
Abstract
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system (CNS) characterized by varying degrees of secondary neurodegeneration. Retinal ganglion cells (RGC) are lost in MS in association with optic neuritis but the mechanisms of neuronal injury remain unclear. Complement component C3 has been implicated in retinal and cerebral synaptic pathology that may precede neurodegeneration. Herein, we examined post-mortem MS retinas, and then used a mouse model, experimental autoimmune encephalomyelitis (EAE), to examine the role of C3 in the pathogenesis of RGC loss associated with optic neuritis. First, we show extensive C3 expression in astrocytes (C3+/GFAP+ cells) and significant RGC loss (RBPMS+ cells) in post-mortem retinas from people with MS compared to retinas from non-MS individuals. A patient with progressive MS with a remote history of optic neuritis showed marked reactive astrogliosis with C3 expression in the inner retina extending into deeper layers in the affected eye more than the unaffected eye. To study whether C3 mediates retinal degeneration, we utilized global C3–/– EAE mice and found that they had less RGC loss and partially preserved neurites in the retina compared with C3+/+ EAE mice. C3–/– EAE mice had fewer axonal swellings in the optic nerve, reflecting reduced axonal injury, but had no changes in demyelination or T cell infiltration into the CNS. Using a C3-tdTomato reporter mouse line, we show definitive evidence of C3 expression in astrocytes in the retina and optic nerves of EAE mice. Conditional deletion of C3 in astrocytes showed RGC protection replicating the effects seen in the global knockouts. These data implicate astrocyte C3 expression as a critical mediator of retinal neuronal pathology in EAE and MS, and are consistent with recent studies showing C3 gene variants are associated with faster rates of retinal neurodegeneration in human disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The role of the immune system in mediating CNS tissue injury is increasingly being examined not only in neuro-inflammatory diseases but also in classical primary neurodegenerative processes. While peripheral immune cells may extravasate into the CNS, it is also clear that resident glia often lose their homeostatic functions and become dysregulated as part of reactive gliosis [1, 4]. Indeed, glial activation and aberrant expression of inflammatory and potentially neurotoxic mediators have been described and are postulated to be pathogenic in numerous neurodegenerative diseases [2, 8, 25, 27, 28, 34]. The anterior visual pathway affords an opportunity to study the mechanisms by which inflammatory demyelination in the optic nerve mediates retinal ganglion cell (RGC) loss in the retina. RGC layer thinning measured by optical coherence tomography (OCT) following acute optic neuritis and chronic optic neuropathy are well described in people with MS, and one study has documented loss of nucleated cells in the ganglion cell layer of MS post-mortem retinas [10, 33].
Seminal work showing expression of early complement pathway molecules, C1q and C3, on neuronal synapses in the retina and the thalamus during development and in disease has led to the hypothesis that aberrant complement expression by microglia (C1q) and astroglia (C3+) may be neurotoxic [40]. More recently, it has been suggested that C1q and C3 activation fragments such as C3b and C3d opsonize adult neuronal synapses leading to their pruning by activated microglia that express the C3d receptor CD11b, thereby mediating neuronal degeneration in neurodegenerative diseases including MS [15, 25, 32, 38, 43].
Pathological studies of MS cortical, thalamic, and hippocampal gray matter lesions have shown increased complement transcription and deposition, with variable expression in both active and chronic lesions across studies [17, 29,30,31, 44]. The opsonin complement activation product, C3b, was shown to be increased on neurons and glia in MS cortical and deep grey matter as compared to healthy controls [44]. Both C1q and C3 activation were increased in the CA3/2 and CA1 subfield of MS hippocampi, which also exhibited a marked decrease in synapses, indicating a correlation between the complement pathway and synaptic changes [35, 44]. Clusters of C3d+ microglia have been described in chronic MS tissue and are thought to be involved in debris clearance at late stages of disease [29]. In addition, we recently showed that C3 gene variants are associated with more rapid rates of retinal neurodegeneration in people with MS [7]. Our MS GWAS analyses suggest that C3 gene variants may predispose people with MS to develop more rapid and severe retinal neurodegeneration, and we hypothesize that these variants lead to increased C3 expression. There has been a paucity of post-mortem MS retina to examine for C3 expression. Herein, we examine C3 expression and its cellular source in post-mortem MS and non-MS control retina. We show extensive C3 expression in astrocytes in MS retinas associated with RGC loss.
Several recent publications support a critical role of C3 in mediating synaptic pathology in the MS animal model experimental autoimmune encephalomyelitis (EAE) [5, 13, 19, 41, 45]. We and others have shown RGC loss following optic neuritis in MOG35-55 induced EAE, indicating that it is a useful model to study anterior visual pathway pathology as occurs in MS optic neuropathy. To better understand the mechanism of RGC loss and optic neuritis and explore the possible role of C3, we induced EAE in C3 sufficient (C3+/+) and C3 knockout (C3–/–) C57BL/6 J mice by immunization with MOG35-55. Global C3 deficiency partially ameliorated RGC loss in mice without affecting infiltration of inflammatory cells into the CNS, paralysis score, demyelination or gliosis. Using a novel C3-tdTomato reporter mouse line we show that astrocytes are a major source of C3 expression in the retina and optic nerve. Conditional deletion of C3 in astrocytes also partially protected RGCs and adjacent neurites. These data definitively implicate astrocyte expression of C3 in mediating neuronal injury in EAE and support the notion that C3 inhibition may be beneficial to people with MS.
Materials and methods
Human retina staining by immunofluorescence
Multiple sclerosis and control retinas were obtained from a rapid autopsy protocol at the NIH or from the Johns Hopkins Ocular Pathology Laboratory and post-mortem delay was less than 24 h. Paraffin embedded human retinas were sectioned at 8 µm using a microtome. Sections were deparaffinized by immersing into Xylene, 100% ethanol, 95% ethanol, 90% ethanol and 50% ethanol sequentially for 3 min for each step. Sections were washed with PBS before antigen retrieval. Citrate buffer was used for antigen retrieval by heating to 80 °C for 50 min. After antigen retrieval, sections were permeabilized with PBS containing 0.4% of Triton X-100 for 5 min and then blocked with PBS containing 5% normal donkey serum, 5% normal goat serum, and 0.1% Triton X-100 for 30 min at room temperature. Sections were then incubated with the primary antibody of interest overnight at 4 °C. The primary antibodies that were used are shown in Table 1. The sections were then incubated for 3 h at room temperature with anti-species-specific secondary antibodies directly conjugated to Alexa fluorophores (1:1000, Invitrogen) followed by nuclei staining with Hoechst. Aqua poly/mount reagent (Polysciences, Warrington, PA, USA) was used to mount sections. Images were captured using a Zeiss Axio Observer Z1 epifluorescence microscope and Zeiss 710 confocal microscope with the appropriate excitation and emission filters. RGCs were quantified by enumerating the number of RNA binding protein with multiple splicing (RBPMS)+ cells per unit length of retina across the entire length of each retina. To quantify GFAP and C3 levels, twelve equidistant regions along each retina were selected. ImageJ software (National Institutes of Health) was used to quantify mean fluorescent intensity (MFI) in the ganglion cell layers of each region, and the twelve regions were then averaged to represent the GFAP and C3 MFIs for each subject.
