
Abstract
Messenger RNA (mRNA) translation is the terminal step in protein synthesis, providing a crucial regulatory checkpoint for this process. Translational control allows specific cell types to respond to rapid changes in the microenvironment or to serve specific functions. For example, neurons use mRNA transport to achieve local protein synthesis at significant distances from the nucleus, the site of RNA transcription. Altered expression or functions of the various components of the translational machinery have been linked to several pathologies in the central nervous system. In this review, we provide a brief overview of the basic principles of mRNA translation, and discuss alterations of this process relevant to CNS disease conditions, with a focus on brain tumors and chronic neurological conditions. Finally, synthesizing this knowledge, we discuss the opportunities to exploit the biology of altered mRNA translation for novel therapies in brain disorders, as well as how studying these alterations can shed new light on disease mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Francis Crick, a pioneer in molecular biology, had the fortuitous intuition that RNA molecules worked as “adaptors” in the transfer of genetic information from DNA to amino acids (Crick 1957, On protein synthesis). Subsequent work would confirm this notion, and led to the unraveling of details into the structure, function, and regulation of the translation machinery [76]. In light of recent advances, alterations in mRNA translation are increasingly recognized in human pathogenesis. Indeed, it is estimated that translation control determines ~ 50% of protein levels, making this process a far better predictor of protein levels than mRNA expression [156]. Although informative publications discuss various aspects of mRNA translation in synaptic function and brain diseases, a comprehensive review focusing on the role of mRNA translation control in central nervous system disorders is currently lacking. Although biologically very different diseases, and typically studied by distinct research communities, neurodegenerative disorders and brain tumors are both characterized by alterations of the translational machinery. As discussed in detail below, such alterations are typically disease specific, but we believe that a better understanding of translational modifications in different diseases will have cross-cutting benefits to the research community. With this as the underlying theme, this review outlines major findings regarding translation control across different brain pathologies and discusses their therapeutic implications.
An overview of mRNA translation and its regulatory mechanisms
Protein synthesis is regulated at multiple steps, including transcription (amounts of mRNA generated) and translation (efficiency of mRNA translation), in addition to protein stability and posttranslational modifications. Given the relatively long half-life (> 6 h) of mRNAs in mammalian cells [156], translation control is extremely relevant particularly when cells face the need to quickly respond to rapid microenvironmental changes via new protein synthesis [168]. In addition, translational control is exploited by specific cell types, such as neurons, to achieve timely and spatially heterogeneous protein synthesis.
Traditionally, studies into the regulation of eukaryotic translation have focused primarily at the initiation step, as mediated by reactions involving the ribosomal subunits, eukaryotic initiation factors (eIFs) such as eIF3 and eIF4F (comprising the cap-binding complex), and engagement of the start codon [165]. Emerging evidence suggests that subsequent reactions during nascent polypeptide elongation also act as critical regulatory steps, with important implications for adaptation to cell stress and neurological diseases [39, 62]. A detailed overview of the translation machinery and its regulation is beyond the scope of this work, and the reader is referred to notable reviews published elsewhere [73, 84]. Briefly, three consecutive steps, each comprising multiple molecular interactions and reactions, constitute mRNA translation, namely (1) initiation, (2) elongation, and (3) termination.
Translation initiation and its canonical regulation
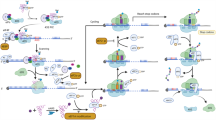
A crucial first step in translation initiation is the formation of the ternary complex between a methionyl transfer RNA (Met-tRNAi), encoding the first amino acid, Met, and the GTP bound eIF2 (eIF2-GTP, Fig. 1a). Once formed, the ternary complex binds to eIF1A and the 40S ribosomal subunit in complex with eIF3, constituting the so-called 43S preinitiation complex (PIC; Fig. 1b). At the same time, the 5′ cap of an mRNA to be translated is bound by members of the eIF4F complex (Fig. 1b), constituted by the DEAD-box RNA helicase eIF4A, the cap-binding protein eIF4E, and the scaffold protein, eIF4G. As a result, eIF4F bound to the target message associates with the preinitiation complex, forming the initiation complex (Fig. 1c). The initiation complex then scans the bound mRNA 5’ untranslated region (5′-UTR) until the initial AUG start codon is recognized, followed by recruitment of the 60S ribosomal subunit, allowing Met-tRNAi engagement with the start codon to initiate translation. This, coupled with the hydrolysis of eIF5-bound GTP to GDP, constitutes an irreversible step, such that ribosomal subunits will not dissociate until protein synthesis is complete (Fig. 1d).

