
Abstract
Oncogenic osteomalacia is a rare paraneoplastic syndrome of acquired hypophosphatemic osteomalacia, resulting from a deficit in renal tubular phosphate reabsorption, in which fibroblast growth factor 23 (FGF23) seems to be implicated. This condition is usually associated with a phosphaturic mesenchymal tumor of mixed connective tissue located in the bone or soft tissue. The clinical and the radiologic findings are the same as those seen in osteomalacia, and the biochemical features include renal phosphate loss, low serum phosphate and 1,25-(OH)2 vitD3 levels, increased alkaline phosphatase, and normal calcium, PTH, calcitonin, 25-OH-vitD3 and 25,25-(OH)2 vitD3. We present two cases of oncogenic osteomalacia associated with phosphaturic mesenchymal tumors, which were histologically similar, but presented a completely different evolution. In the first patient, the tumor developed on the sole of the foot. Following removal of the mass, the symptoms resolved and biochemical and radiological parameters returned to normal. However, in the second patient, a liver tumor developed and resection did not resolve the disease. Multiple lesions appeared in several locations during follow-up. This disease usually remits with complete tumor resection. Nevertheless, if this is not possible, oral treatment with phosphate, calcium and calcitriol can improve the symptoms. If scintigraphy of the tumor shows octreotide receptors, patients may respond partially to therapy with somatostatin analogs, with stabilization of the lesion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oncogenic osteomalacia is a rare paraneoplastic syndrome of acquired hypophosphatemic osteomalacia, resulting from a deficit in renal tubular phosphate reabsorption, in which fibroblast growth factor 23 (FGF23) seems to be implicated. This condition is usually associated with a phosphaturic mesenchymal tumor of mixed connective tissue located in the bone or soft tissue. The clinical and the radiological findings are the same as those seen in osteomalacia, and the biochemical features include renal phosphate loss, low serum phosphate and 1,25-(OH)2 vitD3 levels, increased alkaline phosphatase, and normal calcium, PTH, calcitonin, 25-OH-vitD3 and 25,25-(OH)2 vitD3 [1].
We present two cases of oncogenic osteomalacia associated with phosphaturic mesenchymal tumors, which were histologically similar, but presented a completely different evolution.
Case 1
A 34-year-old man consulted 15 years previously for a 1-year history of pain in the left inguinal area radiating to the anterior part of the thigh. It was subsequently associated with rib pain, mechanical low back pain and pain in the left heel while walking. Physical examination disclosed flatfoot and soft tissue occupation of the left plantar arch. There was no family or personal history of bone disease.
Laboratory analyses showed hypophosphatemia with normal calcium levels, increased serum alkaline phosphatase (AP) activity and osteocalcin, and normal PTH, 25-OH-D3 and 1,25-(OH)2-D3. Urine analysis disclosed increased phosphates, normal calcium and increased free hydroxyproline excretion.
Hip radiography showed a 1 cm loss of the lineal continuity in the internal cortical bone of the proximal femur (Fig. 1). Bone densitometry provided evidence of moderate osteoporosis of the lumbar spine and severe osteoporosis of the proximal third of the femur (triangle of Ward). On bone scanning, increased tracer uptake was seen in the pelvis, lesser trochanter of the left femur, rib cage and posterior arches of the left 5th rib, and the right 4th and 5th ribs.

Case 1. Radiographic study of the left hip. A fracture line is evident at the area of the calcar femorale
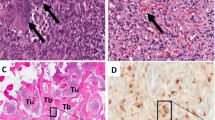
Conservative treatment with 1 year’s rest yielded no improvement. Hence, prophylactic DHS osteosynthesis was carried out (Fig. 2). The clinical symptoms and analytical and radiological parameters persisted [Looser–Milkman pseudofracture in the internal cortical bone (Fig. 2)], and we suspected acquired hypophosphatemic osteomalacia, that is, oncogenic osteomalacia. Treatment with oral phosphorus (40 mL/day) and calcitriol (0.25 mg) was initiated for the osteomalacia and a tumor found in the plantar arch was removed 1 year later (3 years after onset of symptoms). The diagnosis was a mixed connective variant of phosphaturic mesenchymal tumor with the presence of fibroblasts, some thin-walled vessels and nests having a chondroid appearance, with intense dystrophic calcification of the entire tumor.