Animals
C57BL/6J mice (strain 000664) and the global C3-deficient [46] mice (strain 003,641) were purchased from The Jackson Laboratory. An additional C3-deficient strain, generated previously by crossing a floxed allele with a Cre-Deleter strain [22], was used to validate the results observed with the first strain. C3-tdTomato/floxed (C3tdt−fl) reporter mice, which were recently described [22], were used to confirm CNS C3 expression and conditionally deplete C3. GFAP-Cre mice [11] were purchased from the Jackson Laboratory (strain 024,098) and crossed to C3-tdTomato/floxed reporter mice to produce GFAP-Cre+;C3tdT−fl mice. Only Cre+ females were crossed to Cre− males to avoid embryonic recombination previously reported in this Cre strain. All animals were housed in the pathogen-free, temperature-controlled animal facility at the Johns Hopkins University School of Medicine with 12 h/12 h light/dark cycles and fed with standard food and water ad libitum. National Institutes of Health guidelines for the use of experimental animals were exactly followed and all experimental protocols were approved by the Johns Hopkins Institutional Animal Care and Use Committee.
EAE model
EAE was induced as we described previously [19]. All studies were carried out using 10–13 week-old mice. Briefly, mice were injected subcutaneously with 150 μg of MOG33-55 (Johns Hopkins Peptide Synthesis Core) in complete Freund’s adjuvant (CFA) (ThermoScientific, Rockford, IL, USA) containing 600 μg of mycobacterium tuberculosis (BD, Franklin Lakes, NJ) on the lateral abdomen. On the same day as immunization and 2 days later, mice were also injected IP with 300 ng of pertussis toxin (List Biological Labs, Campbell, California, USA). Mice were sacrificed at late disease, PID42, to explore the effect of neuroprotection in long term, and peak stage, PID16, to look at infiltration and possible pathological manifestations related to long-term effects.
Clinical EAE behavioral scores were obtained daily, in a masked manner, using the established standard scoring from 1 to 5; 0 = no signs of disease; 1 = loss of tail tonicity; 2 = loss of tail tonicity and mild paralysis of hindlimbs; 3 = paralysis of hindlimbs; 4 = hindlimbs paralysis and mild paralysis of forelimbs; and 5 = complete paralysis or death.
Mouse retina and optic nerve sample preparation.
Mouse retina and optic nerves were collected as described previously [19]. After anesthetizing, mice underwent cardiac perfusion with 30 mL phosphate buffered saline (PBS). After PBS perfusion, the eyes were removed from the eye socket by blunt enucleation using curved dressing forceps and immediately placed into 4% paraformaldehyde (PFA) for immersion fixation for 4 h then transferred into 30% sucrose for cryoprotection.
Whole flat mount retinas and vertical sectioned retinas were prepared as described using paired eyes from a single mouse as we previously showed RGC counts across retinas are the same within an EAE mouse [19]. Flat mount retinas were processed for RGC labeling with anti-Brn3a (Pou4f1) antibody. After staining, the whole retinas were flat mounted onto slides and imaged for RGC counting. For vertical sectioned retinas, the eyes were placed into TissueTek mold filled with Optimal Cutting Temperature (O.C.T.) compound (VWR, Radnor, PA, USA), snap frozen with 2-methylbutane and then stored at − 80 °C until cryosectioned. The eyes were sectioned at 16 μm vertically and then mounted onto slides (ThermoScientific, Rockford, IL, USA) followed by staining with antibodies.
Optic nerves were dissected out and evenly divided into three parts and then vertically placed into TissueTek mold filled with O.C.T. and snap frozen with 2-methylbutane. Frozen samples were stored at − 80 °C until cryosectioning. Optic nerves were sectioned at 10 μm thickness and then mounted on slides for further immunofluorescence staining.
Immunofluorescence staining
All immunofluorescence staining protocols are previously published [19]. Briefly, sections were permeabilized with PBS containing 0.4% of Triton X-100 then blocked with PBS containing 5% normal donkey serum and 0.1% Triton X-100 for 1 h at room temperature and incubated with primary antibody of interest overnight at 4 °C. The primary antibodies that were used are shown in Table 1. The sections were then incubated for 1 h at room temperature with species-specific secondary antibodies directly conjugated to Alexa fluorophores (1:1000, Invitrogen) followed by nuclei staining with Hoechst. A coverslip was mounted onto sections using aqua poly/mount reagent (Polysciences, Warrington, PA, USA). Images were captured using a Zeiss Axio Observer Z1 epifluorescence microscope and Axiovision software with the appropriate excitation and emission filters.
For vertical retina Tuj quantification, the IPLs of six equidistant regions of interest (ROI) per retina were analyzed using ImageJ software. For optic nerve, three cross sectional regions were quantified as previously described [19]. ImageJ software was used to quantify mean fluorescent intensity (MFI) of the immunofluorescent signal by persons blinded to sample information. For enumerating glia and T cells, Zen Blue software (Carl Zeiss, Oberkochen, Germany) was used.
RGC counts
To count RGC number in whole retina, a mosaic image of the whole mount retina was captured using a Zeiss Axio Observer Z1 epifluorescence microscope with a motorized stage and a z-stack step size of 1 μm. After the images were acquired, each retinal quadrant was segmented into central, middle, and peripheral regions and 4 areas were counted per region, as shown in Fig. 2 and described previously [19]. Our MATLAB-based semi-automated RGC counting algorithm was used to determine RGC number in each mouse [19]. Briefly, selected segments were exported as tagged image file format (TIF) and fed into a custom MATLAB (Mathworks, Natick, MA, USA) size segmentation algorithm to perform automated counts. Cells in selected retinal segments undergo filtering, automatic or user-defined thresholding, and binarization followed by identification of cell boundaries. Labeled RGCs were counted at 20X magnification and a total of twelve regions were averaged to represent RGC density for each mouse retina. All analyses were performed masked to genotype and behavior score and data underwent quality control check.
Flow cytometric analysis
Cell isolation
Resident glia and infiltrating mononuclear cells were harvested from the CNS of PID 16 EAE mice. Mice were euthanized with isoflurane overdose then cardiac perfused with ice cold HBSS. Tissue was collected and chopped with a razor prior to enzymatic digestion with 2 mg/ml Collagenase IV and 100 U/ml DNase (Worthington, Lakewood, NJ, USA) for 30 min at 37 °C. Digested tissue was passed through a 100 µm cell strainer to create a single cell suspension. Cells were separated from myelin with a 40% percoll solution and processed for flow cytometry. Peripheral immune cells were isolated from spleens after perfusion. Spleens were mechanically dissociated over 100 µm cell strainers, and red blood cells were removed by incubation in RBC Lysis Buffer (Biolegend, San Diego, CA, USA).
