
Abstract
Synucleinopathies are a group of neurodegenerative disorders caused by the misfolding and self-templating of the protein α-synuclein, or the formation of α-synuclein prions. Each disorder differs by age of onset, presenting clinical symptoms, α-synuclein inclusion morphology, and neuropathological distribution. Explaining this disease-specific variability, the strain hypothesis postulates that each prion disease is encoded by a distinct conformation of the misfolded protein, and therefore, each synucleinopathy is caused by a unique α-synuclein structure. This review discusses the current data supporting the role of α-synuclein strains in disease heterogeneity. Several in vitro and in vivo models exist for evaluating strain behavior, however, as the focus of this article is to compare strains across synucleinopathy patients, our discussion predominantly focuses on the two models most commonly used for this purpose: the α-syn140*A53T–YFP cell line and the TgM83+/− mouse model. Here we define each strain based on biochemical stability, ability to propagate in α-syn140–YFP cell lines, and incubation period, inclusion morphology and distribution, and neurological signs in TgM83+/− mice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
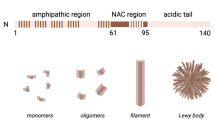
Alpha-synuclein is a 140 amino acid cytoplasmic protein, encoded by the SNCA gene. It is found throughout the body with highest expression in the central nervous system (CNS), where it makes up ~ 1% of all protein expressed in the cytosol [46]. Within the CNS, α-synuclein is predominantly expressed in neurons, with varied reports about expression levels in oligodendrocytes [29]. The precise physiological role of α-synuclein is largely unknown; however, studies suggest that it is involved in the formation of SNARE complexes and neurotransmitter regulation [15, 18, 19]. While α-synuclein is an intrinsically disordered protein, it adopts an α-helical conformation when membrane-bound [16, 18, 31]. Alternatively, the protein can adopt a β-sheet-rich structure, leading to the deposition of pathological aggregates in a group of neurodegenerative diseases called synucleinopathies. Within synucleinopathies, Parkinson’s disease (PD), Parkinson’s disease with dementia (PDD), and dementia with Lewy bodies (DLB) are characterized by α-synuclein aggregating into Lewy bodies (LBs) and Lewy neurites (LNs) in neurons (Fig. 1a) [96]. Alternatively, α-synuclein coalesces into glial cytoplasmic inclusions (GCIs) in oligodendrocytes in multiple system atrophy (MSA; Fig. 1b) [95, 103].

Neuropathological lesions in synucleinopathies. In synucleinopathy patients, misfolded α-synuclein accumulates into neuropathological lesions. a Lewy body pathology is present in patients with PD, PDD, and DLB. b Glial cytoplasmic inclusions are seen in patients with MSA. (Scale bar, 50 µm)
While synucleinopathies are classified as movement disorders, disease heterogeneity extends beyond neuropathological lesions to clinical phenotypes. PD was first described by Dr. James Parkinson in 1817, noting the resting tremor and gait abnormalities he described as a “shaking palsy” [72], and almost a century later, Frederic Lewy identified LBs in the brains of PD patients [33]. PDD and DLB patients share similar neuropathological lesions with PD but differ in diagnosis. Patients presenting with motor impairment followed by dementia are diagnosed with PDD, while those that develop motor deficits concurrently with or after the onset of dementia are diagnosed with DLB. Together, these three diseases are referred to as Lewy body diseases (LBDs). Separate from LBDs, Graham and Oppenheimer introduced the term multiple system atrophy (MSA) to unify the clinical presentations of Shy-Drager syndrome (SDS) [93], striatonigral degeneration (SND) [101], and olivopontocerebellar atrophy (OPCA) [23], which were all characterized by idiopathic orthostatic hypotension and cerebellar ataxia [41]. Lantos and colleagues subsequently defined the GCIs in oligodendrocytes as a hallmark of MSA neuropathology. These lesions were observed across SDS, SND, and OPCA patients, supporting their reclassification into MSA [70]. MSA can be further subdivided into a parkinsonian (MSA-P) or cerebellar (MSA-C) phenotype depending on the predominant symptoms at diagnosis [38, 47]. MSA-P is characterized by rigidity and nystagmus, while MSA-C presents with tremors, gait ataxia, and oculomotor dysfunction [38]. Interestingly, these variations in clinical signs are paired with differences in GCI distribution in the brains of MSA-C and MSA-P patients [1, 56, 70, 71].
Increasing evidence indicates that an underlying cause of disease in synucleinopathy patients is the misfolding and self-templating of the protein α-synuclein, or the formation of α-synuclein prions [39, 48, 78]. The self-templating and spreading of α-synuclein was first observed in human patient samples, leading to additional experimental investigation in rodent models. In the late 1980s, fetal nigral tissue grafts were transplanted into PD patients with the goal of alleviating the deficits caused by the loss of dopaminergic neurons in the substantia nigra [54, 64, 94]. Patients that survived more than 10 years after receiving the grafts developed LB pathology in the fetal tissue [52, 61, 62]. This finding was supported by multiple rodent studies showing a similar spread of α-synuclein pathology to grafted tissue [45, 53], indicating a potential host-to-graft spread of pathogenic protein. Research from multiple investigators has since used cell culture models to establish that α-synuclein propagates from cell-to-cell, supporting the hypothesis that misfolded α-synuclein becomes a prion that self-templates to spread disease [26, 35, 45, 82, 83].
Following the discovery that α-synuclein is linked to both PD [77, 96] and MSA [95, 103], a key area of investigation has focused on understanding the ability of a single protein to cause a spectrum of clinical phenotypes and neuropathologies (Fig. 1). Over the last 5 years, the strain hypothesis has emerged as the leading explanation of the clinical variability among synucleinopathy patients. Across biology, strains have classically been defined by differences in their genetically coded information; viral and bacterial strains are known to differ in pathogenicity and clinical symptoms as a result of variations in their genetic codes. In the absence of a nucleic acid, the prion field grappled with the need to explain the heterogeneity of diseases caused by misfolding of the prion protein (PrP). The field landed on the strain hypothesis, positing that strains are encoded by the conformation of the protein itself, with each strain resulting from a unique structure of misfolded PrP. Although it is not well-understood how these distinct conformations arise, decades of biochemical, biological, and structural data support this hypothesis [7]. This same explanation is now used to understand the clinical heterogeneity and variation in neuropathological lesions seen across synucleinopathy patients. In the following sections, we will discuss the mounting evidence to support those conclusions. While multiple laboratories have utilized a variety of model systems to investigate the role of α-synuclein misfolding in disease, this review will focus on the models that have been used most frequently to compare α-synuclein across diseases.
Cell models
Cell models have facilitated the rapid growth of a body of work investigating the maintenance of α-synuclein strain properties in vitro. Human embryonic kidney (HEK293T) cells stably expressing a YFP-tagged substrate were first used to study propagation of the protein tau [88]. In this approach, a positive cell infection results in the formation of fluorescent puncta, or YFP-positive protein aggregates. The assay was adapted to express full-length wild-type (WT) and A53T-mutated human α-synuclein (α-syn140–YFP and α-syn140*A53T–YFP, respectively) to detect α-synuclein prions isolated from synucleinopathy patient samples [110]. Alpha-synuclein prions isolated from MSA patient samples induced YFP-positive puncta in the α-syn140*A53T–YFP cells (Fig. 2), whereas α-synuclein prions isolated from PD, PDD, and DLB had no effect [110]. Interestingly, the lack of infectivity was not due to differences in the amino acid sequence as all patient samples were WT at position 53. Using a similar biosensor cell line expressing mutant α-synuclein*A53T tagged with CFP and YFP, Marc Diamond’s group demonstrated that detergent-insoluble preparations from PD patient samples induced small YFP-positive inclusions, while detergent-soluble and insoluble fractions from MSA patient samples induced filamentous inclusions throughout the cytoplasm (a summary of α-synuclein infectivity data across cell and mouse experiments is shown in Fig. 3) [111]. Notably, these differences were maintained upon serial passaging in the biosensor cells. Together, these studies suggest that α-synuclein misfolds into distinct strains with unique biochemical and biological properties in MSA and PD patients.

