
Abstract
Low-level somatic mutations have been shown to be the major genetic etiology of intractable epilepsy. The extents thereof, however, have yet to be systematically and accurately explored in a large cohort of resected epilepsy brain tissues. Moreover, clinically useful and precise analysis tools for detecting low-level somatic mutations from unmatched formalin-fixed paraffin-embedded (FFPE) brain samples, the most clinically relevant samples, are still lacking. In total, 446 tissues samples from 232 intractable epilepsy patients with various brain pathologies were analyzed using deep sequencing (average read depth, 1112x) of known epilepsy-related genes (up to 28 genes) followed by confirmatory site-specific amplicon sequencing. Pathogenic mutations were discovered in 31.9% (74 of 232) of the resected epilepsy brain tissues and were recurrently found in only eight major focal epilepsy genes, including AKT3, DEPDC5, MTOR, PIK3CA, TSC1, TSC2, SCL35A2, and BRAF. Somatic mutations, two-hit mutations, and germline mutations accounted for 22.0% (51), 0.9% (2), and 9.1% (21) of the patients with intractable epilepsy, respectively. The majority of pathogenic somatic mutations (62.3%, 33 of 53) had a low variant allelic frequency of less than 5%. The use of deep sequencing replicates in the eight major focal epilepsy genes robustly increased PPVs to 50–100% and sensitivities to 71–100%. In an independent FCDII cohort of only unmatched FFPE brain tissues, deep sequencing replicates in the eight major focal epilepsy genes identified pathogenic somatic mutations in 33.3% (5 of 15) of FCDII individuals (similar to the genetic detecting rate in the entire FCDII cohort) without any false-positive calls. Deep sequencing replicates of major focal epilepsy genes in unmatched FFPE brain tissues can be used to accurately and efficiently detect low-level somatic mutations, thereby improving overall patient care by enriching genetic counseling and informing treatment decisions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a neurological disorder common in children and is characterized by recurrent seizure. Approximately, 30% of children with epilepsy fail to achieve seizure control despite adequate treatment with anti-epileptic drugs and are diagnosed with intractable epilepsy [1]. With the advent of next-generation sequencing (NGS), genetic causes underlying intractable epilepsy have been identified [2]. In particular, increasing evidence has indicated that low-level somatic mutations in the affected brain are a major genetic etiology of intractable epilepsy [3,4,5,6]. We and other study groups have reported that somatic mutations in mTOR pathway genes (AKT3, DEPDC5, MTOR, PIK3CA, TSC1, and TSC2) or SLC35A2 are the major genetic causes of hemimegalencephaly (HME), focal cortical dysplasia (FCD), and epilepsy with no lesion [3,4,5,6,7,8,9,10,11]. Meanwhile, BRAF V600E somatic mutation, which is frequently found in ganglioglioma, has been found to contribute to intrinsic epileptogenesis in pediatric brain tumors [12]. Nevertheless, while previous studies have implicated brain somatic mutations with low variant allelic frequency as causative of intractable epilepsy, until now, genetic studies with large cohorts of intractable epilepsy patients with matched brain–peripheral samples are lacking.
Interestingly, low-level somatic mutations with a mutational burden (i.e., variant allelic frequency, VAF) of merely 1% in the affected brain have been shown to be sufficient to cause intractable epilepsy, suggesting that high read depth sequencing (e.g., ~ 1000 × read depth) is required to determine how mutations affect epileptic disorders [4, 5]. Unfortunately, however, the relatively low read depth of conventional NGS, false-positive variants produced by high read depth sequencing, and a lack of research with clinically relevant brain tissues (e.g., formalin fixed paraffin embedded [FFPE] tissues) have precluded clinicians from outlining the impact of somatic mutations on intractable epilepsy and from performing genetic detection thereof.
In the current study, we aimed to document the contribution of somatic mutations in known focal epilepsy-related genes, as well as germline mutations, in a large cohort of intractable epilepsy patients with matched brain–peripheral tissues and with unmatched brain tissues (446 tissues samples from 232 patients with intractable epilepsy). Also, we sought to examine the accuracy of genetic detection of low-level somatic mutations under various tissue conditions: matched brain–peripheral samples or unmatched brain-only samples and freshly frozen (FFZ) or FFPE brain samples. This was undertaken to devise an efficient genetic analysis method of use in the most clinically relevant condition: unmatched FFPE brain samples.