Case 1. Radiographs of the left hip 2 months following surgery: a anteroposterior view, b axial view. The fracture shows no consolidation
Two weeks after removing the tumor, the symptoms and biochemical parameters improved (phosphate had increased to 3.1 mg/dL, AP activity had decreased to 461 U/L, and calcium decreased to 9.1 mg/dL), with complete recovery 1 year later. At that time, the densitometry and radiography features had also considerably improved and the Looser–Milkman fracture was in the process of resolution.
Following resection of the tumor, there was a fall in AP concentrations, with minimum values at 1 month after surgery. After that time point, concentrations of 1,25-(OH)2 vit-D3 increased progressively. The last follow-up visit was 11 years after plantar tumor resection, at which time the patient remained asymptomatic and radiology and biochemical results were normal.
Case 2
A 27-year-old woman had presented generalized muscle pain at the age of 7 years. After 1 year, she additionally experienced pain in the rib cage and thigh muscles, refractory to conventional analgesics. Laboratory testing showed low serum phosphate with normal calcium levels, increased serum AP, decreased 1,25-(OH)2-vitD3 and urinary phosphate loss. The bone series revealed pseudofractures and Looser–Milkman lines. A presumptive diagnosis of hypophosphatemic osteomalacia was established and medical treatment was started with calcium 500 mg/day, phosphate 1000 mg/day and calcitriol (1,25-(OH)2-vitD3) 50 μg/day, with partial clinical and analytical improvement.
After 5 years, the patient presented pain in the right hypochondrium, and CT showed a mass in the fifth segment of the liver (Fig. 3). The hepatic tumor was resected and pathological study identified hemangiopericytoma, a phosphaturic mesenchymal tumor. For this reason, the clinical picture was oriented towards hypophosphatemic osteomalacia, secondary to a mesenchymal tumor.

Case 2. Abdominal CT showing calcified hepatic mass. The hepatic tumor was resected and pathological study identified hemangiopericytoma, a phosphaturic mesenchymal tumor
After 4 years following hepatic tumor resection, there was an onset of pain in the right iliac fossa caused by two new lesions in the right iliac wing and presacrum, which were removed. After 2 years, the patient presented another recurrence consisting of hepatic metastasis, pulmonary nodules and lesions in the 7th and 8th ribs on the right side. These last tumors were resected together with the respective portion of the chest wall, and histological study was consistent with mixed soft tissue sarcoma (hemangiopericytoma). Later in the same year, symptoms of hypoesthesia started in the right T7/T8 dermatomes and bilateral cutaneoplantar extensor response. Magnetic resonance imaging demonstrated an epidural spinal tumor at T6–T9 with spinal canal compromise (70%), which required surgery (en bloc resection with instrumented fusion; Fig. 4) and associated dorsal radiotherapy (DT 44 Gy). A CT follow-up study 9 months later disclosed progression of the liver tumor, which was treated with six cycles of adriamycin, thereby stabilizing the condition. Nonetheless, 2 years later a recurrent hemangiopericytoma appeared in the left breast. Tumor study for somatostatin receptors was negative; hence octreotide analogs were not administered. Treatment with imatinib mesylate at 400 mg/day was started and the disease stabilized again.

Case 2. Magnetic resonance imaging demonstrates an epidural spinal tumor at T6–T9 with spinal canal compromise (70%) that required surgery
Because of the patient’s lengthy treatment with calcium, vitamin D3 and phosphates, 1 year later she developed tertiary hyperparathyroidism with hypocalcemia, hypophosphoremia, hyperphosphaturia and elevated PTH.
After another year, the patient experienced pain in the right thigh with abolition of the ipsilateral patellar reflex. CT performed at that time showed a pelvic mass displacing the femoral vascular nerve bundle and bone metastasis in the pelvic ring, both femurs and several vertebral bodies. She was hospitalized to carry out palliative resection of the right iliac wing tumor. The CT study performed 6 months later showed no changes: pulmonary nodules, 2.5 cm lesion in the left breast, multiple hepatic space-occupying lesions, and masses in the iliac pelvis, Douglas pouch and spleen.
