysis, no significant differences were observed in protein level of PAI-1 in keloid fibroblasts treated shRNANC and transfection reagent.

Analysis of plasminogen activator inhibitor-1 by RNA interference in keloid fibroblasts. shRNANC or shRNA2 (50 nM) were introduced into keloid fibroblasts, respectively, at subconfluence using GenePORTER. In parallel wells, fibroblast cells were transfected with transfection reagent alone (Mock). Forty-eight hours after transfection, RNA was extracted to verify that the PAI-1 gene was effectively silenced. a Real time RT-PCR for PAI-1 was performed. b Conditioned media samples were used to measure PAI-1 production. Results are mean ± SEM and are standardized as a percentage of control. **P < 0.05 vs. negative control (shRNANC). ***P < 0.01 vs. negative control (shRNANC)
To analysis the antiproliferative and increased apoptosis effects of shRNA2, we transiently transfected shRNA2 into keloid fibroblasts. As shown in Fig. 3a, shRNA2 transfected keloid fibroblasts began to show a decreased viability at 48 h after transfection (P < 0.05). The trend deteriorated by 72 h when the viability decreased to as low as 28.5% for shRNA transfected keloid fibroblasts, as compared with the negative control (Fig. 3a). Also the apoptosis detection showed that the keloid fibroblasts apoptosis were increased by shRNA2 (Fig. 3b). At 48 h, the apoptosis increased to the absolute value of 32.3% (P < 0.05) and at 72 h, the value further increased to 52.8% (P < 0.01). These results demonstrated that keloid fibroblasts treated with shRNA2 was obviously inhibited compared with the negative control group and mock group.

Inhibition of keloid fibroblasts proliferation and increased apoptosis by pGC shRNA2 in vitro. shRNANC or shRNA2 (50 nM) were introduced into keloid fibroblasts, respectively, at subconfluence using GenePORTER. In parallel wells, fibroblast cells were transfected with transfection reagent alone (Mock). Keloid fibroblasts were subjected to MTT assay (a) and apoptosis detection (b) for every 24 h in a successive. Results are mean ± SEM and are standardized as a percentage of control. **P < 0.05 vs. negative control (shRNANC). ***P < 0.01 vs. negative control (shRNANC)
Discussion
Although a multiplicity of treatment modalities including intralesional and topical steroids, intralesional interferon, shave excision, silicone sheeting, pulsed dye laser, combination of intralesional steroids and excision, and, in severe cases, radiation therapy are currently available for keloids, there is lack of a universally effective treatment. Previous reports have revealed that, histologically, keloids show an increased blood vessel density compared with normal dermis or normal scars [6, 18, 29]. However, the abundance of blood vessels at the periphery of keloids may indicate that keloid fibroblasts induce angiogenesis at this site via production of VEGF. There have been some studies on the relationship between keloids and VEGF [28, 11, 8, 19]. Steinbrech et al. performed a comparative study of VEGF production by keloid fibroblast under hypoxic and normoxic conditions using ELISA. They stated that VEGF was increased in keloid by hypoxia relative to normoxia [24]. Wu et al. reported that VEGF was expressed at higher levels in keloid tissues and keloid fibroblasts compared with associated normal skin using Western blot and Northern blot analyses [28]. Gira et al. performed in situ hybridization for VEGF on keloid and normal skin, which showed strong expression of VEGF mRNA by epidermal keratinocytes overlying keloid compared with normal skin [8]. Le et al. revealed that an accumulation of VEGF in capillary lining cells and fibroblasts located in the papillary dermis in keloid was increased as compared with normal skin by immunohistochemical studies of these tissue sections using an antibody against VEGF [11].