In vitro detection of α-synuclein prions. TgM83+/− mice were inoculated with brain homogenate from one control or two MSA patient samples. MSA-inoculated mice were collected after the onset of neurological signs, while control-inoculated animals were terminated 450-day post inoculation. Frozen brain tissue collected from the mice was homogenized and α-synuclein prions were isolated via phosphotungstic acid precipitation. The resulting protein pellets were incubated with HEK293T cells expressing α-syn140*A53T–YFP for 4 days before collecting live-cell images. Representative images are shown from cells infected with mouse-passaged a control sample or b, c MSA patient samples. (Scale bar, 50 µm)

Distinct α-synuclein strains can be defined by in vivo and in vitro biological activity. Alpha-synuclein prion strains can be defined biologically by two parameters: incubation period in TgM83+/− mouse bioassay and propagation in α-syn140*A53T–YFP or α-syn140*E46K–YFP cells. a TgM83+/+ mice develop spontaneous α-synuclein prions that induce neurological disease in TgM83+/− animals in ~ 210-day post inoculation (dpi) and propagate in α-syn140*A53T–YFP cells. b MSA patient samples transmit disease to TgM83+/− mice in ~ 120 dpi. While the presence of the E46K mutation blocks MSA infection in cells, the A53T mutation supports propagation in vitro. c, d PD and DLB patient samples are unable to transmit disease to TgM83+/− mice over 540 dpi. PD propagation in α-syn140*A53T–YFP cells is preparation-dependent (c), whereas DLB patient samples are unable to propagate regardless of preparation. e PFFs generated de novo induce neurological signs in TgM83+/− with variable incubation periods. They also induce α-synuclein misfolding in both the E46K and A53T-expressing cell lines
To further demonstrate strain differences across synucleinopathy patients, a panel of HEK cell lines expressing α-syn–YFP fusion proteins containing PD-causing mutations was established [106]. The familial E46K mutation, which was identified in a patient cohort from the Basque Country [113], blocked MSA prion replication in vitro (Figs. 3, 4a). Co-expression of the A53T mutation with E46K was unable to rescue this effect, suggesting that α-synuclein misfolds into distinct conformations in MSA and LBD patients [106]. Importantly, WT or mutant (A30P, E46K, or A53T) α-synuclein fibrils infected all cell lines tested, indicating that the observed cell infectivity differences were not due to an inability to induce α-synuclein misfolding in the E46K-expressing cells (Figs. 3, 4c–e). Supporting this finding, Peng et al. used primary oligodendrocytes to propagate detergent-insoluble α-synuclein isolated from PD and MSA patient samples (LB- α-Syn and GCI- α-Syn, respectively) and found that GCI-α-Syn was able to propagate more robustly than LB-α-Syn [75].

a, c–e) Data modified from [106]
Infectivity profiles for α-synuclein strains. Natural and synthetic α-synuclein strains show distinct infectivity profiles across a panel of α-syn140–YFP cell lines. a, b Brain homogenate prepared from one MSA patient and one PD patient sample were used to test infectivity in cells expressing WT or mutant α-synuclein fused to YFP. a The MSA patient sample propagates well in the WT and A53T (T) cell lines, but is unable to propagate in cells expressing the E46K mutation alone (K) or in combination with A53T (KT). b The PD patient sample was unable to infect any of the four cell lines tested. Recombinant protein was used to make (c) WT, (d) E46K, or (e) A53T PFFs. Cells were infected with 25 µg/mL of protein. The WT and A53T PFFs showed a similar preference to propagate in the WT or T cell lines, though were still able to propagate in the K and KT cells. The E46K PFFs exhibited a unique strain profile from the other PFFs, with the most robust propagation in the KT cells.
Recombinant preformed α-synuclein fibrils (PFFs) have been widely used to study strain differences in cell culture models [9, 44, 73, 74]. PFFs created under various conditions were first shown to differ in their ability to cross-seed tau aggregation both in vitro and in vivo [44]. Subsequently, Ronald Melki’s laboratory used different buffer conditions to generate two distinct synthetic α-synuclein polymorphs—fibrils and ribbons—as determined by proteinase K (PK) cleavage pattern, X-ray diffraction, and transmission electron microscopy [9]. The fibrillar α-synuclein was not only more toxic to the neuroblastoma cell line, N2a, but was also able to induce aggregation at a faster rate than the ribbon polymorph. Most recently, Lau et al. created distinct fibrils under salt (S) and no salt (NS) conditions to investigate α-synuclein strain behavior [57]. HEK cells expressing α-syn140*A53T–YFP developed either globular or thread-like aggregates when infected with the S or NS fibrils, respectively (Fig. 3) [57]. Matching this difference in biological activity, the fibrils showed unique biochemical properties, including distinct PK cleavage patterns, resistance to guanidine hydrochloride (GdnHCl) denaturation, and replication kinetics, as monitored by thioflavin T incorporation [57]. Overall, these data support the hypothesis that distinct α-synuclein prion strains, whether natural or synthetic, exhibit differences in biochemical properties that likely contribute to distinctive biological behaviors, such as in vitro propagation kinetics.
Considering these observations, we propose the use of cellular infectivity profiles as one method for defining α-synuclein strains (Fig. 4). The inability of MSA prions to propagate in cells expressing the E46K mutation establishes one important criterion for identifying the MSA strain relative to other natural or synthetic strains. As discussed below, recombinant PFFs can form multiple α-synuclein conformations, which likely encode distinct behavioral differences. To standardize research on α-synuclein strains across laboratories, efforts to use cell-based strain typing to define the strain(s) present in each PFF preparation are warranted. This undertaking would improve the field’s ability to compare findings across laboratories and research models, and better support ongoing drug discovery programs.
Animal models
Data collected by several research groups across multiple animal models have established that α-synuclein prions are pathogenic, transmitting disease or neuropathology following intracerebral (ic) or peripheral inoculation [4, 5, 12, 65, 69, 79, 81, 85, 86, 104, 106, 108, 109]. A variety of transgenic mouse models using CNS-specific promoters to express WT or mutant human α-synuclein have been used to investigate α-synuclein misfolding and spreading in vivo either in oligodendrocytes [50, 92, 112] or neurons [36, 40, 49, 58, 67, 90]. Despite the large number of models developed to study disease progression and etiology, the majority of inoculation studies comparing α-synuclein strain behavior have been conducted in the TgM83 mouse model, which overexpresses human α-synuclein with the A53T mutation [36]. For this reason, the following discussion will briefly touch on studies from other mouse and non-human primate models but will largely focus on studies in the TgM83 mouse.