Different stages of cap-dependent translation initiation. a eIF2-GTP and met-tRNAi (the ternary complex) bind to eIF1A and to the 40S ribosomal subunit complexed with eIF3, constituting the 43S preinitiation complex (b). c The eIF4F complex, bound to the 5′ cap of the mRNA to be translated, associates with the preinitiation complex, forming the initiation complex. d The initiation complex then scans the bound mRNA until the first AUG start codon is recognized. The subsequent recruitment of the large 80S ribosomal subunit, coupled with the hydrolysis of the eIF5 bound GTP to GDP, results in mRNA translation
Several highly conserved mechanisms regulating translation initiation have been well documented, such as phosphorylation of the α subunit of the heterotrimeric eIF2 complex at Ser51. This increases affinity of the eIF2 complex for the guanine exchange factor eIF2B, limiting the conversion of inactive eIF2-GDP to active eIF2-GTP and inhibiting global translation [129]. Four stress activated kinases are known to phosphorylate eIF2α as part of the so-called integrated stress response (ISR), to limit protein synthesis under stress: PKR [protein kinase R, activated in response to double-stranded (ds)RNA], HRI (heme-regulated inhibitor, in response to oxidative stress), GCN2 (general control nonderepressible 2, in response to amino acid deprivation), and PERK (PRKR-like endoplasmic reticulum kinase, in response to ER stress) [129]. Although eIF2α phosphorylation inhibits global translation, a subset of mRNAs containing upstream open-reading frames (uORFs) within their 5′-UTR remains preferentially translated under stress [74] (described further below).
Another critical regulator of translation initiation and protein synthesis is the serine/threonine kinase mammalian target of rapamycin complex 1 (mTORC1), a well-known nutrient/energy sensor. Under nutrient replete conditions, mTORC1 is activated and phosphorylates the eIF4E-binding proteins, 4E-BP1/2, thereby releasing them from eIF4E to promote eIF4E binding to the cap, leading to enhanced cap-dependent translation [165]. In contrast, under nutrient deprivation and certain other stresses, mTORC1 is inactivated and 4E-BPs remain hypophosphorylated, allowing them to sequester eIF4E to limit translation initiation [165].
Recruitment and translation efficiency of a given mRNA is also determined by distinct structures within the 5′-UTR, such as complex stem-loops, 5′ terminal oligopyrimidine (5′ TOP) motifs, and uORFs. These mechanisms have been reviewed in detail elsewhere [74] and are beyond the scope of the current review. It is also well known that a subset of eukaryotic mRNAs can be translated in a 5′ cap-independent manner (Fig. 2), in part through sequences called internal ribosome entry sites (IRESs) that directly bind translation factors called IRES tran-sacting factors (ITAFs, [101, 106]).

Cap-independent translation initiation. A subset of eukaryotic mRNAs can be translated in a 5′ cap-independent way, due to sequences called internal ribosome entry sites (IRESs) that facilitate direct binding of translation initiation factors called IRES-transacting factors (ITAFs)
RNA modifications are also emerging as potentially major regulators of translation initiation. The m6A (6-methyladenosine) modification of mRNA is the best-studied eukaryotic post-transcriptional mRNA modification. In eukaryotes, this modification is dependent on the METTL3 enzyme, whereas the FTO demethylase is a key enzyme for mRNA demethylation [87]. While the biological functions of m6A are incomplete, recent studies have implicated m6A-modified mRNAs (see also below) in cap-independent translation, through direct binding of translation initiation factors, such as eIF3 subunits [124].