After 2 years, the patient’s clinical status worsened, with increasing pain particularly in the lower limbs that made walking difficult. Magnetic resonance imaging of the vertebral spine showed multiple bone metastases that diffusely affected the entire spine, with cord compression at S1–S2 due to a pelvic mass that had increased in size. Local radiotherapy and analgesia were administered. Thoracoabdominal CT showed slight progression of disease in the liver and lower pelvis, the remaining lesions being stable. On the OctreoScan study (Fig. 5), radiotracer uptake was observed in the abdominopelvic, hepatic and left femoral masses, with moderate expression of somatostatin receptors. A decision was made to change the treatment to octreotide 20 mgs and adjust the analgesia. Within a few days of treatment, the pain was controlled and the patient’s general state improved to the point that she was able to walk with the aid of crutches. This improvement was likely due to the combined treatment with analgesics, octreotides, gabapentin and radiotherapy. A PET scan carried out 1 month after beginning treatment with octreotide analogs was compared with the previous CT to assess treatment response. A partial metabolic response that was more evident at the level of the mediastinum and vertebrae was observed, as well as multiple metastases showing tumor viability and post-therapy calcification, with less metabolic activity in both lungs, the left breast, pancreatic area, Douglas pouch and bone, and greater activity in the left proximal femur.

Case 2. On OctreoScan study, radiotracer uptake was observed in the abdominopelvic, hepatic and left femoral masses, with moderate expression of somatostatin receptors
Discussion
Osteomalacia is a generalized mineralization disorder of the osteoid matrix, consisting of a deficit in calcium and phosphate incorporation. Accumulation of non-mineralized osteoid decreases bone resistance, and this situation manifests clinically as pain and musculoskeletal deformity, muscle weakness and hypocalcemia. In the growing skeleton, the metaphyses are also affected, causing anomalous growth (rickets) [1].
In 1947, McCane described the first case of hypophosphatemic connective tissue tumor in a 17-year-old adolescent girl with an osteoid tumor in the femur associated with osteomalacia. She improved with resection of the lesion.
Oncogenic osteomalacia is a rare entity (around 100 cases described), although the prevalence of the association may be underestimated because the tumors are small (usually 1–4 cm) and can occur at unusual sites. The most frequent are bone tumors (55% of cases) occurring in the long bones of the lower limbs, such as the femur and tibia, with 30% arising in the head and neck (mandible, maxilla, ethmoidal bone, and cranial dome) and 20% in the upper limbs. Those occurring in soft tissues (45%, including the skin) are also mainly found in the lower limbs (66%). Another reason why these tumors go unnoticed is because they are quite indolent. Some patients diagnosed with idiopathic hypophosphatemic osteomalacia may actually have this syndrome. Most of the tumors are small and benign (only 10% are malignant and 5% multiple), although the symptoms may be generalized [2, 4].
Oncogenic osteomalacia is considered a consequence of an underlying neoplasm. Patients show proximal tubular malfunction similar to that occurring in Fanconi syndrome (phosphaturia, glucosuria and amino-aciduria) due to a humoral factor produced by the tumor and related to phosphate metabolism. This factor is now considered to be fibroblast growth factor FGF23 (previously known as phosphatonin), an 8- to 25-kD protein labile to heat, encoded by the homonym gene (FGF23: chromosome 12, locus 12p13.3) [4, 12].
The results of Stewart et al. [12] support the concept that FGF23 is an important regulator of phosphate metabolism. Administration of synthetic FGF23 and genetic overexpression of FGF23 in mice produce hypophosphatemia and renal phosphate loss. Genetic inactivity of FGF23 (in both mice and humans) causes hyperphosphatemia. Moreover, FGF23 production can be regulated by the phosphate both in vitro and in vivo [12].
In some studies [12, 17], FGF23 levels were determined by immunoassay before and after tumor resection. These efforts concluded that FGF23 is increased in many patients with a tumor responsible for osteomalacia, and levels above normal in the postoperative period correlate with persistence of the tumor. When the mass is totally resected, the patient’s renal function and serum FGF23 levels usually return to normal in the first 24 h. Thus, FGF23 determination may help to orient the diagnosis and prognosis of the illness by giving quick information about the existence or absence of residual tumor tissue [17].