Some studies have been demonstrated that down regulation of VEGF via siRNAs or shRNAs would be a potential strategy for several diseases by RNAi technology [12, 17, 22]. Our study shows here for the first time that RNAi of VEGF using a plasmid-based strategy can stably silence VEGF and PAI-1 expression in keloid fibroblasts. To test the efficacy of the VEGF siRNA sequence selected in this study, we constructed several plasmid-based expression systems in which sense and antisense strands of anti-VEGF siRNA sequences were transcribed into hairpin structures under the control of a U6 promoter and then processed into functional siRNAs by double-strand-specific RNase called Dicer inside the cells. Transfection efficiency, estimated by transfecting keloid fibroblasts with green fluorescent protein plasmids (data not show), was 81%, and hence paralleled the reduction in expression of VEGF. Our study showed that in transient transfection and transient transfection with selection of keloid fibroblasts, shRNA2 down the expression of VEGF mRNA expression dramatically by 78 and 90% in a sequence-specific manner, with no significant difference among the controls. VEGF protein secretion was similarly inhibited by transfection of shRNAs expressing plasmid as quantitated by ELISA. Furthermore keloid fibroblasts growth treated with shRNA2 was obviously inhibited compared with the negative control group and mock group. These data demonstrated that the current VEGF siRNA sequence can nearly accomplish complete inhibition of VEGF mRNA expression.
Besides its pivotal role in angiogenesis, VEGF also contributes to extracellular matrix metabolism via regulating the expression and activity of plasminogen activators (uPA and tPA), urokinase receptor, and PAI-1 in endothelial cells [14, 15]. Previous studies have reported an intrinsically high level of PAI-1 in keloid fibroblasts and concluded that there is a possible role of PAI-1 in the impaired collagen degradation [26, 27, 30, 31]. In this study, both mRNA and protein of PAI-1 in shRNA2 treated keloid fibroblasts was also significantly reduced as determined by real-time RCR and western bolt. This agrees with previous related studies [11, 28]. Wu et al. and Le et al. also demonstrated that VEGF stimulates the expression of PAI-1 at mRNA, protein, and enzyme activity levels in keloid fibroblast. These findings suggest that modulating PAI-1 may contribute to the process of keloid formation and tissue fibrosis via enhancing the antiproteolytic activity of the extracellular matrix metabolism equilibrium toward a state of an impaired degration. Taken together, RNAi of VEGF can be an effective strategy for keloid.
However, before clinical application can be considered, a number of problems need to be solved about RNAi of keloid. One major problem is the selection of effective and highly efficient target sites, and how to achieve a complete inhibition using a simple method. In this study, the downregulation of VEGF expression was not achieved a complete of gene function, which might be associated with GenePORTER transfection efficiency in keloid fibroblasts. The other mechanism of this might be related to the death of a part of keloid fibroblasts for the toxicity of GenePORTER. The third might be the specific siRNA sequence. So, in this study, using previously published selection criteria for siRNAs, three siRNA sequences were chosen, and one was found to be highly effective in knockdown of VEGF expression. Investigators also continue to publish additional criteria that can be used in algorithms to select siRNA sequences and to increase the efficiency of RNAi [18, 10]. Thus, how to improve the transfection efficiency in keloid fibroblasts by GenePORTER has become the objective of our further research. Another problem is how to efficiently and specifically deliver shRNA-expression vectors to target cells. Until recently, two approaches have been developed to introduce extra siRNA into cells, namely transient delivery of chemically or enzymatically synthesized siRNA and expression of siRNA intracellularly from plasmid DNA [2]. Vector-based RNAi has particular advantages, considering the likelihood of RNAse contamination and the great costs associated with chemically or enzymatically synthesized siRNA [2]. In addition, transgenic vector-based RNAi could allow for large scale production and function as an alternative method of gene silencing in vivo [1]. Most importantly, vector-based RNAi may stably express siRNA in mammalian cells [23]. And when delivered by lentiviral [9] or adeno-associated virus (AAV) [24] in vivo, it was able to serve as a promising tool for gene therapy. Our ongoing work is currently focusing on this area. Advances in RNAi techniques may thus provide a promising strategy for gene therapeutic applications to prevent keloids.