Methods
Subject ascertainment
Children with epileptic disorder who had undergone epilepsy surgery since 2004 at Severance Children’s Hospital were identified. Prior to surgery, patients underwent extensive evaluations with video electroencephalography (EEG) monitoring, high-resolution MRI, fluorodeoxyglucose (FDG)-PET, and subtraction ictal single-photon emission computed tomography (SPECT) co-registered to MRI (SISCOM) to localize anatomic lesions. We excluded patients with a history of trauma, ischemic injury, or intracranial hemorrhage. Pre-surgical evaluation and surgical intervention procedures have been described in a previous article [13]. The resection margin for primary seizure foci was defined according to the following: (1) focal lesion exhibiting massive and exclusive ictal-onset confirmed by intracranial EEG; (2) interictal intracranial EEG findings, including repetitive spikes > 3/s, runs of repetitive spikes, slow wave discharges, localized or spindle shaped fast activities, and electrodecremental fast activities; and (3) absence of the eloquent cortex. Complete resection was achieved when all areas deemed to be seizure-focus and irritative zones on intracranial EEG were removed. Fifteen FCDII cases with unmatched FFPE brain tissues were obtained from the archives of the Department of Neuropathology of the Amsterdam UMC (Amsterdam, The Netherlands) and the University Medical Center Utrecht (UMCU, The Netherlands). According to recent recommendations from the International League against Epilepsy (ILAE) Diagnostic Methods Commission and European Epilepsy Brain Bank (EBBB) Consortium, pathological diagnoses were reconfirmed and sub-grouped into four categories as described in previous articles [14, 15]. This study was performed and all human tissues were obtained with informed consent in accordance with protocols approved by Severance Hospital and the KAIST Institutional Review Board and Committee on Human Research.
DNA extraction from tissue samples
From FFZ brain samples, genomic DNA was extracted using QIAamp DNA Mini kits (Qiagen, USA). From FFPE brain samples, genomic DNA was extracted using QIAamp DNA FFPE kits (Qiagen, USA). For peripheral blood samples, QIAamp DNA Blood Mini kits (Qiagen, USA) were used. For saliva samples, prepIT·L2P purification kits (DNA Genotek, USA) were used.
Selection of genes associated with epilepsy and targeted gene hybrid capture sequencing
For genetic analysis, we selected several genes shown to be associated with epileptic disorders in recent studies. For 102 patients, we designed a targeted gene hybrid capture sequencing panel for 28 genes using SureDesign online tools (SureSelect DNA standard wizard). For 56 patients, we applied a targeted gene hybrid capture sequencing panel for 13 genes designed and manufactured by Celemics Inc. (Celemics, Korea). We performed library preparation according to the manufacturers’ protocol. The final libraries of hybrid capture were sequenced on a HiSeq 2500 sequencer (Illumina, USA) by Macrogen (Korea) and on a Miseq Dx sequencer (Illumina, USA) by Sovargen (Korea).
Bioinformatic analysis
For all sequencing files, we used the “Best Practices” workflow suggested by the Broad Institute. We aligned raw sequences from Fastq files to the hg19/GRCh37 assembly of the human genome reference sequence using BWA-MEM. For matched sample analysis, we utilized MuTect, Strelka, and GATK4 MuTect2, which are designed to analyze sequencing data sets for matched brain–blood (or saliva) samples [16, 17]. For unmatched brain-only sample analysis, we used three variant callers: MuTect STD mode, Lofreq [18], and Agilent’s software SureCall (optimized for variants with a variant allele frequency as low as 1%) [19]. Brain tissue sample sequencing data were used alone for unmatched sample analysis. All identified mutations were annotated using the snpEFF program [20]. For replicated sample analysis, we utilized Mutect STD mode and RePlow, a variant caller designed to detect low-level somatic mutations from replicated brain sample data [21]. We then excluded (1) registered mutations in a public database (common dbSNP147); (2) mutations with a putative low snpEFF impact score; (3) mutations with a PolyPhen & SIFT ≠ Damaging, phastCons score < 0.9; and (4) mutations with an allele frequency > 0.1% in the ExAC database for minor allele frequencies and East Asian populations [22]. To detect germline mutations, we used GATK HaplotypeCaller. All identified mutations were annotated and filtered as mentioned above. In the analysis of germline mutations, we included single nucleotide variants resulting in protein truncation (e.g., stop-gain mutation) and variants reported as pathogenic according to the American College of Medical Genetics and Genomics (ACMG) guidelines. According to the ACMG guidelines, which are particularly useful for interpretation of germline sequence variants, missense variants cannot be assumed to be pathogenic if there is no evidence from functional studies or no confirmation of de novo mutations [23]. Based on these criteria, we excluded germline variants with uncertain pathogenicity. For PTEN and DEPDC5, single-nucleotide variants resulting in protein truncation or loss of function were considered as pathogenic because their pathogenic mechanism is loss of function. We excluded germline mutations in TSC1 or TSC2 reported as benign or with conflicting interpretation according to the ACMG guidelines: several studies have reported that various missense variants in TSC1 or TSC2 can be classified as neutral because they do not increase the activity of the mTOR pathway [24]. For analysis of post-zygotic mosaicism, we utilized Mutect STD mode for each matched brain and peripheral tissue specimen. After extracting the intersection of the results, we applied the filtering process as described above. Additionally, we excluded the variants with VAFs of more than 30%. Lastly, we manually investigated variants in the Integrative Genomics Viewer (IGV), applying the following exclusion criteria: (1) adjacent alterations, not germline mutations, found only in reads with target mutations; (2) target mutations detected in the read in one direction; and (3) one or more biological similar sequence regions calculated by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (Supplementary Fig. 1 Online Resource) [25].