Flow cytometry
Following preparation of single cell suspensions, cells were stained with a fixable viability dye (Live/Dead Aqua ThermoScientific, Rockford, IL, USA. Zombie NIR, Biolegend, San Diego, CA, USA) for 20 min at room temperature, then Fc blocked (Biolegend, San Diego, CA, USA) for 10 min at 4 °C, and then surface stained for 30 min at room temperature in FACS buffer (2% FBS in PBS with 2 mM EDTA). When intracellular staining (ICS) was performed after surface staining, the cells were then fixed and permeabilized using IC Fixation Buffer and Permeabilization Buffer (ThermoScientific, Rockford, IL, USA) according to manufacturer’s recommendations. ICS staining was then performed in permeabilization buffer for 1 h at room temperature. Cells were then washed twice and resuspended in FACS buffer. When measuring cytokines, cells were re-stimulated with Cell Stimulation Cocktail and Protein Transport Inhibitor (ThermoScientific, Rockford, IL, USA) for 5 h at 37 °C in complete media prior to beginning the staining. Samples were run on a MACSQuant X flow cytometer (Miltenyi Biotec, Bergisch Gladbach, Germany) or a Cytek Aurora (Cytek Biosciences, Fremont, CA, USA). Fluorescence-minus-one (FMOs) staining controls were used for gating where necessary. Data were analyzed in FlowJo (BD Biosciences, Franklin Lakes, NJ, USA).
Statistical analysis
Statistical analysis was conducted using GraphPad Prism software version 8 (GraphPad, San Diego, CA, USA). Unpaired two-tailed Student's t test was used to analyze data. Results were considered significant if the p value was < 0.05. Error bars indicate SEM in all figures.
Results
C3 is highly expressed in GFAP+ cells in MS patient retinas
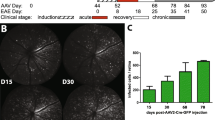
Given our previously reported genetic findings linking C3 gene variants with more rapid rates of retinal degeneration in MS, we examined C3 expression and its cellular source in post-mortem MS retinas (Fig. 1). We obtained the retinas from a 60-year-old woman with progressive MS (MS-1) who passed away after a 6-month history of pancreatic cancer (not treated with checkpoint inhibitors). She had a longstanding history (signs present in 2009) of optic neuropathy (OS) with relative afferent pupillary defect and her retinal optical coherence tomography (OCT) scans performed in 2019 a year prior to her death showed marked thinning of the retinal nerve fiber layer (RNFL) and ganglion cell layer in the left eye (OS) but with only mild subclinical thinning in the right eye (OD) (Figs. 1a and S1, online resource). At post mortem, we immunostained retinal cross-sections to examine expression of astrocytes, neurons, and C3 (Fig. 1b). The left retina (OS) exhibited marked reactive astrogliosis and C3 expression, which was extensive but regionally variable (Fig. 1c, d). The C3 expression was almost exclusively colocalized to GFAP+ cells. The unaffected eye had much less astrogliosis and C3 expression, although areas of GFAP/C3 expression were also seen despite no clinical symptoms or signs of optic neuropathy. To extend these observations, we examined 3 additional MS and 4 non-MS retinas (Table 2), and the same expression pattern with regions of high C3+/GFAP+ cells was seen repeatedly in the MS cases examined but was significantly less although also noted in some of our aged control cases (Fig. 1e, f). Across these cases, the MS patients expressed higher levels of C3 in the GCL than the non-MS controls. There was no significant difference in GFAP expression (Fig. 1g). Finally, we enumerated RBPMS+ cells in the ganglion cell layer and found, as has been previously reported using H&E stain [10], that RGC counts are lower in MS cases compared to age-matched controls (Fig. 1h, i). These data provide support for the concept that neurodegeneration, especially in the retina is common in MS and is frequently associated with an altered phenotype of reactive, potentially neurotoxic glia [2, 10, 25].

MS retinas exhibit enhanced regional C3 expression and astrogliosis. a Retinal optical coherence tomography (OCT) scans from a 60-year-old woman with progressive MS and optic neuropathy, showing marked thinning of the ganglion cell layer in the left eye (OS) but not in the right eye (OD). Red indicates thickness value is less than 1st percentile for age matched controls. b Schematic diagram of retinal cross-section and immunofluorescence staining of individual retinal layers. GCL ganglion cell layer, INL inner nuclear layer, IPL inner plexiform layer, OPL outer plexiform layer, ONL outer nuclear layer, RPE Retinal pigment epithelium. c Representative immunofluorescence images of paraffin-embedded retinal cross-sections from the left eye (OS) of the patient presented in panel A. Magnification 20x; Scale bar = 200 μm. Enlarged images of the boxed region are shown to the right; Scale bar = 50 μm. d Representative immunofluorescence images of paraffin-embedded retinal cross-sections from the right eye (OD) of the patient presented in panel A. Magnification 20x; Scale bar = 200 μm. Enlarged images of the boxed region are shown to the right. Scale bar = 50 μm. e Representative images of non-MS and additional MS retina stained with GFAP and C3; Scale bar = 50 μm. f Confocal image of human MS retina stained with GFAP and C3, showing colocalization of C3 and GFAP staining; Scale bar = 10 μm. g Quantification of C3 and GFAP expression in the GCL from MS and non-MS retinas, presented as mean fluorescent intensity (MFI). AU arbitrary unit. (h) Representative images of RBPMS+ cells in retinal cross sections from 2 non-MS and 2 MS individuals; Scale bar = 50 μm. i Quantitation of the number of RBPMS+ cells/ mm length of retina from MS (n = 4) vs non-MS (n = 4) cases. The average length of retinas quantified for RBPMS + cells were 27.82 ± 6.47 mm for non-MS and 23.33 ± 2.13 mm for MS cases. The number of RBPMS+ cells for the retinas shown in panel c–d: OS#4, OD#18, average #11 presented in the plot as one of the MS cases. Significance between groups was assessed by unpaired t-test. Error bars represent SEM.