In conclusion, on the basis of our in vitro evidence we can state that VEGF targeted inhibition of angiogenesis may be an appropriate therapeutic strategy for the management of keloid. We are presently striving to establish an effective method that can stably express shRNA in order to investigate the possibility of long-term analysis of gene function and provide foundation for further animal and clinical experiments.
Abbreviations
- VEGF :
-
Vascular endothelial growth factor
- SiRNA :
-
Short interfering RNA
- PAI-1 :
-
Plasminogen activator inhibitor-1
- RNAi :
-
RNA interference
- DsRNA :
-
Double-stranded RNA
- ShRNA :
-
Short hairpin RNA
References
Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550–553
Brummelkamp TR, Bernards R, Agami R (2002) Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2:243–247
Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407:249–257
Detwiller KY, Fernando NT, Segal NH, Ryeom SW, D’Amore PA, Yoon SS (2005) Analysis of hypoxia-related gene expression in sarcomas and effect of hypoxia on RNA interference of vascular endothelial cell growth factor A. Cancer Res 13:5881–5889
Diegelmann RF, Cohen IK, McCoy BJ (1979) Growth kinetics and collagen synthesis of normal skin, normal scar and keloid fibroblasts in vitro. J Cell Physiol 98:341–346
Ehrlich HP, Desmouliere A, Diegelmann RF, Cohen IK, Compton CC, Garner WL, Kapanci Y, Gabbiani G (1994) Morphological and immunochemical differences between keloid and hypertrophic scar. Am J Pathol 145:105–113
Ferrara N, Houck K, Jakeman L, Leung DW (1992) Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev 13:18–32
Gira AK, Brown LF, Washington CV, Cohen C, Arbiser JL (2004) Keloids demonstrate high-level epidermal expression of vascular endothelial growth factor. J Am Acad Dermatol 50:850–853
Hasuwa H, Kaseda K, Einarsdottir T, Okabe M (2002) Small interfering RNA and gene silencing in transgenic mice and rats. FEBS Lett 532:227–230
Henschel A, Buchholz F, Habermann B (2004) DEQOR: a web-based tool for the design and quality control of siRNAs. Nucleic Acids Res 32:W113–W120
Le AD, Zhang Q, Wu Y, Messadi DV, Akhondzadeh A, Nguyen AL, Aghaloo TL, Kelly AP, Bertolami CN (2004) Elevated vascular endothelial growth factor in keloids: relevance to tissue fibrosis. Cells Tissues Organs 176:87–94
Matsumoto G, Kushibiki T, Kinoshita Y, Lee U, Omi Y, Kubota E, Tabata Y (2006) Cationized gelatin delivery of a plasmid DNA expressing small interference RNA for VEGF inhibits murine squamous cell carcinoma. Cancer Sci 97:313–321
Miyagishi M, Taira K (2002) U6-promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression inmammalian cells. Nat Biotechnol 20:497–500
Murray JC (1994) Keloids and hypertrophic scars. Clin Dermatol 2:27–37
Oloffson B, Korpelainen E, PeppersMS, Mandriota SJ, Aase K, Kumar V, Gunji Y, Jeltsch MM, Shibuya M, Alitalo K, Eriksson U (1998)Vascular endothelial growth factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc Natl Acad Sci USA 95:11709–11714
Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS (2002) Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev 16:948–958
Paul CP, Good PD, Winer I, Engelke DR (2002) Effective expression of small interfering RNA in human cells. Nat Biotechnol 20:505–508
Pepper MS, Ferrara N, Orci L, Montesano R (1991) Vascular endothelial growth factor (VEGF) induces plasminogen activators and plasminogen activator inhibitor-1 in microvascular endothelial cells. Biochem Biophys Res Commun 181:902–906