Non-human primates
While the ease of genetically manipulating mice makes the model system useful for investigating both neuronal development and disease, non-human primates can offer advantages over mouse models. The increased similarity in brain structure between non-human primates and humans can offer a more pertinent model for investigating neurodegenerative disease progression and pathology. For example, there is evidence of dopaminergic neuron loss in the substantia nigra pars compacta (SNpc) paired with age-related increases in α-synuclein deposition in non-human primates [21, 51]. In addition, the cellular environment between humans and non-human primates may be more analogous as primate brains contain more myelination than rodent brains [13, 32, 59, 80]. As a result, primate models have been used to test transmission of α-synuclein prions isolated from synucleinopathy patient samples [81, 91]. Following injection of human LB extracts into rhesus monkeys, Recasens et al. observed nigrostriatal dopaminergic degeneration and α-synuclein pathology in the putamen, SNpc, precentral gyrus, globus pallidus, superior frontal gyrus, and entorhinal area in the temporal cortex [81]. Similar studies in marmosets [91] and cynomolgus monkeys [22] demonstrated that PFF inoculation into the striatum also induces α-synuclein pathology that spreads to the SNpc, causing degeneration of dopaminergic neurons. While both of these studies demonstrate the ability of α-synuclein prions to propagate and spread in a non-human primate host, the relevance of PFFs as a source of α-synuclein prions has come into question in light of structural differences between natural and synthetic α-synuclein strains discussed in following sections. These recent revelations may limit the conclusions that can be drawn about α-synuclein strain behavior from studies using PFFs as inocula. As such, the number and type of human synucleinopathy samples tested should be expanded to fully assess strain-specific α-synuclein propagation in non-human primates.
TgM83 mice
Around 1 year of age, homozygous TgM83+/+ mice develop a spontaneous synucleinopathy characterized by hindlimb paralysis [36]. Transgene expression in these mice is regulated by the neuron-specific Prnp promoter, resulting in neuronal α-synuclein pathology and astrogliosis predominantly within the subcortical midbrain and brainstem. Mougenot et al. first showed that disease onset in these animals is accelerated following ic inoculation of brain homogenate from aged, symptomatic TgM83+/+ mice [69]. This finding was subsequently repeated by others, confirming the observation of α-synuclein prion transmission to the TgM83+/+ model [65]. In contrast, hemizygous TgM83+/− mice do not develop neurological disease after more than 2 years of age, removing the complication that inoculation studies may simply accelerate the onset of spontaneous disease [104]. In 2013, Watts et al. used the hemizygous animals to test ic inoculation of brain homogenate from symptomatic TgM83+/+ mice and reported disease onset ~ 216-days post inoculation (dpi) paired with robust neuronal α-synuclein deposition and astrogliosis in the brainstem (Fig. 3) [104]. Similar to the spontaneous symptoms observed in the TgM83+/+ mice, disease in the TgM83+/+-inoculated hemizygous mice presented as weight loss, ataxia, hindlimb shaking, circling behavior, dysmetria, and proprioceptive deficits. Importantly, inoculation with control samples (either DPBS or tissue from control patients) had no effect on the animals. However, inoculation of brain homogenate from two MSA patient samples induced neurological disease in ~ 120 dpi (Fig. 3). Disease in these animals presented as ataxia, circling, hindlimb paralysis, and bradykinesia. This divergence in incubation periods suggests conformational differences exist in the α-synuclein prion strain present in the two inocula. The ability of the TgM83+/− mice to propagate multiple α-synuclein strains has resulted in its frequent use for in vivo transmission studies.
Subsequent studies comparing disease onset in TgM83+/− mice injected with a variety of inocula have yielded key insights into α-synuclein strain biology. Inoculation of brain homogenate from PD patient samples revealed that PD α-synuclein is unable to induce disease in TgM83+/− mice (Fig. 3) [79]. However, 14 MSA samples from 3 different continents transmitted neurological disease to the mice in ~ 120 dpi. This stark difference in biological activity in vivo was replicated in the cell culture model, as described above [110]. In contrast to natural α-synuclein strains, PFFs have also been used as inocula in TgM83+/− mice, resulting in variable incubation periods spanning from ~ 90 to 330 dpi across laboratories, suggesting preparation-specific differences may induce the formation of distinct α-synuclein strains [4, 5, 12, 85, 86].
A hallmark of classic prion diseases is the ability to induce neurological disease following peripheral exposure. Testing the ability of α-synuclein prions to retain strain properties after non-ic inoculation, Benoit Giasson’s laboratory injected PFFs into the hindlimb muscle of TgM83+/+ and TgM83+/− mice. Inoculated animals developed bilateral foot drop and progressive hindlimb paralysis concomitant with neuronal α-synuclein pathological lesions and astrogliosis within 56 and 129 dpi, respectively [86]. Expanding on this work, the same group then compared hindlimb injections of WT, E46K, H50Q, G51D, and A53E α-synuclein fibrils in TgM83+/− mice [85]. Mice injected with H50Q, G51D, and A53E fibrils developed motor impairment between 100 and 135 dpi, while WT injections produced a longer incubation period of 126–168 dpi. E46K fibrils yielded the greatest variability, with the earliest signs developing at 124 dpi, and 2 of 9 animals remaining asymptomatic > 375 days. While inclusion morphology and distribution were similar across all inocula, the reduced transmission efficiency of WT and E46K PFFs suggest the two sequences favored distinct fibril conformations. This hypothesis was supported by studies comparing WT and E46K PFFs unilaterally inoculated into the sciatic nerve of TgM83+/− mice [5]. All mice injected with WT fibrils were symptomatic by 135 dpi, but the E46K fibrils induced a delayed disease onset ranging from ~ 140 to 330 dpi, with some mice remaining asymptomatic until the experiment was terminated at 365 days.
Building on these observations, recent work from Joel Watts and colleagues described two distinct α-synuclein fibril strains, S and NS, which they used to inoculate TgM83+/− mice (Fig. 3) [57]. Mice injected with S fibrils had a shorter incubation period (~ 142 dpi) and developed hindlimb paralysis and bradykinesia, while NS-injected mice had a longer incubation period (~ 375 dpi) with disease characterized by mild kyphosis, weight loss, and hindlimb shaking. Interestingly, the S- and NS-induced clinical signs and incubation periods mirrored those caused by MSA or TgM83+/+ inoculation, respectively, and were maintained upon serial passage in TgM83+/− mice, a common property of prion strains. Moreover, the morphology and distribution of α-synuclein neuropathology, PK cleavage patterns, and GdnHCl stability profiles varied between mice inoculated with either the S or NS fibril strains. However, these characteristics were similar in S fibril- and MSA-inoculated mice, as well as NS fibril- and TgM83+/+-inoculated mice, suggesting the S and NS fibrils share strain properties with the two natural α-synuclein strains. Finally, Lau et al. used protein misfolding cyclic amplification (PMCA) to create a third PFF strain, which exhibited a distinct cleavage pattern after PK digestion compared to S and NS fibrils. However, inoculation with the PMCA-derived fibrils produced similar results as S fibrils following animal bioassay. These data demonstrate that TgM83+/− mice support propagation of distinct α-synuclein prion strains in vivo, but there may be a limitation to the number of PFF conformations that are efficiently propagated. Alternatively, it is also possible that unlike the S and NS fibrils, PMCA-induced PFFs may contain multiple conformations or sub-strains, and the propagation of a mixture of conformations in the TgM83+/− mice results in strain selection.