Deletion of C3 (C3–/–) partially ameliorated RGC loss and preserved neurites in the inner retina of experimental autoimmune encephalomyelitis (EAE) mice
To ascertain whether C3 expression was deleterious or protective following optic neuritis, we next examined retinas from EAE mice with or without global deletion of C3. We prepared whole flat mount sections of the retina at 42 days post-immunization (PID42, late EAE) to quantify RGCs. RGCs were stained with the specific marker, Brn3a (Pou4f1) and were quantified using semi-automated software as described previously [19]. C3+/+ and C3–/– CFA control mice had similar amounts of RGCs (Fig. 2a, b). As expected, C3+/+ mice with EAE had significant loss of RGCs relative to their CFA controls. While still experiencing significant RGC loss compared to CFA controls, C3–/– mice with EAE had significantly more RGCs relative to C3+/+ EAE mice, suggesting C3–/– was moderately protective against RGC loss though not required for normal RGC development. We also examined vertical sections of the retina to assess layer specific pathology. C3–/– mice had significantly less neuritic loss (measured using ßIII-tubulin (Tuj) staining) in the IPL than C3+/+ mice (Fig. 2c, d). The increased density of RGC neurites in C3–/– mice with EAE provided further confirmation of a protective effect of global C3 deletion on RGC health. There was no difference in mean fluorescence intensity (MFI) of ßIII-tubulin on neurites between C3+/+ and C3–/– CFA mice.

C3–/– EAE mice have less RGC loss and partially preserved neurite projections in the inner retina. a Representative images of Brn3a staining in flat-mount whole retina; Scale bar = 1 mm, 20X magnification. Brn3a+ RGC are shown from a central segment in whole mount retina from CFA and EAE groups at PID42. Images were acquired with 20 × magnification; Scale bar = 50 µm. b Quantification of Brn3a+ RGC in whole mount retina from different groups at PID42. Each dot represents a mouse RGC count, which was derived from an average of 12 segments as shown in panel A. c Representative images of Tuj staining in the retina of CFA and EAE mice. Dotted white lines indicate IPL ROI quantified in d. Images were acquired with 40 × magnification; Scale bar = 20 µm. d Quantification of IPL Tuj staining in CFA and EAE mice, presented as Mean Fluorescent Intensity (MFI). AU arbitrary unit. Significance between groups was assessed by unpaired t-test. Error bars represent SEM
C3 deletion did not suppress EAE behavioral score, effector T cell infiltration into the optic nerve or demyelination
To determine if C3–/– mice had RGC protection due to less inflammation, we first examined the incidence and severity of EAE using behavioral score. The incidence of EAE in C3+/+ and C3–/– mice was similar (C3+/+: C3–/– = 84.1%: 81.4%), and the average motor behavioral score was not different between C3+/+ and C3–/– EAE (2.7 vs 2.5 (Fig. 3a)). Since behavioral score predominately reflects spinal cord pathology and it has been previously shown that C3 expression is more upregulated in the optic nerves, we also measured immune cell infiltration into the CNS in EAE C3+/+ mice vs C3–/– mice [18]. We examined the CD4+ T-cell infiltration into optic nerve in EAE at PID16 (peak stage), and PID42 (late stage) by IHC. At PID16, both C3+/+ and C3–/– EAE mice had CD4+ T-cell infiltrates of similar amounts in the optic nerve (Fig. 3b, c). At PID42, although the absolute numbers were reduced (Fig. 3b, c), again no differences were seen between C3+/+ and C3–/– EAE mice. To determine whether global C3 depletion might affect myeloid cell infiltrate or gliosis in the optic nerve, we stained for IBA1 (infiltrating myeloid and microglia) and GFAP in PID16 optic nerves (Fig. 3d–f) and saw no differences. Immune cell infiltration into the brain and spinal cord was also examined by flow cytometry at PID16. No differences were observed between C3+/+ and C3–/– mice in T-cell infiltration (CD3+;CD4+), myeloid infiltration (CD11b + ;P2RY12-), microgliosis (CD11b + ;P2RY12 +), or T-cell cytokine production (IFNγ+ and IL-17a+ in CD3+;CD4+ cells)(Fig. 3g–i, Fig. S3, online resource). Given contradictory reports previously published on the effect of C3 deletion on the EAE behavioral score [5, 41], we also induced EAE in a second independent C3 deficient strain and again observed no behavioral differences (Fig. S4, online resource).

C3 deletion does not affect EAE behavioral score, effector T cell infiltration into the optic nerve, or demyelination but reduces axonal swellings. a Behavior score of EAE mice over time from one representative experiment (C3+/+ EAE, n = 8; C3–/– EAE, n = 6). The experiment was replicated 5 times (across all 5 replicate experiments, C3+/+ EAE, n = 108; C3–/– EAE, n = 91). b Representative images of CD4+ T-cell staining in the cross sections of optic nerve from EAE mice, 20 × magnification; Scale bar = 50 μm. c Quantification of CD4+ T-cells in the optic nerves of EAE mice at PID16 and PID42. d Representative images of GFAP and IBA1 staining in optic nerves of EAE mice at PID16, 20 × magnification, Scale bar = 50 μm. e–f Quantification of IBA1 e and GFAP f staining in optic nerves of EAE mice at PID16, presented as MFI. AU arbitrary unit. g–i The percentage of CD3+CD4+ T cells g, infiltrating myeloid cells (CD11b + ;P2RY12-) h, and microglia (CD11b + ;P2RY12 +) i in the brain of EAE mice at PID16, quantified by flow cytometry in the population of viable cells. j Representative images of SMI31/MBP and SMI32/MBP staining in the optic nerve of EAE mice at PID16. The last row shows the magnified images of SMI32; Scale bar = 50 μm. k–l Quantification of MBP and SMI31 staining in the optic nerve of EAE mice at PID16, presented as MFI. m The counts of SMI32+ spheroids in the optic nerve of EAE mice at PID16. Significance between groups was assessed by unpaired t-test. Error bars represent SEM. In all quantifications, each dot represents a unique mouse
C3–/– EAE mice had reduced axonal spheroids in the optic nerve as compared to C3+/+ EAE mice
Since C3 deletion protected against RGC soma and neurite loss, we expected it would also alleviate axonal injury in the optic nerve. We examined myelination and axonal integrity in the optic nerve by staining with myelin basic protein (MBP) and phosphorylated (SMI31, intact axons) and non-phosphorylated (SMI32, reversibly injured axons) neurofilament markers at PID16. C3–/– had no effect on myelination (Fig. 3j, k), nor on the expression of SMI31 (Fig. 3j, l). However, C3–/– EAE mice had fewer SMI32+ spheroids in the optic nerve as compared to C3+/+ EAE mice (Fig. 3j, m), indicating that C3 deletion reduced the early axonal degeneration in EAE mice.
C3-tdTomato reporter mice revealed astrocytes as a major cellular source of C3 in the inflamed EAE CNS
Complement component C3 is well recognized as a critical component of the peripheral immune system [14]. Previously, its expression in the CNS had been thought to be negligible and/or limited to neuronal synapses such as on RGCs [40]. More recently, neurotoxic astrocytes were implicated as a major cellular source of C3 in a number of human neurodegenerative diseases, as well as MS, and related animal models [19, 25, 42, 48]. However, microglia, which are innate immune cells, have also been shown to express C3 in MS autopsy studies [36]. We used a recently published mouse line in which a floxed tdTomato (tdT-fl) cassette is inserted towards the end of the endogenous C3 locus. In these mice, Cre mediated recombination deletes the tdTomato cassette along with the last 5 exons of the C3 transcript and leads to a shortened unstable C3 mRNA [22]. This reporter system has the added advantage of clearly pinpointing which cells have induced C3 gene expression and is not dependent on antibodies, which also bind to secreted C3 and its degradation products (C3b and C3d).