Validation sequencing for candidate variants
For all candidate variants from each analysis, validation sequencing was performed as previously described [5]. Briefly, for somatic mutations and post-zygotic mosaicism mutations, we designed region-specific primers with the Illumina Nextera single index (Illumina, USA). Target sequences were PCR amplified using PrimeSTAR GXL DNA polymerase (Takara). For a second PCR amplification, 20 ng of purified PCR products from the first amplification was annealed with both Illumina adaptor and index sequences. To verify fragment sizes and the quality of the amplified libraries, individual aliquots were run on a 2100 Bioanalyzer (Agilent, USA). Libraries were pooled and sequenced on a MiSeq or HiSeq sequencer (Illumina, USA). For germline mutations, we manually investigated whether mutations were present in both brain and peripheral sequencing data using IGV (Supplementary Fig. 2 Online Resource).
Results
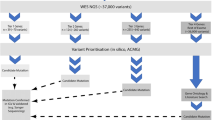
For more comprehensive analysis of a large cohort, we included deep sequencing data from individuals with matched brain–peripheral tissues on whom we previously reported (Fig. 1, Supplementary Table 1 Online Resource): deep sequencing (average read depth, 633X) of six mTOR pathway genes (AKT3, MTOR, PIK3CA, PIK3R2, TSC1, and TSC2) in 43 FCDII patients [4, 5] and deep sequencing of 28 genes (AKT3, DEPDC5, MTOR, PIK3CA, PIK3R2, TSC1, TSC2, PTEN, BRAF, etc., Supplementary Table 2 Online Resource) plus SLC35A2-specific targeted gene hybrid capture sequencing in 31 patients with mild malformation of cortical development (MCD) or epilepsy with no lesion [10]. Among 158 novel intractable epilepsy cases additionally included in the current study, 102 individuals were subjected to deep sequencing (average read depth, 1256X) of 28 genes, the same panel applied in the previous study, while the remaining 56 were subjected to deep sequencing (average read depth, 1224X) of 13 genes (AKT1, AKT3, DEPDC5, MTOR, NPRL2, NPRL3, PIK3CA, PIK3R2, PTEN, TSC1, TSC2, SLC35A2, and BRAF) (Supplementary Table 3 Online Resource). In total, we analyzed deep targeted gene hybrid capture sequencing data on focal epilepsy-related genes in 446 tissues samples from 232 individuals with intractable epilepsy: 214 with matched brain–peripheral samples and 18 with unmatched brain samples. According to histopathological diagnosis, 160 patients were diagnosed with MCD, nine with hippocampal sclerosis, 20 with epilepsy-related tumor, and 43 with no lesion characterized by means of microscopic inspection (Table 1) [15].

A schematic diagram of the study design. For more comprehensive analysis of a large cohort, we included deep sequencing data for individuals on whom we previously reported. In total, we analyzed 214 matched cases and 18 unmatched cases (140 freshly frozen tissues and 92 FFPE tissues)
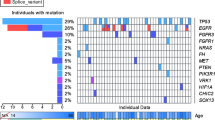
After confirmatory targeted amplicon sequencing, we identified 53 pathogenic somatic mutations and 23 pathogenic germline mutations from the 232 patients. Our deep targeted gene hybrid capture sequencing of focal epilepsy-related genes revealed somatic mutations, two-hit mutations, and germline mutations in 22.0% (51 of 232), 0.9% (2 of 232), and 9.1% (21 of 232) of the patients, respectively. Eight of the focal epilepsy-related genes in particular, including AKT3, DEPDC5, MTOR, PIK3CA, TSC1, TSC2, SLC35A3, and BRAF, were able to cover all of the pathogenic somatic and germline mutations found in our large cohort of 232 individuals (Fig. 2a, Supplementary Table 4 Online Resource). Among 53 individuals with pathogenic somatic mutations, one hemimegalencephaly (HME) patient (HME20) carried a somatic mutation in both brain and blood tissues (MTOR p.L1460P, VAFs of 15.2% in brain and 2% in blood) suggestive of post-zygotic mosaicism. One FCDIIB patient (FCD112) and one tuberous sclerosis complex (TSC) patient (TSC19) carried both a germline and somatic mutation (germline DEPDC5 p.F1508fs plus somatic DEPDC5 p.T1390fs for FCDII; germline TSC2 c.2355 + 2 T > A plus somatic DEPDC5 p.R949C for TSC) consistent with a two-hit model [26,27,28]. Also, the majority of pathogenic somatic mutations (62.3%, 33 of 53) showed low mutational burdens with VAFs of less than 5%, which are unlikely to be detected with conventional NGS and Sanger sequencing (Fig. 2b). In relation to histopathological diagnosis, we identified somatic mutations, two-hit mutations, and germline mutations in 24.4% (39 of 160), 1.3% (2 of 160), and 12.5% (20 of 160) of all MCD patients, respectively. In more detail, somatic mutations accounted for 