One criterion often used to define or characterize prion strains is tropism, or the regional deposition of pathogenic protein in terminal animals [7]. This concept is often referred to in the synucleinopathy literature as selective vulnerability, or the idea that specific cell types and/or brain regions consistently develop neuropathological lesions in patients with a specific disease. Experiments testing peripheral inoculation of MSA patient samples in TgM83+/− mice demonstrated that intralingual, intraperitoneal, and intramuscular injections induced neurological disease in the animals with a lesion profile consistent with mice inoculated ic [107]. Regardless of inoculation route, mice developed extensive α-synuclein pathology in the midbrain and pons, and to a lesser extent in the thalamus and hypothalamus. More recently, stereotactic inoculation studies compared the standard free-hand ic inoculation protocol with stereotactic injections of MSA prions into the hippocampus, thalamus, or hypothalamus of TgM83+/− mice [109]. Remarkably, the neuropathological lesion profile was independent of the inoculation site. Similar to the mice receiving peripheral inoculations, all TgM83+/− mice developed moderate pathology in the thalamus and hypothalamus and robust pathology in the midbrain and pons. Most notably, mice receiving hippocampal stereotactic inoculations did not develop hippocampal pathology. This finding is not due to an inability of the TgM83+/− mice to develop α-synuclein inclusions in the hippocampus. In the comparison of S and NS fibril strains by Lau et al., the authors reported the induction of hippocampal pathology following inoculation of either the NS or TgM83+/+ α-synuclein strains [57]. As mentioned previously, these lesion profiles were maintained upon serial passaging. Combined, these data indicate that α-synuclein prions display strain tropism, which is an important criterion that can be used to differentiate between strains in vivo.
Knockout mouse background
Allelic interference is well documented in transgenic mice expressing both mouse and human prion protein, resulting in the inability to propagate human PrP prion strains when mice also express endogenous Prnp [98, 99]. To avoid these complications in transmission experiments, mouse models with the endogenous gene knocked out are commonly used in combination with overexpression of the human PRNP gene to investigate human PrP prion strains. Taking the known species barrier for PrP into consideration, α-synuclein transmission studies were performed in TgM83+/− mice bred onto an α-synuclein knockout background (Snca0/0) [79]. Notably, incubation periods were unaltered in mice with Snca0/0, Snca0/+, and Snca+/+ backgrounds, eliminating allelic interference as a source of strain selectivity in the TgM83+/− model.
In addition to the TgM83+/− studies, α-synuclein inoculations have been tested in a second group of mouse models expressing human protein on a mouse knockout background. The Tg(SNCA+/+)Snca0/0 mouse model uses an artificial chromosome to express WT human α-synuclein, and lacks expression of endogenous mouse α-synuclein [55]. Using these animals, Bernis et al. found the mice could support propagation of pathogenic α-synuclein from detergent-extracted MSA and probable incidental LBD (iLBD) patient samples [8]. Notably, while both MSA and iLBD induced similar patterns of pathological lesions in the CNS, the mice failed to exhibit clinical signs [8]. Other studies using crude brain homogenate from MSA patients found that ic injections also failed to induce clinical disease up to 1-year post-inoculation in the same mouse model; however, these studies also found an absence of detectable α-synuclein deposition [79, 108].
In addition to the WT-expressing mouse model, mice expressing the A30P and A53T mutations were generated using the artificial chromosome on the Snca0/0 background [55]. While MSA patient samples were unable to propagate in Tg(SNCA*A30P+/+)Snca0/0 mice following ic inoculation, Tg(SNCA*A53T+/+)Snca0/0 mice supported propagation of MSA α-synuclein prions [108]. In the absence of neurological signs consistent with a movement disorder, α-synuclein lesions were present predominantly in glia and some neurons in the hippocampus (HC), piriform cortex (Pir), and parahippocampal cortex (PHC) of the mice. Conservation of strain-specific phenotypes over serial passaging across models or species is a key criterion of prions [20, 34, 102]. After passaging MSA prions in Tg(SNCA*A53T+/+)Snca0/0 mice, brain homogenate from animals positive for α-synuclein prions from two different MSA patients were used to inoculate TgM83+/− mice [108]. The TgM83+/− mice developed neurological signs, consistent with those described previously, accompanied by neuronal α-synuclein lesions and robust astrogliosis in the hindbrain. Interestingly, when the same MSA patient samples were first passaged in the TgM83+/− mice prior to ic inoculation in the Tg(SNCA*A53T+/+)Snca0/0 mice, the animals failed to develop neurological disease after 300 dpi, but did support α-synuclein prion propagation and developed both neuronal and glial pathology in the HC, Pir, and PHC. Data collected across these mouse models suggest that, unlike PrP prions, expression of endogenous mouse protein does not interfere with the propagation of α-synuclein, nor does the presence of mouse α-synuclein alter MSA strain properties.
Structural data
The overarching prion strain hypothesis states that strain properties are encoded by the conformation of the pathogenic protein. While the biological and biochemical data discussed above support the existence of α-synuclein prion strains, the field has only recently been able to generate the structural findings that overwhelmingly support this hypothesis. It has been known for over 20 years that the non-amyloid-β component, or NAC region, is necessary for the aggregation of pathological α-synuclein [30, 37, 66], but recent advances in cryo-electron microscopy (cryo-EM) are only now enabling structural biologists to begin determining the misfolded conformation unique to each disease.
Over the last 5 years, the structures of several misfolded α-synuclein aggregates have been reported using synthetic PFFs derived from either protein fragments [43, 84] or full-length protein [3, 10, 11, 42, 60, 63, 100]. In 2015, Rodriguez et al. demonstrated that aggregates made from an 11-residue segment termed the NACore of α-synuclein are highly cytotoxic to PC12 cells [84]. Micro-electron diffraction analysis of the NACore fragment revealed the α-synuclein fragments aggregated into protein stacks with in-register β-sheet structure forming steric-zipper protofilaments. In 2016, Tuttle et al. reported the first solid-state NMR structure of full-length α-synuclein PFFs, which were capable of inducing neuronal inclusions in rodents [100]. The authors described a parallel, in-register β-sheet structure with residues E46, Q79, K80, I88, A91 and F94 stabilizing a Greek key motif. Similarities to this initial structure, including the salt bridge between residues E46 and K80 as well as aspects of the Greek key conformation, have since been reported by others using cryo-EM to investigate WT PFF [42, 43, 60, 63] or mutant PFF structures [10]. For example, Li et al. used cryo-EM to describe the twister and rod polymorphs found in WT PFF preparations [60]. Both polymorphs contain the Greek key motif and share a common protofilament kernel comprised of residues H50-V77, which interact with the NAC region of α-synuclein. These studies also showed two protofilaments forming in symmetrical assembly around the NAC region; however, the polymorphs differed by the packing of the protofilament kernels. Notably, the authors reported that the familial mutations E46K, H50Q, G51D, and A53E/T, which are known to cause PD, would disrupt the protofilament packing in the rod polymorph, but would not impede packing in the twister polymorph.
More recently, Strohäker et al. used PMCA to amplify α-synuclein prions from PD or MSA patient samples, as well as a variety of in vitro generated polymorphs [97]. Employing multiple methods of analysis, including NMR spectroscopy, fluorescent probes, and electron paramagnetic resonance, the authors demonstrated that brain-derived α-synuclein prions were structurally different than the in vitro polymorphs. While the authors found overlap between the MSA and PD-derived structures, there was greater structural variability across the PD1 and PD2 fibrils. Critically, the patient-derived fibrils showed distinct profiles from the in vitro PFFs by hydrogen–deuterium exchange and fluorescence shift assays using the dyes curcumin, HS-68, and the Congo red-derivative FSB, indicating that synthetic α-synuclein prions are inconsistent with natural strains found in synucleinopathy patients.