We first validated that the tdTomato signal colocalized with cells that were C3+ by antibody immunohistochemistry in EAE mice at PID16 since this was the time point that C3 was previously shown to be highly activated (Fig. 4). Having established that the reporter was faithful, we utilized its brighter signal and the greater reliability of a genetically driven reporter system to examine C3 expressing cells in the optic nerve and retina. We found that in the optic nerve, tdTomato+ expressing cells were ~ 80% GFAP+ cells and 20% IBA1+ cells (Fig. 4c), suggesting that a greater proportion of C3 expressing cells are astrocytes than microglia/monocytes in the optic nerve of EAE mice at this timepoint. Next, we utilized the reporter to examine C3 expression in the retina (Fig. 5). There were clear examples of RGCs expressing tdTomato although this was much less common than tdTomato+/GFAP+ cells (~ 25% vs ~ 75% respectively) (Fig. 5a, b). We very rarely observed reporter tdTomato+/IBA1+ in the neural retina (Fig. 5c). tdTomato expression was also observed on radial (Müller) glia in the IPL, inner nuclear layer (INL), and outer plexiform layer (OPL) (Fig. 5d) consistent with a possible role for C3 in deep retinal inflammatory pathology. We also examined spinal cords and found C3 expression in astrocytes there as well, but no difference in NeuN (Fig S5, online resource).

Astrocytes are the major C3 expressing cell in the optic nerve of EAE mice at PID16. a GFAP and C3 staining of optic nerves from EAE mice expressing tdTomato under the control of C3 promoter (C3 reporter mice); Scale bar = 50 μm. b IBA1 and C3 staining in the optic nerve of C3 reporter mice with EAE. c Quantification of GFAP+/tdTomato+ cells or IBA1+/tdTomato+ cells in the optic nerve of C3 reporter mice with EAE at PID16. d Enlarged figure for the box area in panel a. e Enlarged figure for the box area in panel b

C3-tdTomato cells are predominantly astrocytes in the retina. a tdTomato+/GFAP+ cells and tdTomato+/NeuN+ cells in cross sectioned retina of a tdTomato reporter mouse with EAE at PID16; Scale bar = 20 μm. b Quantification of tdTomato+/GFAP+ cells, tdTomato+/NeuN+ cells, and tdTomato/IBA1+ cells in the cross sections of retina from tdTomato reporter mice at PID16. Each dot represents one mouse. c Representative image of a cross sectioned retina showing tdTomato+GFAP+ cell while IBA1+ cells are negative for tdTomato; Scale bar = 20 μm. d tdTomato expression in Müller glia in the inner nuclear layer (INL) shown by a white arrow; Scale bar = 20 μm
C3tdT−fl x GFAP-Cre mice specifically depleted C3 in astrocytes and exhibited retinal neuroprotection in EAE
Given the robust expression of C3 in astrocytes, we then performed a series of EAE experiments in C3tdT−fl mice crossed with GFAP-Cre mice to specifically deplete C3 in astrocytes. Cre+ animals had an overall 50% reduction of tdTomato signal in PID16 optic nerve (Fig. 6a, b). Cell specific markers revealed the reduction was specific to Sox9 + astrocytes and not microglia/myeloid cells (IBA1) (Fig. 6c–f). To confirm that GFAP-Cre+ mice had no peripheral effects on C3 expression we examined spleens of PID16 EAE mice and saw no differences in tdTomato signal (Fig. S6a-d, online resource). Again, as with the global knockout EAE mice, we did not observe any change in the behavioral score of Cre+ mice as compared to Cre− mice with EAE (Fig. S6e, online resource). RGC numbers in PID42 EAE mice were higher in the C3tdT−fl Cre+ mice as compared to the C3tdT−fl Cre− controls (Fig. 7a, b). Further, neurites were significantly higher at PID42 EAE in C3tdT−fl Cre+ mice as compared to the C3tdT−fl Cre− controls (Fig. 7c, d). These data are consistent with astrocytes as a major cellular source of C3 and directly implicate C3+ astrocytes as mediators of RGC loss that occurs following optic neuritis in this model.

GFAP-Cre expression specifically deletes C3 from astrocytes in EAE PID16 optic nerves. a Representative images of tdTomato expression in the optic nerve of C3tdT−fl x GFAP-Cre mice at EAE PID16; Scale bar = 50 μm. b Quantification of tdTomato expression in the optic nerve of C3tdT−fl x GFAP-Cre mice at EAE PID16, presented as MFI; AU, arbitrary unit. c Representative images of tdTomato expression in SOX9+ astrocytes in the optic nerve of C3tdT−fl x GFAP-Cre at EAE PID16. d The percentage of tdTomato+ SOX9+ astrocytes in the optic nerve of C3tdT−fl x GFAP-Cre mice at EAE PID16, quantified by IHC. e Representative images of tdTomato expression in IBA1 + cells in the optic nerve of C3tdT−fl x GFAP-Cre mice at EAE PID16; Scale bar = 20 μm. f The percentage of tdTomato+/IBA1+ cells in the optic nerve of C3tdT−fl x GFAP-Cre mice at EAE PID16, quantified by IHC. Significance between groups was assessed by unpaired t-test. Error bars represent SEM

C3 depletion from astrocytes protects RGC and neurite projections in the retina. a Representative images of Brn3a staining in flat-mount whole retina from C3tdT−fl x GFAP-Cre mice at EAE PID42. Magnification 20x; Scale bar = 50 µm. b. Quantification of Brn3a + RGC number from different groups at PID42. c Representative images of Tuj staining in the retina of C3tdT−fl x GFAP-Cre mice at EAE PID42. Magnification 40x; Scale bar = 20 μm. d Quantification of Tuj staining in C3tdT−fl x GFAP-Cre mice with EAE, presented as MFI. Error bars represent SEM. AU arbitrary unit
Discussion
Several studies have shown that complement C3 activation is associated with synaptic pathology in MS and EAE [5, 10, 13, 19, 33, 41, 45]. C3 gene variants are associated with more rapid rates of ganglion cell layer thinning on longitudinal OCT in persons with MS and C3 expression by neurotoxic glia has been specifically implicated as playing a direct role in CNS and retinal degeneration [2, 7, 12, 25]. We first examined post mortem retinas donated by people with MS and saw marked reactive astrogliosis that was regionally heterogeneous. The majority of these astrocytes in the inner retina had high levels of C3 expression. Remarkably, in a patient who had progressive MS with a more than ten-year history of established optic neuropathy, the affected eye with extensive documented (in vivo OCT) ganglion cell layer thinning had more extensive inner retinal C3 astrogliosis as compared to the clinically unaffected eye. Interestingly, there were regions of the affected eye in which the reactive astrogliosis extended into the deeper retina. Nonetheless, the unaffected eye also had C3 astrogliosis, but this was less severe and regionally limited presumably accounting for the clinical quiescence. The patterns observed in this patient were seen to varying degrees in three other MS retinas. While age matched controls without confounding variables are hard to obtain, we did examine four control eyes and found less C3 expression in the inner retina than in MS.