Rahban SR, Garner WL (2003) Fibroproliferative scars. Clin Plast Surg 30:77–89
Reich SJ, Fosnot J, Kuroki A, Tang W, Yang X, Maguire AM, Bennett J, Tolentino MJ (2003) Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol Vis 9:210–216
Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A (2004) Rational siRNA design for RNA interference. Nat Biotechnol 22:326–330
Sayah DN, Soo C, Shaw WW, Watson J, Messadi D, Longaker MT, Zhang X, Ting K (1999) Down-regulation of apoptosis-related genes in keloid tissues. J Surg Res 87:209–216
Shi Y (2003) Mammalian RNAi forthe masses. Trends Genet 19:9–12
Steinbrech DS, Mehrara BJ, Chau D, Rowe NM, Chin G, Lee T, Saadeh PB, Gittes GK, Longaker MT (1999) Hypoxia upregulates VEGF production in keloid fibroblasts. Ann Plast Surg 42:514–519
Sui G, Soohoo C, Affar el B, Gay F, Shi Y, Forrester WC, Shi Y (2002) A DNA vector-based RNAi technology to suppress expression in mammalian cells. Proc Natl Acad Sci USA 99:5515–5520
Takei Y, Kadomatsu K, Yuzawa Y, Matsuo S, Muramatsu T (2004) A small interfering RNA targeting vascular endothelial growth factor as cancer therapeutics. Cancer Res 64:3365–3370
Thomas KA (1996) Vascular endothelial growth factor, a potent and selective angiogenic agent. J Biol Chem 271:603–606
Tiscornia G, Singer O, Ikawa M, Verma IM (2003) A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci USA 110:1844–1848
Tuan TL, Nichter LS (1998) The molecular basis of keloid and hypertrophic scar formation. Mol Med Today 4:19–24
Tuan TL, Zhu JY, Sun B, Nichter LS, Nimni ME, Laug WE (1996) Elevated levels of plasminogen activator inhibitor-1 may account for the altered fibrinolysis by keloid fibroblasts. J Invest Dermatol 106:1007–1011
Tuan TL, Wu HY, Hang EY, Chong SS, Laug W, Messadi D, Kelly P, Le A (2003) Increased plasminogen activator inhibitor-1 in keloid fibroblasts may account for their elevated collagen accumulation in fibrin gel cultures. Am J Pathol 162:1579–1589
Wu Y, Zhang Q, Ann DK, Akhondzadeh A, Duong HS, Messadi DV, Le AD (2004) Increased vascular endothelial growth factor may account for elevated level of plasminogen activator inhibitor-1 via activating ERK1/2 in keloid fibroblasts. Am J Physiol Cell Physiol 286:C905–C912
Yoshimoto H, Ishihara H, Ohtsuru A, Akino K, Murakami R, Kuroda H, Namba H, Ito M, Fujii T, Yamashita S (1999) Overexpression of insulin-like growth factor-1 (IGF-I) receptor and the invasiveness of cultured keloid fibroblasts. Am J Pathol 154:883–889
Zhang Q, Wu Y, Chau C, Ann DK, Bertolami CN, Le AD (2004) Cross talk of hypoxia mediated signaling pathways in upregulation plasminogen activator inhibitor-1 expression in keloid fibroblasts. J Cell Physiol 199:89–97
Zhang QZ, Wu YD, Ann DK, Messadi DV, Tuan TL, Kelly AP, Bertolami CN, Le AD (2003) Mechanisms of hypoxic regulation of plasminogen activator inhibitor-1 gene expression in keloid fibroblasts. J Invest Dermatol 121:1005–1012
Acknowledgments
This work was funded by National Nature Science Foundation of China 30400471.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Guo-You Zhang and Cheng-Gang Yi contributed equally to this work.
Rights and permissions
About this article
Cite this article
Zhang, GY., Yi, CG., Li, X. et al. Inhibition of vascular endothelial growth factor expression in keloid fibroblasts by vector-mediated vascular endothelial growth factor shRNA: a therapeutic potential strategy for keloid. Arch Dermatol Res 300, 177–184 (2008). https://doi.org/10.1007/s00403-007-0825-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00403-007-0825-y