Some hereditary hypophosphatemic illnesses, such as X-linked hypophosphatemia and dominant autosomal hypophosphatemic rickets, also show increased serum FGF23. This fact suggests that these diseases may share the same pathophysiology as oncogenic osteomalacia, although the mechanism responsible for the FGF23 increase is different in each of them [1, 2, 4, 7].
Serum FGF23 is also high in other tumors, such as chronic lymphatic leukemia, other leukemias and B lymphocyte lymphomas, myeloma and monoclonal gammapathy, the last two being additionally associated with increased levels of paraprotein and beta-2 microglobulin. In contrast to oncogenic osteomalacia, hypophosphatemia is not present in patients with these tumors, which suggests that the FGF23 produced by B cells is not functional or that there is another factor that contributes to maintain normal phosphate levels in these cases [1, 2, 4, 7, 12, 17].
Oncogenic osteomalacia is likely the most common cause of acquired hypophosphatemic osteomalacia. The condition usually manifests in the adult age (33 years on average, with a range of 7–73 years) with no predilection for either sex. Our patients are 17 and 34, within this age range. The symptoms precede the diagnosis by 5 years (with a range of 3 months to 17 years) [2, 8]. The signs and symptoms are typical of osteomalacia except for those caused by low calcium, since patients with oncogenic osteomalacia have normal calcium concentrations. More than 90% of the patients report bone pain, particularly in weight-bearing bones (lower limbs and spinal column). Around 66% show deep muscle weakness (proximal myopathy) [2], myalgia, fatigue, multiple fractures due to minimal trauma (particularly rib and vertebral crushing) and progressive deformities (increased dorsal kyphosis and bowing of the extremities). On physical examination, bone palpation is painful, there is muscular hypotonia, and osteotendinous reflexes are preserved [1].
The characteristic biochemical findings of oncogenic osteomalacia are decreased serum and urinary phosphate concentrations, increased serum alkaline phosphates and decreased 1,25-(OH)2-vit D. The phosphaturia and hypophosphatemia of these patients are not mediated by PTH or by PTH-related peptide (PTHrp), since concentrations of these factors and serum calcium are normal [8]. Other analytical parameters, such as calcitonin, 25-OH-vitD and 24,25-(OH)2 vitD are also normal. A decrease or inhibition of the enzyme 25-OH-vitD-1α-hydroxylase is the most striking finding, considering the severity of the hypophosphatemia, which should be a powerful stimulus for converting 25-OH-vitD into 1,25-(OH)2-vit D [8].
The typical radiological features of oncogenic osteomalacia are “pseudofractures” or Looser–Milkman lines, as were seen in our cases, which correspond to stress fractures. Densitometry usually shows osteopenia. On bone scanning, multiple foci of increased uptake are seen, which should not be misinterpreted as metastasis.
Bone biopsy shows osteomalacia with increased osteoclastic bone resorption, which probably explains maintenance of normal levels of calcium and PTH in blood [3).
In 1985, Weidner et al. [14, 15] described a group of mesenchymal tumors causing oncogenic osteomalacia. Approximately 87% of these tumors contained multinucleated giant cells (in some cases with ultrastructural features of conventional osteoclasts) or osteoclast-like multinucleated giant cells, and 80% had extensive vascularization. Both these components may be related to the pathogenesis or metabolic consequences of osteomalacia/rickets . Despite this fact, neither Yoshikawa nor Weidner were able to clearly define the specific features that allow a unified diagnosis because of the rarity of the syndrome, which makes it impossible to carry out morphological analysis of a large number of these tumors in a single center.
Although they had very different histological features, after analyzing 17 tumors in 1987, Weidner proposed a subclassification into four patterns according to their similarities [9, 15]. The first group, preferentially located in soft tissues, included phosphaturic mesenchymal tumor and mixed connective tissue variant [5]. The three other groups were located in bone, and were classified according to their resemblance to typical bone tumors: osteoblastoma-like variant [2], nonossifying-like fibroma variant [1], and ossifying-like fibroma variant [4]. This grouping seemed to confirm that tumors causing oncogenic osteomalacia do not have a homogeneous morphological pattern.