Critical strides toward understanding the structural differences between α-synuclein strains were recently made when Schweighauser et al. reported the misfolded conformations of α-synuclein isolated from MSA patient samples determined by cryo-EM [89]. Unlike the conformations reported from PFFs, the two protofilaments in these structures are asymmetrical, forming inclusions made of two different protofilament structures. Intriguingly, despite critical differences when compared to the synthetic PFF structures, aspects of the Greek key motif are present in the MSA conformation. Moreover, α-synuclein folding in these new conformations is again stabilized by a salt bridge between E46 and K80. This finding suggests that in PD patients harboring the E46K mutation, α-synuclein must misfold into a distinct conformation as lysines at residues 46 and 80 would no longer support the salt bridge, ultimately destabilizing the reported structures (Fig. 5). This finding is consistent with the cell assay data demonstrating MSA prions cannot propagate in cells expressing the E46K mutation (Fig. 4) [106]. It also indicates that α-synuclein must misfold into distinct conformations to give rise to MSA and PD or other LBDs. This conclusion is supported by the finding that α-synuclein filaments from patients with DLB are thinner and exhibit less twisting than filaments isolated from MSA patients [89], similar to earlier analyses of fibrils isolated from DLB patient samples [95]. Lastly, unlike the naturally occurring filaments in MSA, most PFFs consist of two identical symmetric protofilaments or a single protofilament, underscoring the structural differences between natural and synthetic α-synuclein prion strains. This observation supports the conclusions by Markus Zweckstetter’s laboratory demonstrating structural differences between the PD- and MSA-derived fibrils and PFFs [97], as well as the biological and biochemical data revealing differences between MSA and PFFs in cell models and protein stability assays [106].

The α-synuclein strain in MSA is incompatible with the E46K PD mutation. a Ribbon overlay of the Cα backbone for the three α-synuclein structures in MSA reported by Schweighauser et al. [89]. PDB structures 6XYO (in yellow), 6XYP (in white), and 6XYQ (in blue) show variability in subunit A but a consistent arrangement of subunit B. Each conformation likely contains two salt bridges between residues E46 and K80, despite differences in geometry. White box identifies this interaction in subunit B in all three conformations, which is shown in (b–d). b Molecular surface from the cryo-EM structure identifying hydrophilic regions in purple and hydrophobic areas in green. c, d Model of the interaction between residues E46 and K80. c In the MSA prion conformation, a salt bridge between residues E46 and K80 stabilize the Greek key in subunit B. d The PD-causing E46K mutation would disrupt the salt bridge with residue K80, destabilizing the MSA conformation
While current data strongly indicate that the misfolded protein conformation is largely responsible for α-synuclein strain properties, the cellular environment may also play a key role in strain maintenance [75]. Within a given cellular environment, multiple cofactors (proteins, lipids, etc.) may be available to interact with α-synuclein. Peng et al. demonstrated the influence cellular environment may have on α-synuclein strain formation by identifying differences in the ability of neurons and oligodendrocytes to support the conversion of pathogenic α-synuclein [75]. Similarly, work from Surachai Supattapone’s laboratory has aimed to elucidate the contribution of cofactors in prion strain maintenance and infectivity [14, 24, 25, 68]. This body of work suggests that cofactor utilization may occur in a strain-dependent and species-dependent manner [24, 25]. The cryo-EM structures of α-synuclein isolated from MSA patient samples revealed the presence of a non-proteinaceous density located in between the two protofilaments [89]. While the makeup of the density is still unknown, it is possible that this non-protein molecule may play an important role in strain formation and/or maintenance as a cofactor. At this time, it is unknown if a similar density is present in misfolded α-synuclein isolated from LBD patient samples, but none of the reported PFF structures share this feature with MSA.
As additional structures of α-synuclein fibrils have been reported, the data have increasingly identified critical differences between bona fide α-synuclein prions and recombinant PFFs. This insight should also challenge how the field approaches biochemical analysis of strains. Currently, it is standard in the PrP prion field to use resistance to denaturation and degradation as an indirect measurement of protein conformation, despite the lack of a clearly defined relationship between the two. It has been hypothesized that aggregate size and ease of fracturing may be responsible for determining incubation periods of disease. For example, the conformational stability of murine PrP prion strains correlates with incubation period, but an inverse relationship exists for hamster and human PrP prion strains [6, 17, 76, 87]. The conflicting data regarding stability and incubation period suggest that resistance to denaturation is not measuring the disaggregation propensity of any given strain. As a result, conformational stability assays may be useful as strain “fingerprinting,” but protease cleavage patterns are a more direct measurement of the structural differences between strains. Therefore, to better bridge biological and structural investigations of α-synuclein strains, a universal method for defining α-synuclein conformations via specific protease cleavage patterns is needed.
Conclusion
The data discussed in this review provide evidence of α-synuclein strains as the source of heterogeneity observed among synucleinopathy patients. To define α-synuclein strains, we evaluated differences in biochemical stability and biological activity, which included cell infectivity profiles and incubation period, pathological lesion tropism, and neurological signs in transgenic mice. An important caveat is that data have been reported using multiple animal and cellular model systems, creating a challenge for interpreting strain differences across studies. The use of multiple models to investigate α-synuclein strains muddles the interpretation of data that may actually be derived from differences in transgene expression profile or off-target effects of transgene insertion, and the role of these differences in altering the propagation or distribution of pathogenic α-synuclein strains in vivo. In addition, interpreting data collected using PFFs versus α-synuclein isolated from synucleinopathy patient samples increases variability across strain studies. How do we interpret an ever-expanding body of data when we cannot directly compare results? What are the relevant forms of α-synuclein prions to investigate as inoculum in transmission experiments? Do SNCA overexpression models lead to the spontaneous formation of α-synuclein prion strains that more closely mimic patient-derived α-synuclein prions or PFFs? Should PFFs be used to answer questions about strain differences if the structures do not exist in human disease? Understanding the source of α-synuclein strain differences requires investigators to collaborate within model systems to enable the data comparisons needed to drive the field towards the universal cell and animal models most appropriate for the widespread study of synucleinopathies.
The data generated by Lau et al. utilizing both PFFs and bona fide α-synuclein prions suggests that there may be overlap in synthetic and natural α-synuclein conformations, but that overlap is preparation-dependent [57]. While the synthetic strains generated under S or NS conditions appear similar to natural α-synuclein prion strains, there may be subtle differences beyond the resolution of current experimental models and assays that are important to understanding α-synuclein strain biology. There is also the possibility that using multiple methods for producing PFFs induces the formation of multiple α-synuclein conformations. Lau et al. reported two protocols for generating homogeneous S and NS fibrils, but variability within other preparations and between laboratories may add to the difficulty of interpreting or repeating findings across research groups. It is currently unknown how often the same structures are created synthetically because the same biochemical criteria, as well as infection studies, are not performed within a single model system.
As the field begins to narrow in on a universal model to investigate α-synuclein strain biology, debate has focused on the use of WT or Tg mice for inoculation studies. PFFs made from either the mouse or human protein sequence induce phosphorylated α-synuclein pathology in WT mice, but the animals do not develop neurological disease [27, 79, 86]. Additionally, the inability of MSA patient samples to transmit either pathology or neurological signs to WT mice has been used as an argument to suggest that α-synuclein does not form a bona fide prion [105]. However, it is important to view these findings in the greater context of prion diseases. As previously mentioned, species barriers, which arise from differences in the primary amino acid sequence, prevent human prion strains from transmitting to WT mice. While some animal strains, such as scrapie, have been adapted to propagate efficiently in mice, propagation of human prion diseases, such as Creutzfeldt-Jakob disease (CJD), are not supported in WT mice [28, 98]. Instead, expression of human PRNP is required for efficient CJD transmission [98, 99]. Notably, even in many PRNP-expressing models, the PrP prion strain in Gerstmann-Sträussler-Scheinker syndrome patients fails to propagate well [2]. In this context, it is unsurprising that the human α-synuclein prions in MSA patient samples require SNCA expression to support transmission in vivo. It is, therefore, important for the field to consider which Tg models are most useful for investigating the biology of human-derived α-synuclein prion strains.