25.4% of FCDIIA, 31.3% of FCDIIB, 57.1% of HME, and 31.3% of mild MCD cases. Two-hit mutations accounted for 2.1% of FCDIIB cases and 4.5% of TSC cases. Germline mutations accounted for 6.8% of FCDIIA, 6.3% of mild MCD, and 68.2% of TSC cases. In patients diagnosed with epilepsy-related tumor, we found that a somatic mutation in BRAF p.V600E accounted for 44.4% (8 of 18) patients with ganglioglioma. In patients diagnosed with no lesion, we identified somatic mutations and germline mutations in 9.3% (4 of 43) and 2.3% (1 of 43), respectively (Fig. 2c). No somatic or germline mutations were found in cases of FCDI, polymicrogyria, or hippocampal sclerosis. Taken together, these results suggest that low-level brain somatic mutations in eight major focal epilepsy genes contribute substantially to intractable epilepsy associated with FCDII, HME, mild MCD, no lesion, and ganglioglioma.

Landscape of somatic and germline mutations identified in intractable epilepsy patients. a Signaling pathways for all of the mutated genes identified in this study. Bold: somatic mutation, Regular: germline mutation. b The distribution of variant allelic frequencies (VAFs) of identified somatic mutations. c The detecting rate and types of identified mutations according to histopathology. Yellow: somatic mutations, green: two-hit mutations, grey: germline mutations
Although deep sequencing at a high read depth (e.g., 1000X) increases the sensitivity of detecting low-level somatic mutations, it leads to a substantial amount of artifactual somatic mutations with low VAFs, which may significantly lower positive predictive values (PPVs) [29]. Additionally, the method used to preserve patient tissues, such as FFZ or FFPE, can also be associated with the generation of artifactual somatic mutations with low VAFs [16]. In the research laboratory, a low PPV due to artifactual somatic mutations can be overcome by rigorous validation sequencing of all candidate somatic mutations. However, low PPV or limited access to matched brain–peripheral tissues can hamper the clinical use of deep targeted gene hybrid capture sequencing in practice, wherein unmatched FFPE brain samples are the most common and available tissue condition. To evaluate the accuracy of deep targeted gene hybrid capture sequencing under various tissue conditions, we first measured the PPV of deep targeted gene hybrid capture sequencing for 28 focal epilepsy-related genes in matched brain–peripheral tissues from 133 intractable epilepsy patients (31 previously reported cases and 102 new cases) for whom low-level brain somatic mutations could be detected with the highest possible precision by comparing brain and peripheral tissues. Our bioinformatic analysis combining Mutect, Strelka, and MuTect2, followed by strict in-house filtering, initially resulted in 77 candidate somatic mutations (Supplementary Table 5 Online Resource). We then rigorously performed site-specific amplicon sequencing of all of these candidate somatic mutations, validating 17, for a PPV of 22.1% (17 of 77) (Fig. 3a). Regarding tissue preservation, we found that FFZ and FFPE brain tissues showed PPVs of 34.3% (12 of 35) and 11.9% (5 of 42), respectively, in analysis of matched brain–peripheral tissues, suggesting that FFPE tissues are likely to produce more false-positive calls and decrease PPV (Fig. 3b, c). Next, we measured the PPV and sensitivity of analysis of unmatched brain tissues. To do this, we considered the 17 mutations validated by site-specific amplicon sequencing as a true-positive call set and any other variants as false-positive variants. We then analyzed a singleton of deep sequencing data for brain-only samples using various mosaic mutation callers, including Mutect STD mode, Lofreq, and SureCall single analysis mode (Agilent). Among these callers, Mutect STD showed the best performance with the highest PPV and sensitivity, regardless of tissue preservation type (Supplementary Fig. 3 Online Resource): Using Mutect STD, analysis of all unmatched brain tissues yielded a PPV of 17.5% (14 of 80) and a sensitivity of 82.4% (14 of 17) (Fig. 3a). In FFZ tissue, analysis of unmatched brain tissue with Mutect STD showed a PPV of 22.7% (10 of 44) and a sensitivity of 83.3% (10 of 12) (Fig. 3b). In FFPE tissue, the analysis showed a PPV of 11.1% (4 of 36) and a sensitivity of 80.0% (4 of 5) (Fig. 3c). Taken together, our results suggest that both deep sequencing of matched brain–peripheral tissues and unmatched brain-only tissue for 28 epilepsy-related genes shows relatively low PPV and that unmatched FFPE brain tissues, in particular, show much lower detecting accuracy.