This condition should be suspected in teenagers and adults with phosphaturic normocalcemic osteomalacia in the absence of a family history of bone disease. An increase in serum FGF23 supports the diagnosis. When oncogenic osteomalacia is suspected, it is important to carefully search for tumor foci with pentetreotide [9] or octreotide scintigraphy, since the tumors often go unnoticed on conventional tests. 99Tc is mainly used to detect areas of active osteomalacia rather than to locate the tumor [2, 10]. Once the tumor has been detected, the definitive diagnosis is established by biopsy and pathological study.
The differential diagnosis includes diseases that cause osteomalacia and rickets (Table 1), particularly those producing osteomalacia and hypophosphatemia [1, 4, 8].
Apart from phosphaturic mesenchymal tumor, other types of tumors are also associated with oncogenic osteomalacia, such as prostate carcinoma of endodermic origin (likely due to osteoblastic metastasis) and oat cell carcinoma of neuroectodermal origin. Other associated tumors include sarcoma, myeloma, malignant fibrous histiocytoma, neurofibromatosis and neurinoma [8].
Recognizing phosphaturic mesenchymal tumor is of vital importance because complete resection cures the oncogenic osteomalacia in more than 80% of patients [5]. Moreover, between 24 h and 1 year after tumor removal, symptoms resolve in 90% of symptomatic patients: renal tubular phosphate reabsorption returns to normal decreasing urine phosphate loss, and serum phosphate levels normalize in 24–72 h. Levels of 1,25(OH)2vit D3 return to normal in 24–48 h, whereas alkaline phosphatase and osteocalcin take longer. After tumor removal, hypocalcemia can occur secondary to rapid osteoid remineralization, and hyperparathyroidism secondary to hypocalcemia. The skeletal changes, and densitometry and bone biopsy features take months to normalize [3]. The pseudofractures (Looser lines) disappear on radiography and may give rise to an area of sclerosis. If the tumor recurs, the syndrome generally reappears. Our first case had good resolution with the resection of the tumor, but in our second case, the syndrome reappeared representing this evolution. Perhaps the clinical evolution with recurrence of symptoms could suggest a malignant case.
When a tumor is not found, or cannot be completely resected, combined oral treatment with phosphate (2–3 mg/day), calcium, and calcitriol (1,25-(OH)2-vitD3) is recommended, with phosphate monitoring, a treatment similar to that used in X-linked hypophosphatemia. The cause of the dismal evolution of our second case is unknown. The persistent metastatic disease occurring in this patient maintained the analytical alterations and ultimately was the cause of her death. Bone pain and muscle weakness decrease with this therapy, although the symptoms do not completely disappear. In patients with gastrointestinal intolerance to oral phosphate, potassium phosphate iv can be administered, together with oral calcium and vitamin D [2, 3, 11, 16]. However, the risk–benefits of this approach should be assessed because iv phosphate can cause thrombophlebitis, hypocalcemia, right ventricular calcified thrombus and nephrocalcinosis. Another complication of treatment with phosphate and vitamin D administered for lengthy periods is tertiary hyperparathyroidism, which develops in 5% of cases. The proposed mechanism for this complication is that exogenous phosphate stimulates parathyroid activity through calcium sequestering [6, 13]. Administration of massive doses of 1α-OH-D3 and dihydrotachysterol and isolated administration of either phosphate or 1,25-(OH)2-vit D3 are rarely effective.
Unresected tumors that express somatostatin receptors (easily determined with octreotide scan) may be treated with somatostatin analogs, which result in a slight improvement of symptoms, and according to some studies even correct the phosphate and alkaline phosphatase levels. Seufert et al. [10] have reported that some tumor samples analyzed with RT-PCR showed overexpression of somatostatin receptor subtype 2 mRNA and weak or absent expression of subtypes 1, 3, 4 and 5. The discovery of somatostatin receptor subtype 2 expression provides the molecular basis for the positive status on octreotide scanning and the clinical response to octreotide therapy. This is because subtype 2 shows the highest affinity for octreotide among the five isoforms. The remission of renal phosphate loss with octreotide therapy may suggest that FGF23 secretion by the tumor is modulated through somatostatin receptors [10].