Further complicating this debate, the cellular environment may act as a selective pressure in favor of one conformation over another. This selection creates the potential for conformations that evade disease-modifying therapeutics and diagnostics if these tools are developed for conformation-specific targets. An additional challenge is the possibility that α-synuclein may utilize cofactors in a species-dependent manner, which could alter the conformation, and, therefore, the strain, that is observed as the outcome in animal bioassay experiments. Notably, the non-proteinaceous density present in the reported cryo-EM structures of MSA α-synuclein supports the idea that cofactors may be supporting protein misfolding or propagation [89]. This possibility should be considered as a potential explanation for the failure of many drug trials after observing success in mouse models of disease.
Synucleinopathies exist as a spectrum of neurodegenerative disorders that differ in their age of onset, clinical presentation, and neuropathology. Increasing evidence indicates that an underlying cause of this heterogeneity is the presence of distinct α-synuclein prion strains in patient samples. Alpha-synuclein isolated from MSA and LBD samples has repeatedly been shown to exhibit differences in biological activity in cultured cells and animal models. These differences are underscored by unique biochemical properties, including resistance to protease degradation and denaturation by chaotropic agents. Recent advances in structural biology methods are now pairing these observations with conformationally distinct structures of misfolded α-synuclein. Understanding the relationship between the conformational differences and biological activity of each α-synuclein strain is central to developing successful disease-specific diagnostics and therapeutics for synucleinopathy patients.
References
Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T et al (2012) Review: the neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol 38:4–24. https://doi.org/10.1111/j.1365-2990.2011.01234.x
Asante EA, Grimshaw A, Smidak M, Jakubcova T, Tomlinson A, Jeelani A et al (2015) Transmission properties of human PrP 102L prions challenge the relevance of mouse models of GSS. PLoS Pathog 11:e1004953. https://doi.org/10.1371/journal.ppat.1004953
Atsmon-Raz Y, Miller Y (2015) A proposed atomic structure of the self-assembly of the non-amyloid-β component of human α-synuclein as derived by computational tools. J Phys Chem B 119:10005–10015. https://doi.org/10.1021/acs.jpcb.5b03760
Ayers JI, Brooks MM, Rutherford NJ, Howard JK, Sorrentino ZA, Riffe CJ et al (2017) Robust central nervous system pathology in transgenic mice following peripheral injection of alpha-synuclein fibrils. J Virol. https://doi.org/10.1128/JVI.02095-16
Ayers JI, Riffe CJ, Sorrentino ZA, Diamond J, Fagerli E, Brooks M et al (2018) Localized induction of wild-type and mutant alpha-synuclein aggregation reveals propagation along neuroanatomical tracts. J Virol. https://doi.org/10.1128/JVI.00586-18
Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC (2011) The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog 7:e1001317. https://doi.org/10.1371/journal.ppat.1001317
Bartz JC (2017) Prion strain diversity. In: Prusiner SB (ed) Prion diseases. Cold Spring Harbor Perspect. Med. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 31–44. https://doi.org/10.1101/cshperspect.a024349
Bernis ME, Babila JT, Breid S, Wüsten KA, Wüllner U, Tamgüney G (2015) Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun 3:75. https://doi.org/10.1186/s40478-015-0254-7
Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R (2013) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4:2575. https://doi.org/10.1038/ncomms3575
Boyer DR, Li B, Sun C, Fan W, Sawaya MR, Jiang L, Eisenberg DS (2019) Structures of fibrils formed by alpha-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat Struct Mol Biol 26:1044–1052. https://doi.org/10.1038/s41594-019-0322-y
Boyer DR, Li B, Sun C, Fan W, Zhou K, Hughes MP, Sawaya MR, Jiang L, Eisenberg DS (2020) The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc Natl Acad Sci USA 117:3592–3602. https://doi.org/10.1073/pnas.1917914117
Breid S, Bernis ME, Babila JT, Garza MC, Wille H, Tamgüney G (2016) Neuroinvasion of α-synuclein prionoids after intraperitoneal and intraglossal inoculation. J Virol 90:9182–9193. https://doi.org/10.1128/JVI.01399-16
Brosamle C, Schwab ME (2000) Ipsilateral, ventral corticospinal tract of the adult rat: ultrastructure, myelination and synaptic connections. J Neurocytol 29:499–507. https://doi.org/10.1023/a:1007297712821
Burke CM, Walsh DJ, Steele AD, Agrimi U, Di Bari MA, Watts JC, Supattapone S (2019) Full restoration of specific infectivity and strain properties from pure mammalian prion protein. PLoS Pathog 15:e1007662. https://doi.org/10.1371/journal.ppat.1007662
Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329:1663–1667. https://doi.org/10.1126/science.1195227
Bussell R Jr, Ramlall TF, Eliezer D (2005) Helix periodicity, topology, and dynamics of membrane-associated alpha-synuclein. Protein Sci 14:862–872. https://doi.org/10.1110/ps.041255905
Cescatti M, Saverioni D, Capellari S, Tagliavini F, Kitamoto T, Ironside J et al (2016) Analysis of conformational stability of abnormal prion protein aggregates across the spectrum of Creutzfeldt-Jakob disease prions. J Virol 90:6244–6254. https://doi.org/10.1128/JVI.00144-16
Chandra S, Chen X, Rizo J, Jahn R, Südhof TC (2003) A broken α-helix in folded α-synuclein. J Biol Chem 278:15313–15318. https://doi.org/10.1074/jbc.M213128200
Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC (2005) α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123:383–396. https://doi.org/10.1016/j.cell.2005.09.028
Chien P, Weissman JS (2001) Conformational diversity in a yeast prion dictates its seeding specificity. Nature 410:223–227. https://doi.org/10.1038/35065632
Chu Y, Kordower JH (2007) Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson's disease? Neurobiol Dis 25:134–149. https://doi.org/10.1016/j.nbd.2006.08.021
Chu Y, Muller S, Tavares A, Barret O, Alagille D, Seibyl J et al (2019) Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain 142:3565–3579. https://doi.org/10.1093/brain/awz296
Dejerine JJ, Thomas A (1900) L’atrophie olivo-ponto-cérébelleuse. Nouvelle iconographie de la Salpêtrière 13:330–370
Deleault NR, Kascsak R, Geoghegan JC, Supattapone S (2010) Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49:3928–3934. https://doi.org/10.1021/bi100370b
Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J et al (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci USA 109:E1938–1946. https://doi.org/10.1073/pnas.1206999109
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L et al (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 106:13010–13015. https://doi.org/10.1073/pnas.0903691106
Dhillon J-S, Trejo-Lopez JA, Riffe C, Levites Y, Sacino AN, Borchelt DR et al (2019) Comparative analyses of the in vivo induction and transmission of α-synuclein pathology in transgenic mice by MSA brain lysate and recombinant α-synuclein fibrils. Acta Neuropathol Commun 7:80. https://doi.org/10.1186/s40478-019-0733-3
Dickinson AG (1976) Scrapie in sheep and goats. In: Kimberlin RH (ed) Slow virus diseases of animals and man. North-Holland Publishing Company, Amsterdam, pp 209–241
Djelloul M, Holmqvist S, Boza-Serrano A, Azevedo C, Yeung MS, Goldwurm S et al (2015) Alpha-synuclein expression in the oligodendrocyte lineage: an in vitro and in vivo study using rodent and human models. Stem Cell Rep 5:174–184. https://doi.org/10.1016/j.stemcr.2015.07.002
El-Agnaf OM, Jakes R, Curran MD, Middleton D, Ingenito R, Bianchi E et al (1998) Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett 440:71–75. https://doi.org/10.1016/s0014-5793(98)01418-5