Positive predictive values (PPVs) and sensitivities of deep sequencing for detecting low-level somatic mutations according to tissue conditions, number of genes, and analysis methods. a The PPV and sensitivity of matched and unmatched analysis (Mutect STD mode) from all tissue types, including both FFZ and FFPE specimens. Matched analysis provided a PPV of 22.1% (17 of 77). We considered variants validated in matched analysis as a true-positive call set and other variants as false-positive variants. Unmatched analysis showed a PPV of 17.5% (14 of 80) and a sensitivity of 82.4% (14 of 17). b The PPV and sensitivity from the analysis of FFZ brain samples. Matched analysis showed a PPV of 34.3% (12 of 35) and a sensitivity of 100%. Unmatched analysis (Mutect STD mode) provided a PPV of 22.7% (10 of 44) and a sensitivity of 83.3% (14 of 17). c The PPV and sensitivity from the analysis of FFPE brain samples. Matched analysis showed a PPV of 11.9% (5 of 42) and a sensitivity of 100%. Unmatched analysis (Mutect STD mode) provided a PPV of 11.1% (4 of 36) and a sensitivity of 80.0% (4 of 5). d The PPV and sensitivity from deep sequencing of eight major focal epilepsy genes. Matched analysis showed a PPV of 36.2% (17 of 45) and a sensitivity of 100%. Unmatched analysis (Mutect STD mode) provided a PPV of 32.6% (14 of 43) and a sensitivity of 83.3% (14 of 17). e The PPV and sensitivity from the analysis of technical replication of deep sequencing, compared to that of singleton deep sequencing in unmatched brain tissues. The replication of library preparation followed by deep sequencing of eight major focal epilepsy genes was performed for 22 FCDII individuals. In these FCDII cases, matched analysis showed a PPV of 44.4% (4 of 9) and a sensitivity of 100%; Mutect STD intersection provided a PPV of 100% (3 of 3) and a sensitivity of 75% (3 of 4); and RePlow showed a PPV of 50% (4 of 8) and a sensitivity of 100% (4 of 4)
In light of the above, we sought ways to improve detection accuracy such that precise detection of low-level somatic mutations could be possible in clinical settings. Since all somatic mutations in the current study were identified in eight major epilepsy-related genes (AKT3, DEPDC5, MTOR, PIK3CA, TSC1, TSC2, BRAF, and SLC35A2), we narrowed the list of target genes to these eight and measured the resultant PPV and sensitivity. Matched sample analysis provided a PPV of 36.2% (17 of 47; 12 of 20 from FFZ and 5 of 27 from FFPE). Using the Mutect STD mode, the performance of which was best among the singleton analysis tools, provided a PPV of 32.6% (14 of 43; 10 of 19 from FFZ and 4 of 24 from FFPE) without any loss of sensitivity (Fig. 3d). In addition, since our recent study demonstrated that technical replication of deep sequencing could robustly eliminate artifactual somatic mutations with low VAFs and increase the accuracy of detecting low-level somatic mutations, we performed additional deep targeted gene hybrid capture sequencing in brain samples from 22 FCDII individuals who had enough genomic DNA for additional library preparation. Among them, four individuals had pathogenic somatic mutations with VAFs of less than 5%. In analysis of replicates, we utilized the RePlow variant caller, which is designed for detecting somatic single-nucleotide variants from a replicated set of high-depth sequencing data, as well as the Mutect STD mode from which we extracted the intersection of results from each replicate. In these 22 FCDII individuals, analysis of matched brain–peripheral tissues revealed a PPV of 44.4% (4 of 9). The intersection of the Mutect STD mode when using replicates provided a PPV of 100% (3 of 3) and a sensitivity of 75% (3 of 4). RePlow resulted in a PPV of 50% (4 of 8) and a sensitivity of 100% (4 of 4) (Fig. 3e). Furthermore, we examined whether the use of deep sequencing replicates for the eight major focal epilepsy genes would yield a high PPV in an independent intractable epilepsy cohort with different ethnicities, comprising 15 FCDII individuals with only unmatched FFPE brain tissues (Fig. 4a). The replicated data were analyzed using RePlow and Mutect STD mode intersection as described above. After confirmatory site-specific amplicon sequencing, we found that RePlow revealed somatic mutations in 33.3% (5 of 15) of the individuals without any false-positive calls; Mutect STD intersection detected somatic mutations in 20.0% (3 of 15) (Fig. 4b). These performances were similar to the somatic mutation detecting rates in total FCDII individuals (28.0%, 30 of 107) and previously reported cases [4, 5, 30]. Surprisingly, although Mutect STD mode intersection missed two pathogenic somatic mutation, both analysis tools provided a PPV of 100% (Fig. 4c). Taken together, these results supported the use of technical replication of deep sequencing of eight major focal epilepsy genes in FFPE brain samples to robustly increase PPV without a significant loss in sensitivity, thereby allowing for the precise genetic detection of low-level somatic mutations underlying intractable epilepsy in clinical settings.