In the majority of patients with osteogenic osteomalacia, the prognosis is excellent, reflecting the benign nature of the process in most cases. Nonetheless, one of our patients with this condition presented recurrent metastatic disease with a fatal outcome.
References
Braunwald E, Fauci AS, Dennis LK, Hauser SL, Longo DL, Jameson JL (2002) Principios de medicina interna, 15th edn. McGraw-Hill, Madrid, p 2568, pp 2574–2576
Carpenter TO (2003) Oncogenic osteomalacia: a complex dance of factors [editorial]. N Engl J Med 348(17):1705–1708
Cheung FM, Ma L, Wu WC, Siu TH, Choi PT, Tai YP (2006) Oncogenic osteomalacia associated with an occult phosphaturic mesenchymal tumour: clinico-radiologico-pathological correlation and ultrastructural studies. Hong Kong Med J 12(4):319–321
Felig P, Frohman LA (2001) Endocrinology and metabolism, 4th edn. McGraw-Hill, New York, pp 1409–1423
Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY, Econs MJ, Inwards CY, Jan de Beur SM, Mentzel T, Montgomery E, Michal M et al (2004) Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol 28(1):1–30
Huang QL, Feig DS, Blackstein ME (2000) Development of tertiary hyperparathyroidism after phosphate supplementation in oncogenic osteomalacia. J Endocrinol Invest 23(4):263
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K et al (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348(17):1656–1663
Larsen PR, Kronenberg HM, Melmed S, Polonsky KS (2003) Endocrinology, 10th edn. Saunders, Philadelphia, pp 1349–1351, 1393–1395 and 1847
Park YK, Unni KK, Beabout JW, Hodgson SF (1994) Oncogenic osteomalacia: a clinicopathologic study of 17 bone lesions. J Korean Med Sci 9(4):289–298
Seufert J, Ebert K, Müller J et al (2001) Octreotide therapy for tumor-induced osteomalacia. N Engl J Med 345:1883–1888
Shane E, Parisien M, Henderson JE, Dempster DW, Feldman F, Hardy MA, Tohme JF, Karaplis AC, Clemens TL (1997) Tumor-induced osteomalacia: clinical and basic studies. J Bone Miner Res 12(9):1502–1511
Stewart I, Roddie C, Gill A, Clarkson A, Mirams M, Coyle L, Ward C, Clifton-Bligh P, Robinson BG, Mason RS, Clifton-Bligh RJ (2006) Elevated serum FGF23 concentrations in plasma cell dyscrasias. Bone 39(2):369–376
Tartaglia F, Minisola S, Sgueglia M, Blasi S, Brunelli D, Degli Effetti E, Maturo A, Cola A, Custureri F, Campana FP (2006) Tumor-induced hypophosphatemic osteomalacia associated with tertiary hyperparathyroidism: a case report. G Chir 27(1–2):9–13
Weidner N, Bar RS, Weiss D, Strottmann P (1985) Neoplastic pathology of oncogenic osteomalacia/rickets. Cancer 55:1691–1705
Weidner N, Santa Cruz D (1987) Phosphaturic mesenchymal tumors: a polymorphous group causing osteomalacia or rickets. Cancer 59:1442–1454
Yeung SJ, McCutcheon IE, Schultz P, Gagel RF (2000) Use of long-term intravenous phosphate infusion in the palliative treatment of tumor-induced osteomalacia. J Clin Endocrinol Metab 85(2):549–555
Zimering MB, Caldarella FA, White KE, Econs MJ (2005) Persistent tumor-induced osteomalacia confirmed by elevated postoperative levels of serum fibroblast growth factor-23 and 5-year follow-up of bone density changes. Endocr Pract 11(2):108–114
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Seijas, R., Ares, O., Sierra, J. et al. Oncogenic osteomalacia: two case reports with surprisingly different outcomes. Arch Orthop Trauma Surg 129, 533–539 (2009). https://doi.org/10.1007/s00402-008-0808-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00402-008-0808-2