To explore the cellular mechanism underlying RGC loss following optic neuritis, we used the EAE model, in which there is extensive anterior visual pathway pathology associated with glial C3 expression [19]. We, and others, have reported that there is loss of RGCs following optic neuritis in EAE [16, 19, 23, 24, 26]. Here we show that global C3–/– EAE mice have significantly improved RGC survival at PID42 as compared to WT mice. Both neuritic processes in the IPL and optic nerve axons were also protected confirming that global C3 deletion mediated partial retinal neuroprotection.
Remarkably, this effect did not appear to be the sequelae of reduced inflammation since the incidence of EAE and behavioral scores were not altered. Further, we found no evidence that peripheral infiltration by effector T cells was affected by C3 deletion in EAE. In addition, the amount of infiltrating myeloid cells, the number of CD4+ T cell infiltrates into the CNS, and the Th1 and Th17 profile were unchanged by IHC and flow cytometric measurements. The absence of an effect on behavioral scores and early inflammatory mediators of EAE suggested to us that the effects of C3 deletion on reducing RGC loss in EAE were related to CNS resident cells, such as neurotoxic astrocytes.
We next utilized a novel reporter mouse in which the C3 promoter drives expression of a tdTomato reporter. This reporter line was recently used to examine C3 expression in infiltrating immune cells in the skin but has not been used in a CNS inflammatory model. The tdTomato expression was faithful, mirroring C3 Ab immunohistochemistry but the signal was brighter and more specific allowing more accurate colocalization with CNS resident cells. In the optic nerve, we found 80% of C3-expressing cells were astrocytes as compared to 20% myeloid lineage (microglia or macrophages). In the retina, astrocytes were again the predominant C3 expressing cell, but RGCs also expressed C3, albeit less robustly. We also saw astrocytic expression of C3 in the inflamed spinal cord.
In order to definitively implicate astrocyte production of C3 in mediating RGC loss, we crossed C3tdT−fl mice with GFAP-Cre to conditionally deplete C3 specifically from astrocytes and examine whether this would also protect RGCs and neurites. Remarkably, C3 depletion in astrocytes resulted in RGC protection, equivalent to what we saw in the global knockout, further implicating a neurotoxic role of C3+ astrocytes.
Our data are consistent with a model in which aberrant expression of C3 during chronic inflammation leads to opsonization of synapses, which are then phagocytosed by C3 receptor CD11b-expressing microglia [13, 45]. Excessive synaptic stripping may then cause neuronal demise. Two studies have shown that global C3–/– mice induced with EAE have a reduction of synaptic pathology and improved function. Our study extends knowledge gained from these reports in several ways. Here, using a genetic reporter we show that C3 is mostly expressed in astrocytes both in the optic nerve and retina. We show C3 deficiency is associated with retinal ganglion cell protection. Prior studies logically focused on synaptic pathology but were performed in areas of the brain in which quantifying neurons is more challenging. Finally, we show astrocyte specific depletion is also protective.
Some aspects of our results differ as compared to prior studies and may be related to details of our model and the anterior visual pathway as a CNS site. A notable difference in our results was the lack of a significant effect of C3 deletion on the EAE behavioral score. The literature is discrepant on the effects of C3–/– on murine EAE behavioral score. Two groups [13, 41] showed that C3 was required for maximal development of EAE, and C3–/– mice had reduced behavioral scores with a decrease in CNS infiltrating effector T cells. However, another group found no improvement in C3–/– EAE [5]. In our studies of approximately one hundred C3–/– animals (C56/B6J breeder pair originally purchased from Jackson Lab in 2016), we did not see alteration of the disease onset or course in EAE. In addition, we also replicated our findings in a completely different strain of C57/BL6N C3 deficient mice and saw no effect on behavioral score (Fig. S4, online resource). EAE is known to be variable in different centers due to subtle genetic strain differences, as well as environmental factors such as the gut microbiome and seasonality [9, 37]. The observed absence of an effect on EAE priming is advantageous since interventions that affect early EAE will cause a reduction in secondary CNS pathology making it difficult to know what is a primary effect of C3 in the brain and what is a secondary effect of C3 deficiency in the periphery that results in decreased immune cell infiltration. Our data showing neuroprotection, despite no change in the peripheral immune infiltrate, confirm and extend the conclusions in previous studies.
The discordance between the effects on EAE behavioral score, which reflect spinal cord pathology, and the anterior visual pathway pathology are notable. This may be explained by a recent study examining regional expression profiles of astrocytes in EAE in which complement system upregulation was differentially increased in the anterior visual pathway and brain-derived astrocytes as compared to those from the spinal cord [18]. Nonetheless, we did see C3 + reactive gliosis in the spinal cord of EAE mice, however, in EAE there is extensive axonal pathology in the spinal cord that may reflect mechanisms of injury such as axonal transections that are independent of C3.
Our study has some limitations. First, we only examined a small number of post mortem MS and control retinas. While a number of brain banks exist, only a small subset of patients donate their eyes to science and there are very few repositories for MS and even control eye tissue. Nonetheless, the C3 reactive gliosis we observed in MS retinas was significantly greater than in age matched controls and revealed marked regional heterogeneity. Regarding the animal modeling, it is well described that EAE is variable in different veterinary colonies and within different strains. Nonetheless, we reproduced our findings in multiple replicate experiments in both global and conditional KO mice over the course of a number of years. Although genetic reporters have the added advantage of cellular specificity and avoid over-reliance on non-specific antibodies, since our C3-tdTomato reporter did not create a tdT-C3 fusion protein we could not colocalize C3 at a subcellular level and could not track secreted degradation products. Finally, we elected to use constitutive GFAP-Cre mice since inducible systems require the use of tamoxifen, which can possibly suppress EAE or modulate C3 [3, 6]. Constitutive GFAP-Cre may have had unintended recombination during development, but since the majority of the C3 expression in the CNS was in astrocytes such an effect would likely have been minor. Finally, in the GFAP-Cre mice we only observed partial C3 depletion in astrocytes, likely due to inefficient recombination. Nevertheless, we still saw partial neuroprotection relative to Cre- mice similar to what was observed in the global C3 deficient mice relative to wild type mice, supporting our conclusion that astrocytic expression of C3 is an important mediator of neural pathology in the inflamed anterior visual pathway.