Analysis of deep sequencing replicates of eight focal epilepsy gene in unmatched FFPE brain tissues from an independent FCDII cohort. a A schematic figure of the study design for an independent FCDII cohort with unmatched FFPE brain tissues. b The detecting rates of each analysis tool. RePlow and Mutect STD intersect showed detecting rates of 33.3% (5 of 15) and 20.0% (3 of 15), respectively. c The total raw calls of each analysis tool along with PPVs
Discussion
Herein, we performed deep targeted gene hybrid capture sequencing of 446 tissue samples consisting of matched brain–peripheral tissues or unmatched brain-only tissues from 232 patients with intractable epilepsy and revealed the contribution of somatic mutations, post-zygotic mosaicism, and germline mutations to intractable epilepsy. To our knowledge, the current study includes the largest cohort of intractable epilepsy patients with matched brain–peripheral tissues from which to examine somatic mutations. Through strict and comprehensive analysis, we discovered that all identified mutations causative for intractable epilepsy were in eight major focal epilepsy genes and that somatic mutations, two-hit mutations, and germline mutations in these genes accounted for 22.0% (51 of 232), 0.9% (2 of 232), and 9.1% (21 of 232) of the studied patients, respectively. Somatic mutations of low allelic frequency, especially those at less than 5%, accounted for more than 60% of all identified somatic mutations. We showed that the use of deep sequencing replicates in the eight major focal epilepsy genes dramatically increased detection accuracy without a significant loss of sensitivity, thereby allowing for precise genetic detection of low-level somatic mutations in unmatched FFPE brain tissues (the most common tissue condition in clinical settings) from patients with intractable epilepsy.
Typical genome wide sequencing, such as whole-exome sequencing with a 100-150X read depth and whole genome sequencing with a 30–60X read depth, shows limited capability in detecting mutations of low allelic frequency, especially those at less than 5% [31]. Meanwhile, targeted gene hybrid capture sequencing of disease-specific genes provides a significant increase in sequencing depth for target sites and reduces sequencing cost, allowing for the detection of low-frequency mutations. However, NGS at a high depth dramatically drops the detection accuracy of low-level somatic mutations due to background errors with low VAFs [29]. Also, the condition of brain specimens, such as FFPE specimens, can negatively affect the detection accuracy of low-level somatic mutations [32]. Our results of singleton deep targeted gene hybrid capture sequencing in unmatched FFPE brain tissues showed a maximum PPV of 11.1%. Although false positives can be removed by an orthogonal validation method, such as site-specific amplicon sequencing or digital droplet (dd) PCR in the research laboratory [4, 6], such low PPV in singleton deep targeted gene hybrid capture sequencing hampers the precision of genetic analysis of low-level somatic mutations in the clinical setting. To increase detection accuracy, we narrowed down target genes to eight major focal epilepsy genes and applied the concept of technical replication. By doing so, we were able to increase the PPV of deep targeted gene hybrid capture sequencing up to 100% at maximum without a significant loss of sensitivity in unmatched FFPE brain tissues from FCDII individuals. Through these results, we demonstrated that analysis of deep sequencing replicates of eight major focal epilepsy-related genes offers acceptable performance in the analysis of unmatched FFPE brain tissues, the most clinically relevant tissue condition.
In the present study, our deep targeted gene hybrid capture sequencing of eight major focal epilepsy genes was able to identify pathogenic mutations in approximately 32% of our resected intractable epilepsy brain tissues. The identified somatic mutations were frequently detected in a specific gene set (AKT3, DEPDC5, MTOR, PIK3CA, TSC1, TSC2, BRAF, and SLC35A2). In previous studies, several somatic mutations in these genes have been shown to cause intractable epilepsy using animal models, in vitro mTOR assay, or glycosylation pattern assay [3,4,5, 10, 30, 33, 34]. Accordingly, these genes are considered to be susceptible to pathogenic somatic mutations in patients with intractable epilepsy. Notwithstanding, we were still unable to detect causative or putative mutations in approximately 70% of the patients. We think that three possible scenarios may explain the 70% of cases that were negative for mutations in these genes: (1) extremely low-level somatic mutations with a VAF less than 1% that were still responsible for epileptic seizures [21], (2) somatic mutations in non-coding regions or other unknown genes [35, 36], and (3) epigenetic changes or other environmental etiologies for these patients [37, 38]. Regarding extremely low-level somatic mutations, we may be able to apply more sensitive methods, such as ddPCR or single cell sequencing, or to enrich dysmorphic neurons by laser microdissection, followed by deep sequencing or ddPCR. Regarding somatic mutations in non-coding regions or other unknown genes, we might be able to use deep whole-genome sequencing or deep whole-exome sequencing in a larger cohort. Regarding epigenetic causes, we can investigate DNA methylation and non-coding RNA, such as microRNA or tRNA. In doing so, we may be able to identify other genetic or epigenetic causes underlying mutation-negative epileptic brains.