Eliezer D, Kutluay E, Bussell R Jr, Browne G (2001) Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol 307:1061–1073. https://doi.org/10.1006/jmbi.2001.4538
Firmin L, Field P, Maier MA, Kraskov A, Kirkwood PA, Nakajima K et al (2014) Axon diameters and conduction velocities in the macaque pyramidal tract. J Neurophysiol 112:1229–1240. https://doi.org/10.1152/jn.00720.2013
Forster E, Lewy FH (1912) Paralysis agitans. In: Lewandowsky M (ed) Pathologische Anatomie. Handbuch der Neurologie. Springer, Berlin, pp 920–933
Fraser H, Dickinson AG (1967) Distribution of experimentally induced scrapie lesions in the brain. Nature 216:1310–1311. https://doi.org/10.1038/2161310a0
Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y et al (2012) Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol 72:517–524. https://doi.org/10.1002/ana.23747
Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002) Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 34:521–533. https://doi.org/10.1016/s0896-6273(02)00682-7
Giasson BI, Murray IV, Trojanowski JQ, Lee VM (2001) A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem 276:2380–2386. https://doi.org/10.1074/jbc.M008919200
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ et al (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71:670–676. https://doi.org/10.1212/01.wnl.0000324625.00404.15
Goedert M (2015) Alzheimer's and Parkinson's diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349:1255555. https://doi.org/10.1126/science.1255555
Gomez-Isla T, Irizarry MC, Mariash A, Cheung B, Soto O, Schrump S et al (2003) Motor dysfunction and gliosis with preserved dopaminergic markers in human α-synuclein A30P transgenic mice. Neurobiol Aging 24:245–258. https://doi.org/10.1016/s0197-4580(02)00091-x
Graham JG, Oppenheimer DR (1969) Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry 32:28–34. https://doi.org/10.1136/jnnp.32.1.28
Guerrero-Ferreira R, Taylor NM, Arteni AA, Kumari P, Mona D, Ringler P et al (2019) Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife. https://doi.org/10.7554/eLife.48907
Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R et al (2018) Cryo-EM structure of alpha-synuclein fibrils. eLife 7:e36402. https://doi.org/10.7554/eLife.36402
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B et al (2013) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. https://doi.org/10.1016/j.cell.2013.05.057
Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G et al (2011) alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Investig 121:715–725. https://doi.org/10.1172/JCI43366
Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA et al (1995) The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14:467–475. https://doi.org/10.1016/0896-6273(95)90302-x
Jellinger KA, Seppi K, Wenning GK (2005) Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Mov Disord 20:S29–S36. https://doi.org/10.1002/mds.20537
Jucker M, Walker LC (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21:1341–1349. https://doi.org/10.1038/s41593-018-0238-6
Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A et al (2000) Subcellular localization of wild-type and Parkinson’s disease-associated mutant α-synuclein in human and transgenic mouse brain. J Neurosci 20:6365–6373
Kahle PJ, Neumann M, Ozmen L, Müller V, Jacobsen H, Spooren W et al (2002) Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep 3:583–588. https://doi.org/10.1093/embo-reports/kvf109
Kanaan NM, Kordower JH, Collier TJ (2007) Age-related accumulation of Marinesco bodies and lipofuscin in rhesus monkey midbrain dopamine neurons: relevance to selective neuronal vulnerability. J Comp Neurol 502:683–700. https://doi.org/10.1002/cne.21333
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 14:504–506. https://doi.org/10.1038/nm1747
Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L et al (2011) Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis 43:552–557. https://doi.org/10.1016/j.nbd.2011.05.001
Kordower JH, Freeman TB, Snow BJ, Vingerhoets FJ, Mufson EJ, Sanberg PR et al (1995) Neuropathological evidence of graft survival and striatal reinnervation after the transplantation of fetal mesencephalic tissue in a patient with Parkinson's disease. N Engl J Med 332:1118–1124. https://doi.org/10.1056/NEJM199504273321702
Kuo YM, Li Z, Jiao Y, Gaborit N, Pani AK, Orrison BM et al (2010) Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum Mol Genet 19:1633–1650. https://doi.org/10.1093/hmg/ddq038
Kuzdas-Wood D, Stefanova N, Jellinger KA, Seppi K, Schlossmacher MG, Poewe W et al (2014) Towards translational therapies for multiple system atrophy. Prog Neurobiol 118:19–35. https://doi.org/10.1016/j.pneurobio.2014.02.007
Lau A, So RWL, Lau HHC, Sang JC, Ruiz-Riquelme A, Fleck SC et al (2019) α-Synuclein strains target distinct brain regions and cell types. Nat Neurosci 23:21–31. https://doi.org/10.1038/s41593-019-0541-x
Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS et al (2002) Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 –%3e Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA 99:8968–8973. https://doi.org/10.1073/pnas.132197599
Leenen LP, Meek J, Posthuma PR, Nieuwenhuys R (1985) A detailed morphometrical analysis of the pyramidal tract of the rat. Brain Res 359:65–80. https://doi.org/10.1016/0006-8993(85)91413-1
Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G et al (2018) Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun 9:3609. https://doi.org/10.1038/s41467-018-05971-2
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ et al (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 14:501–503. https://doi.org/10.1038/nm1746
Li W, Englund E, Widner H, Mattsson B, van Westen D, Latt J et al (2016) Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci USA 113:6544–6549. https://doi.org/10.1073/pnas.1605245113
Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z et al (2018) Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res 28:897–903. https://doi.org/10.1038/s41422-018-0075-x
Lindvall O, Widner H, Rehncrona S, Brundin P, Odin P, Gustavii B et al (1992) Transplantation of fetal dopamine neurons in Parkinson's disease: one-year clinical and neurophysiological observations in two patients with putaminal implants. Ann Neurol 31:155–165. https://doi.org/10.1002/ana.410310206
Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VMY (2012) Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 209:975–986. https://doi.org/10.1084/jem.20112457
Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR et al (2009) Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci USA 106:20051–20056. https://doi.org/10.1073/pnas.0908005106
Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A et al (2000) Dopaminergic loss and inclusion body formation in a-synuclein mice: implications for neurodegenerative disorders. Science 287:1265–1269. https://doi.org/10.1126/science.287.5456.1265
Miller MB, Wang DW, Wang F, Noble GP, Ma J, Woods VL Jr et al (2013) Cofactor molecules induce structural transformation during infectious prion formation. Structure 21:2061–2068. https://doi.org/10.1016/j.str.2013.08.025
Mougenot A-L, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L et al (2012) Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 33:2225–2228. https://doi.org/10.1016/j.neurobiolaging.2011.06.022
Papp MI, Kahn JE, Lantos PL (1989) Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 94:79–100. https://doi.org/10.1016/0022-510x(89)90219-0
Papp MI, Lantos PL (1992) Accumulation of tubular structures in oligodendroglial and neuronal cells as the basic alteration in multiple system atrophy. J Neurol Sci 107:172–182. https://doi.org/10.1016/0022-510x(92)90286-t
Parkinson J (1817) An essay on the shaking palsy. Sherwood, Neely, and Jones, London
Peelaerts W, Baekelandt V (2016) α-Synuclein strains and the variable pathologies of synucleinopathies. J Neurochem 139:256–274. https://doi.org/10.1111/jnc.13595
Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M et al (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. https://doi.org/10.1038/nature14547
Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL et al (2018) Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 557:558–563. https://doi.org/10.1038/s41586-018-0104-4
Peretz D, Scott M, Groth D, Williamson A, Burton D, Cohen FE et al (2001) Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 10:854–863. https://doi.org/10.1110/ps.39201
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. https://doi.org/10.1126/science.276.5321.2045
Prusiner SB (2017) An introduction to prion biology. In: Prusiner SB (ed) Prion biology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 1–15
Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB et al (2015) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA 112:E5308–E5317. https://doi.org/10.1073/pnas.1514475112
Ralston DD, Milroy AM, Ralston HJ 3rd (1987) Non-myelinated axons are rare in the medullary pyramids of the macaque monkey. Neurosci Lett 73:215–219. https://doi.org/10.1016/0304-3940(87)90247-3
Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A et al (2014) Lewy body extracts from Parkinson's disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362. https://doi.org/10.1002/ana.24066
Reyes JF, Olsson TT, Lamberts JT, Devine MJ, Kunath T et al (2015) A cell culture model for monitoring α-synuclein cell-to-cell transfer. Neurobiol Dis 77:266–275. https://doi.org/10.1016/j.nbd.2014.07.003
Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E (2014) Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62:387–398. https://doi.org/10.1002/glia.22611
Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D et al (2015) Structure of the toxic core of α-synuclein from invisible crystals. Nature 525:486–490. https://doi.org/10.1038/nature15368
Rutherford NJ, Dhillon JS, Riffe CJ, Howard JK, Brooks M, Giasson BI (2017) Comparison of the in vivo induction and transmission of alpha-synuclein pathology by mutant alpha-synuclein fibril seeds in transgenic mice. Hum Mol Genet 26:4906–4915. https://doi.org/10.1093/hmg/ddx371
Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW et al (2014) Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci USA 111:10732–10737. https://doi.org/10.1073/pnas.1321785111
Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M et al (1998) Eight prion strains have PrPSc molecules with different conformations. Nat Med 4:1157–1165. https://doi.org/10.1038/2654
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. https://doi.org/10.1016/j.neuron.2014.04.047
Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B et al (2020) Structures of α-synuclein fiaments from multiple system atrophy. bioRxiv. https://doi.org/10.1101/2020.02.05.935619
Sharon R, Bar-Joseph I, Mirick GE, Serhan CN, Selkoe DJ (2003) Altered fatty acid composition of dopaminergic neurons expressing α-synuclein and human brains with α-synucleinopathies. J Biol Chem 278:49874–49881. https://doi.org/10.1074/jbc.M309127200
Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M et al (2017) Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol Commun 5:12. https://doi.org/10.1186/s40478-017-0413-0
Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G et al (2005) Neurological and neurodegenerative alterations in a transgenic mouse model expressing human α-synuclein under oligodendrocyte promoter: Implications for multiple system atrophy. J Neurosci 25:10689–10699. https://doi.org/10.1523/JNEUROSCI.3527-05.2005
Shy GM, Drager GA (1960) A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study. Arch Neurol 2:511–527
Spencer DD, Robbins RJ, Naftolin F, Marek KL, Vollmer T, Leranth C et al (1992) Unilateral transplantation of human fetal mesencephalic tissue into the caudate nucleus of patients with Parkinson's disease. N Engl J Med 327:1541–1548. https://doi.org/10.1056/NEJM199211263272201
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett 251:205–208. https://doi.org/10.1016/s0304-3940(98)00504-7
Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M (1997) α-Synuclein in Lewy bodies. Nature 388:839–840. https://doi.org/10.1038/42166
Strohaker T, Jung BC, Liou SH, Fernandez CO, Riedel D, Becker S et al (2019) Structural heterogeneity of alpha-synuclein fibrils amplified from patient brain extracts. Nat Commun 10:5535. https://doi.org/10.1038/s41467-019-13564-w
Telling GC, Scott M, Hsiao KK, Foster D, Yang S-L, Torchia M et al (1994) Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA 91:9936–9940. https://doi.org/10.1073/pnas.91.21.9936
Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE et al (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83:79–90. https://doi.org/10.1016/0092-8674(95)90236-8
Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD et al (2016) Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol 23:409–415. https://doi.org/10.1038/nsmb.3194
van der Eecken H, Adams RD, van Bogaert L (1960) Striopallidal-nigral degeneration. An hitherto undescribed lesion in paralysis agitans. J Neuropathol Exp Neurol 19:159–161
Verges KJ, Smith MH, Toyama BH, Weissman JS (2011) Strain conformation, primary structure and the propagation of the yeast prion [PSI+]. Nat Struct Mol Biol 18:493–499. https://doi.org/10.1038/nsmb.2030
Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H (1998) α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 249:180–182. https://doi.org/10.1016/s0304-3940(98)00407-8
Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM et al (2013) Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci USA 110:19555–19560. https://doi.org/10.1073/pnas.1318268110
Wenning G, Trojanowski JQ, Kaufmann H, Wisniewski T, Rocca WA, Low PA (2018) Is multiple system atrophy an infectious disease? Ann Neurol 83:10–12. https://doi.org/10.1002/ana.25132
Woerman AL, Kazmi SA, Patel S, Aoyagi A, Oehler A, Widjaja K et al (2018) Familial Parkinson’s point mutation abolishes multiple system atrophy prion replication. Proc Natl Acad Sci USA 115:409–414. https://doi.org/10.1073/pnas.1719369115
Woerman AL, Kazmi SA, Patel S, Freyman Y, Oehler A, Aoyagi A et al (2018) MSA prions exhibit remarkable stability and resistance to inactivation. Acta Neuropathol 135:49–63. https://doi.org/10.1007/s00401-017-1762-2
Woerman AL, Oehler A, Kazmi SA, Lee J, Halliday GM, Middleton LT et al (2019) Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol 137:437–454. https://doi.org/10.1007/s00401-019-01959-4
Woerman AL, Patel S, Kazmi SA, Oehler A, Lee J, Mordes DA et al (2020) Kinetics of α-synuclein prions preceding neuropathological inclusions in multiple system atrophy. PLoS Pathog 16:e1008222. https://doi.org/10.1371/journal.ppat.1008222
Woerman AL, Stöhr J, Aoyagi A, Rampersaud R, Krejciova Z, Watts JC et al (2015) Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci USA 112:E4949–E4958. https://doi.org/10.1073/pnas.1513426112
Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song E-S et al (2019) Parkinson's disease and multiple system atrophy have distinct α-synuclein seed characteristics. J Biol Chem 294:1045–1058. https://doi.org/10.1074/jbc.RA118.004471
Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K et al (2005) Mouse model of multiple system atrophy α-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45:847–859. https://doi.org/10.1016/j.neuron.2005.01.032
Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I et al (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. https://doi.org/10.1002/ana.10795
Acknowledgements
We thank Steven H. Olson for his assistance with modeling the E46K mutation and Sarah Pyle for her graphic design work. This work was supported by the University of Massachusetts, Amherst.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Holec, S.A.M., Woerman, A.L. Evidence of distinct α-synuclein strains underlying disease heterogeneity. Acta Neuropathol 142, 73–86 (2021). https://doi.org/10.1007/s00401-020-02163-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-020-02163-5