C3 has been implicated in a number of other neurodegenerative diseases, including Alzheimer’s and retinal pathologies such as macular degeneration. C3 –/– mice have been shown to be protective in Alzheimer’s Disease animal models [15, 38, 47] and in retinal photoreceptor outer segment atrophy [21]. Alternatively, C3 and C3R deletion were recently shown to be deleterious in a retinitis pigmentosa model in which microglial clearance of apoptotic photoreceptors is a necessary homeostatic function [39]. Further studies are needed to understand the balance between excessive C3 production leading to neuronal demise and its basal functions in tissue health and clearance of debris.
The multiple recent studies implicating C3 reactive astrogliosis in neurodegeneration have led to an increased interest in testing C3 inhibitors. The small molecule C3 inhibitor, APL-2, has already shown potential to reduce age related macular degeneration geographic atrophy (GA) in a phase 2 trial but eculizumab, which targets the downstream molecule C5 failed to halt GA progression in a phase 2 study [20]. These results suggest that the beneficial effects of C3 inhibition in protecting against retinal degeneration may not be related to C3 activation of C5b-C9 membrane attack complex (MAC), but rather that C3b mediates opsonization of synapses and neuronal pathology independently of MAC. Alternatively, it is possible that C3 is not acting solely as a secreted extracellular product, but rather it may also be a signaling molecule in glia, and its depletion could then alter the effector profile of astrocytes in a manner that results in neuroprotection.
In summary, RGC loss is a prominent neurological manifestation of neurodegeneration in MS/EAE and may be an important mediator of progressive disability. Complement component C3 expression is extensive in MS post-mortem retinas and in EAE mouse retina with retinal neuronal loss, suggesting neurotoxic glia are a critical mechanism of injury. Further, we show a beneficial effect of C3 global and astrocyte specific deletion in EAE that occurs independently of peripheral immune cell suppression. Continued exploration of the role of C3 activation and astrocyte activation in MS pathogenesis will shed light on the molecular mechanisms of neurodegeneration in MS and may provide novel strategies to protect neuronal function and treat progressive MS.
References
Antel JP, Becher B, Ludwin SK, Prat A, Quintana FJ (2020) Glial cells as regulators of neuroimmune interactions in the central nervous system. J Immunol 204:251–255. https://doi.org/10.4049/jimmunol.1900908
Barbar L, Jain T, Zimmer M, Kruglikov I, Sadick JS, Wang M, et al (2020) CD49f Is a novel marker of functional and reactive human iPSC-derived astrocytes. Neuron https://doi.org/10.1016/j.neuron.2020.05.014
Bebo BF Jr, Dehghani B, Foster S, Kurniawan A, Lopez FJ, Sherman LS (2009) Treatment with selective estrogen receptor modulators regulates myelin specific T-cells and suppresses experimental autoimmune encephalomyelitis. Glia 57:777–790. https://doi.org/10.1002/glia.20805
Brambilla R (2019) The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol 137:757–783. https://doi.org/10.1007/s00401-019-01980-7
Calida DM, Constantinescu C, Purev E, Zhang GX, Ventura ES, Lavi E et al (2001) Cutting edge: C3, a key component of complement activation, is not required for the development of myelin oligodendrocyte glycoprotein peptide-induced experimental autoimmune encephalomyelitis in mice. J Immunol 166:723–726
Elloso MM, Phiel K, Henderson RA, Harris HA, Adelman SJ (2005) Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J Endocrinol 185:243–252. https://doi.org/10.1677/joe.1.06063
Fitzgerald KC, Kim K, Smith MD, Aston SA, Fioravante N, Rothman AM et al (2019) Early complement genes are associated with visual system degeneration in multiple sclerosis. Brain 142:2722–2736. https://doi.org/10.1093/brain/awz188
Gleichman AJ, Carmichael ST (2020) Glia in neurodegeneration: drivers of disease or along for the ride? Neurobiol Dis 142:104957. https://doi.org/10.1016/j.nbd.2020.104957
Godel C, Kunkel B, Kashani A, Lassmann H, Arumugam M, Krishnamoorthy G (2020) Perturbation of gut microbiota decreases susceptibility but does not modulate ongoing autoimmune neurological disease. J Neuroinflammation 17:79. https://doi.org/10.1186/s12974-020-01766-9
Green AJ, McQuaid S, Hauser SL, Allen IV, Lyness R (2010) Ocular pathology in multiple sclerosis: retinal atrophy and inflammation irrespective of disease duration. Brain 133:1591–1601. https://doi.org/10.1093/brain/awq080
Gregorian C, Nakashima J, Le Belle J, Ohab J, Kim R, Liu A et al (2009) Deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J Neurosci 29:1874–1886. https://doi.org/10.1523/jneurosci.3095-08.2009
Guttenplan KA, Stafford BK, El-Danaf RN, Adler DI, Munch AE, Weigel MK et al (2020) Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Rep 31:107776. https://doi.org/10.1016/j.celrep.2020.107776
Hammond JW, Bellizzi MJ, Ware C, Qiu WQ, Saminathan P, Li H et al (2020) Complement-dependent synapse loss and microgliosis in a mouse model of multiple sclerosis. Brain Behav Immun https://doi.org/10.1016/j.bbi.2020.03.004
Hess C, Kemper C (2016) Complement-mediated regulation of metabolism and basic cellular processes. Immunity 45:240–254. https://doi.org/10.1016/j.immuni.2016.08.003
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. https://doi.org/10.1126/science.aad8373
Horstmann L, Kuehn S, Pedreiturria X, Haak K, Pfarrer C, Dick HB et al (2016) Microglia response in retina and optic nerve in chronic experimental autoimmune encephalomyelitis. J Neuroimmunol 298:32–41. https://doi.org/10.1016/j.jneuroim.2016.06.008
Ingram G, Loveless S, Howell OW, Hakobyan S, Dancey B, Harris CL et al (2014) Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol Commun 2:53. https://doi.org/10.1186/2051-5960-2-53
Itoh N, Itoh Y, Tassoni A, Ren E, Kaito M, Ohno A et al (2018) Cell-specific and region-specific transcriptomics in the multiple sclerosis model: focus on astrocytes. Proc Natl Acad Sci 115:E302–E309. https://doi.org/10.1073/pnas.1716032115
Jin J, Smith MD, Kersbergen CJ, Kam TI, Viswanathan M, Martin K et al (2019) Glial pathology and retinal neurotoxicity in the anterior visual pathway in experimental autoimmune encephalomyelitis. Acta Neuropathol Commun 7:125. https://doi.org/10.1186/s40478-019-0767-6
Kassa E, Ciulla TA, Hussain RM, Dugel PU (2019) Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin Biol Ther 19:335–342. https://doi.org/10.1080/14712598.2019.1575358