Interestingly, post-zygotic mosaicism was encountered in a sporadic epilepsy individual (Patient HME20). Regarding the somatic mutation, MTOR p.Leu1460Pro was found in both brain (VAF 15%) and blood (VAF 2%) samples from Patient HME20. We speculate that this mutation might arise at the early stage of development before the ectoderm and mesoderm arise. The HME20 individual did not present any systemic symptoms other than epileptic seizures and, after surgery, this patient has maintained seizure-free status for the past 2 years, achieving Engel classification 1.
Our study showed that deep sequencing replicates of major focal epilepsy genes in unmatched FFPE brain tissues are of use in accurately and efficiently identifying low-level somatic mutations. Although our detection tool is not adequate for detecting new pathogenic genes, it could allow for more comprehensive genetic analysis of low-level somatic mutations in resected epilepsy brain tissue by facilitating the detection of somatic mutations in clinical settings. Moreover, the high PPV and low cost of our precise genetic analysis could help physicians determine which individuals would likely benefit from alternative medical treatments with mTOR inhibitors among patients who do not become seizure-free after epilepsy surgery and are found to have disease-causing mutations in mTOR pathway genes. Taken together, results from our genetic analysis could improve overall patient care by providing more comprehensive genetic counselling and informing decisions on alternative treatment. As a clinical trial of an mTOR inhibitor targeting FCDII with somatic mutations in mTOR pathway genes is underway (NCT03198949), precise genetic detection of low-level somatic mutations will be important not only for molecular genetic analysis of FCDII, but also for providing alternative medical treatment to FCDII patients.
References
Kwan P, Brodie MJ (2000) Early identification of refractory epilepsy. N Engl J Med 342:314–319. https://doi.org/10.1056/NEJM200002033420503
Epi4K consortium; Epilepsy Phenome/Genome Project (2017) Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol 16:135–143. https://doi.org/10.1016/S1474-4422(16)30359-3
Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A et al (2012) De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 44:941–945. https://doi.org/10.1038/ng.2329
Lim JS, Gopalappa R, Kim SH, Ramakrishna S, Lee M, Kim WI et al (2017) Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am J Hum Genet 100:454–472. https://doi.org/10.1016/j.ajhg.2017.01.030
Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK et al (2015) Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 21:395–400. https://doi.org/10.1038/nm.3824
Winawer MR, Griffin NG, Samanamud J, Baugh EH, Rathakrishnan D, Ramalingam S et al (2018) Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann Neurol 83:1133–1146. https://doi.org/10.1002/ana.25243
Baulac S, Ishida S, Marsan E, Miquel C, Biraben A, Nguyen DK et al (2015) Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol 77:675–683. https://doi.org/10.1002/ana.24368
Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK et al (2012) Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74:41–48. https://doi.org/10.1016/j.neuron.2012.03.010
Ribierre T, Deleuze C, Bacq A, Baldassari S, Marsan E, Chipaux M et al (2018) Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Invest 128:2452–2458. https://doi.org/10.1172/JCI99384
Sim NS, Seo Y, Lim JS, Kim WK, Son H, Kim HD et al (2018) Brain somatic mutations in SLC35A2 cause intractable epilepsy with aberrant N-glycosylation. Neurol Genet 4:e294. https://doi.org/10.1212/NXG.0000000000000294
Weckhuysen S, Marsan E, Lambrecq V, Marchal C, Morin-Brureau M, An-Gourfinkel I et al (2016) Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 57:994–1003. https://doi.org/10.1111/epi.13391
Koh HY, Kim SH, Jang J, Kim H, Han S, Lim JS et al (2018) BRAF somatic mutation contributes to intrinsic epileptogenicity in pediatric brain tumors. Nat Med 24:1662–1668. https://doi.org/10.1038/s41591-018-0172-x
Kim YH, Kang HC, Kim DS, Kim SH, Shim KW, Kim HD et al (2011) Neuroimaging in identifying focal cortical dysplasia and prognostic factors in pediatric and adolescent epilepsy surgery. Epilepsia 52:722–727. https://doi.org/10.1111/j.1528-1167.2010.02950.x
Blumcke I, Aronica E, Miyata H, Sarnat HB, Thom M et al (2016) International recommendation for a comprehensive neuropathologic workup of epilepsy surgery brain tissue: a consensus Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 57:348–358. https://doi.org/10.1111/epi.13319
Blumcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG et al (2017) Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med 377:1648–1656. https://doi.org/10.1056/NEJMoa1703784
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C et al (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31:213–219. https://doi.org/10.1038/nbt.2514
Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK (2012) Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28:1811–1817. https://doi.org/10.1093/bioinformatics/bts271
Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH et al (2012) LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201. https://doi.org/10.1093/nar/gks918
D'Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM et al (2017) Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep 21:3754–3766. https://doi.org/10.1016/j.celrep.2017.11.106
Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L et al (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. https://doi.org/10.4161/fly.19695
Kim J, Kim D, Lim JS, Maeng JH, Son H, Kang HC et al (2019) The use of technical replication for detection of low-level somatic mutations in next-generation sequencing. Nat Commun 10:1047. https://doi.org/10.1038/s41467-019-09026-y
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. https://doi.org/10.1038/nature19057
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Hoogeveen-Westerveld M, Wentink M, van den Heuvel D, Mozaffari M, Ekong R, Povey S et al (2011) Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with Tuberous Sclerosis Complex. Hum Mutat 32:424–435. https://doi.org/10.1002/humu.21451
Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. https://doi.org/10.1093/bib/bbs017
Green AJ, Smith M, Yates JR (1994) Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet 6:193–196. https://doi.org/10.1038/ng0294-193
Niida Y, Stemmer-Rachamimov AO, Logrip M, Tapon D, Perez R, Kwiatkowski DJ et al (2001) Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am J Hum Genet 69:493–503. https://doi.org/10.1086/321972
Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL et al (2014) Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol 75:782–787. https://doi.org/10.1002/ana.24126
Kroigard AB, Thomassen M, Laenkholm AV, Kruse TA, Larsen MJ (2016) Evaluation of nine somatic variant callers for detection of somatic mutations in exome and targeted deep sequencing data. PLoS ONE 11:e0151664. https://doi.org/10.1371/journal.pone.0151664
Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ, Hiatt JB, Roden WH et al (2015) PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 138:1613–1628. https://doi.org/10.1093/brain/awv045
Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA et al (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45:1113–1120. https://doi.org/10.1038/ng.2764
Kerick M, Isau M, Timmermann B, Sultmann H, Herwig R, Krobitsch S et al (2011) Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genom 4:68. https://doi.org/10.1186/1755-8794-4-68
D'Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM et al (2015) Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol 77:720–725. https://doi.org/10.1002/ana.24357
Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H et al (2015) Somatic Mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol 78:375–386. https://doi.org/10.1002/ana.24444
Beal JC, Cherian K, Moshe SL (2012) Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr Neurol 47:317–323. https://doi.org/10.1016/j.pediatrneurol.2012.06.002
Mefford HC, Zemel M, Geraghty E, Cook J, Clayton PT, Paul K et al (2015) Intragenic deletions of ALDH7A1 in pyridoxine-dependent epilepsy caused by Alu-Alu recombination. Neurology 85:756–762. https://doi.org/10.1212/WNL.0000000000001883
Hogg MC, Raoof R, El Naggar H, Monsefi N, Delanty N, O'Brien DF et al (2019) Elevation in plasma tRNA fragments precede seizures in human epilepsy. J Clin Invest 130:2946–2951. https://doi.org/10.1172/JCI126346
Miller-Delaney SF, Bryan K, Das S, McKiernan RC, Bray IM, Reynolds JP et al (2015) Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain 138:616–631. https://doi.org/10.1093/brain/awu373
Acknowledgements
This work was supported by grants from the Suh Kyungbae Foundation (to J.H.L.), the National Research Foundation of Korea (NRF) grant funded by the Korea government, Ministry of Science and ICT (No. 2019R1A3B2066619 to J.H.L), Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (H15C3143 and H16C0415 to J.H.L.; HI15C1601 and HI18C0586 to H.C.K.), and Netherlands Organisation for Health Research and Development (ZonMW, Programma Translationeel Onderzoek, Project number: 95105004; AE, CM to E.A.).
Author information
Authors and Affiliations
Contributions
NSS and JHL organized the project, and NSS performed genetic studies. SHK performed pathological study. NSS performed data analysis and bioinformatics analysis. DSK, KS, and JSK performed surgeries and collected patient samples. HK, AK, HDK, and JSL managed patient information. EA, CM, and WGMS collected patient samples, patient information, and tissue samples. NSS, WKK and HYK managed tissue samples and sequencing data. NSS and JHL wrote the manuscript. DSK, HK, and JHL led the project and oversaw the manuscript preparation.
Corresponding authors
Ethics declarations
Conflict of interest
J.H.L is a co-founder of SoVarGen, Inc., which seeks to develop new diagnostics and therapeutics for brain disorders. The remaining authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sim, N.S., Ko, A., Kim, W.K. et al. Precise detection of low-level somatic mutation in resected epilepsy brain tissue. Acta Neuropathol 138, 901–912 (2019). https://doi.org/10.1007/s00401-019-02052-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-019-02052-6