Katschke KJ Jr, Xi H, Cox C, Truong T, Malato Y, Lee WP et al (2018) Classical and alternative complement activation on photoreceptor outer segments drives monocyte-dependent retinal atrophy. Sci Rep 8:7348. https://doi.org/10.1038/s41598-018-25557-8
Kolev M, West EE, Kunz N, Chauss D, Moseman EA, Rahman J et al (2020) Diapedesis-induced integrin signaling via LFA-1 facilitates tissue immunity by inducing intrinsic complement C3 expression in immune cells. Immunity 52(513–527):e518. https://doi.org/10.1016/j.immuni.2020.02.006
Larabee CM, Desai S, Agasing A, Georgescu C, Wren JD, Axtell RC et al (2016) Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol Vis 22:1503–1513
Larabee CM, Hu Y, Desai S, Georgescu C, Wren JD, Axtell RC et al (2016) Myelin-specific Th17 cells induce severe relapsing optic neuritis with irreversible loss of retinal ganglion cells in C57BL/6 mice. Mol Vis 22:332–341
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Liu Q, Li H, Yang J, Niu X, Zhao C, Zhao L et al (2017) Valproic acid attenuates inflammation of optic nerve and apoptosis of retinal ganglion cells in a rat model of optic neuritis. Biomed Pharmacother 96:1363–1370. https://doi.org/10.1016/j.biopha.2017.11.066
Lucin KM, Wyss-Coray T (2009) Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64:110–122. https://doi.org/10.1016/j.neuron.2009.08.039
Madore C, Yin Z, Leibowitz J, Butovsky O (2020) Microglia, lifestyle stress, and neurodegeneration. Immunity 52:222–240. https://doi.org/10.1016/j.immuni.2019.12.003
Michailidou I, Naessens DM, Hametner S, Guldenaar W, Kooi EJ, Geurts JJ et al (2017) Complement C3 on microglial clusters in multiple sclerosis occur in chronic but not acute disease: Implication for disease pathogenesis. Glia 65:264–277. https://doi.org/10.1002/glia.23090
Michailidou I, Willems JG, Kooi EJ, van Eden C, Gold SM, Geurts JJ et al (2015) Complement C1q–C3-associated synaptic changes in multiple sclerosis hippocampus. Ann Neurol 77:1007–1026. https://doi.org/10.1002/ana.24398
Nytrova P, Potlukova E, Kemlink D, Woodhall M, Horakova D, Waters P et al (2014) Complement activation in patients with neuromyelitis optica. J Neuroimmunol 274:185–191. https://doi.org/10.1016/j.jneuroim.2014.07.001
Pauly D, Agarwal D, Dana N, Schafer N, Biber J, Wunderlich KA et al (2019) Cell-type-specific complement expression in the healthy and diseased retina. Cell Rep 29(2835–2848):e2834. https://doi.org/10.1016/j.celrep.2019.10.084
Petzold A, Balcer LJ, Calabresi PA, Costello F, Frohman TC, Frohman EM et al (2017) Retinal layer segmentation in multiple sclerosis: a systematic review and meta-analysis. Lancet Neurol 16:797–812. https://doi.org/10.1016/S1474-4422(17)30278-8
Pluvinage JV, Wyss-Coray T (2020) Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat Rev Neurosci 21:93–102. https://doi.org/10.1038/s41583-019-0255-9
Ramaglia V, Dubey M, Malpede MA, Petersen N, de Vries SI, Ahmed SM et al (2021) Complement-associated loss of CA2 inhibitory synapses in the demyelinated hippocampus impairs memory. Acta Neuropathol. https://doi.org/10.1007/s00401-021-02338-8
Ramaglia V, Hughes TR, Donev RM, Ruseva MM, Wu X, Huitinga I et al (2012) C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci 109:965–970. https://doi.org/10.1073/pnas.1111924109
Reynolds JD, Case LK, Krementsov DN, Raza A, Bartiss R, Teuscher C (2017) Modeling month-season of birth as a risk factor in mouse models of chronic disease: from multiple sclerosis to autoimmune encephalomyelitis. FASEB J 31:2709–2719. https://doi.org/10.1096/fj.201700062
Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE et al (2015) Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci 35:13029–13042. https://doi.org/10.1523/JNEUROSCI.1698-15.2015
Silverman SM, Ma W, Wang X, Zhao L, Wong WT (2019) C3- and CR3-dependent microglial clearance protects photoreceptors in retinitis pigmentosa. J Exp Med 216:1925–1943. https://doi.org/10.1084/jem.20190009
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. https://doi.org/10.1016/j.cell.2007.10.036
Szalai AJ, Hu X, Adams JE, Barnum SR (2007) Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol Immunol 44:3132–3136. https://doi.org/10.1016/j.molimm.2007.02.002
Tassoni A, Farkhondeh V, Itoh Y, Itoh N, Sofroniew MV, Voskuhl RR (2019) The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci Rep 9:10010. https://doi.org/10.1038/s41598-019-46232-6
Tenner AJ, Stevens B, Woodruff TM (2018) New tricks for an ancient system: physiological and pathological roles of complement in the CNS. Mol Immunol 102:3–13. https://doi.org/10.1016/j.molimm.2018.06.264
Watkins LM, Neal JW, Loveless S, Michailidou I, Ramaglia V, Rees MI et al (2016) Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflammation 13:161. https://doi.org/10.1186/s12974-016-0611-x
Werneburg S, Jung J, Kunjamma RB, Ha SK, Luciano NJ, Willis CM et al (2020) Targeted complement inhibition at synapses prevents microglial synaptic engulfment and synapse loss in demyelinating disease. Immunity 52(167–182):e167. https://doi.org/10.1016/j.immuni.2019.12.004
Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC (1995) Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A 92:11490–11494. https://doi.org/10.1073/pnas.92.25.11490
Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S et al (2019) Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep 28(2111–2123):e2116. https://doi.org/10.1016/j.celrep.2019.07.060
Yun SP, Kam T-I, Panicker N, Kim S, Oh Y, Park J-S et al (2018) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 24:931–938. https://doi.org/10.1038/s41591-018-0051-5
Acknowledgements
The authors wish to thank Kyle Martin, Arthur Anthony Reyes, and Hannah-Noelle Lord for assistance with scoring EAE mice. The authors also thank the JHU Ross Flow Cytometry Core for use of a Cytek Aurora with a yellow laser to assess tdT expression.
Funding
This study was funded by; R01 NS041435 (PAC), National MS Society (NMSS) grants RG-1907–34756 (PAC) and SI-2004–36590 (DSR), the Division of Intramural Research of the National Institute of Neurological Disorders and Stroke (NINDS) and the National Heart, Lung, and Blood Institute (NHLBI), NIH (ZIANS003119 to DSR and ZIAHL006223 to CK), Fonds de recherche du Québec—Santé (FRQS)# 270746 to MG, and NIH S10OD026859 to JHU.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gharagozloo, M., Smith, M.D., Jin, J. et al. Complement component 3 from astrocytes mediates retinal ganglion cell loss during neuroinflammation. Acta Neuropathol 142, 899–915 (2021). https://doi.org/10.1007/s00401-021-02366-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-021-02366-4