Translation elongation and its regulation
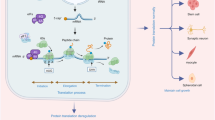
Following initiation, peptide chain extension occurs in a process known as translation elongation. This process is relatively simple compared to initiation, and mainly mediated by eukaryotic elongation factors 1 and 2 (eEF1 and eEF2). Briefly, once the Met-tRNA has been delivered to the P site of the ribosome, the next aminoacyl-tRNA bound to eEF1-GTP comes to the A site. GTP hydrolysis leads to a conformational change in the ribosome, and Met forms a peptide bond to aminoacyl-tRNA in the P site. Ribosome translocation to the next codon is regulated by the hydrolysis of GTP bound to eEF2 (Fig. 3a). This cycle of events repeats as the ribosome moves along the mRNA being translated, and the nascent polypeptide chain elongates. Translation terminates when a stop codon (UAA, UGA, or UAG) enters the ribosome and is recognized by release factors such as eFR1 [43], in a step called translation termination. Subsequent reactions result in separation of the polypeptide chain from the tRNA and protein release.

eEF2 kinase controls the elongation phase of mRNA translation by phosphorylating eEF2 on Thr56. a During translation elongation, aminoacyl-tRNA located on the A site (aminoacyl or acceptor site) forms a peptide bond with the peptidyl-tRNA occupying the P site (peptidyl or polypeptide site), resulting in the attachment of the growing peptide chain to the tRNA in the A site. Then, GTP bound eEF2 mediates translocation of peptidyl-tRNA from the A site (dashed cartoon) to the P site (solid cartoon), and the now deacetylated-tRNA leaves the ribosome on E site (exit site). The growing peptide chain is depicted as beads on a string with methionine (met) as the first amino acid in the chain. eEF2K phosphorylates and inactivates eEF2, preventing its binding to the ribosome. b Phosphorylation of eEF2 Thr56 in neurons, reflecting neuronal eEF2K activity is increased in response to several stimuli. Negative regulators of eEF2 phosphorylation (Thr-56)/eEF2K activity include mTORC1, p70RS6K, and p90RSK. We reported that human amyloid-β (Aβ) or alpha-synuclein also increase eEF2 Thr56 phosphorylation, suggesting increased eEF2K activation [85, 86]
Elongation is traditionally under-appreciated in translational control, compared to the control of initiation. However, emerging evidence highlights an important regulatory role for control of ribosome transit (elongation) [39, 62, 83, 109]. Eukaryotic translation elongation is mainly regulated by eukaryotic elongation factor 2 kinase (eEF2K, also known as calcium/calmodulin-dependent kinase III, CamKIII) [96], a crucial molecule linking the energy sensors AMP-Activated Protein Kinase (AMPK) and mTORC1 to translation. Activation of the AMPK–eEF2K pathway slows polypeptide chain elongation, as eEF2K inactivates eEF2 by phosphorylation of its Thr56 residue (Fig. 3a) [152]. AMPK is a master cellular energy sensor, as increased AMP:ATP ratios lead to AMPK activation [61]. In addition, three upstream kinases, LKB1 (liver kinase B1), CaMKKβ (Ca2+/calmodulin-dependent kinase kinase β), and TAK1 (transforming growth factor β-activated kinase 1), activate AMPK to fine tune AMPK-eEF2K activity [97]. Activated AMPK suppresses energy consuming processes (e.g., protein and fatty acid synthesis) and promotes energy production via glycolysis, lipid metabolism, and increased glucose uptake [20]. Regulation of eEF2K is also mediated by calmodulin binding in response to changes in cellular calcium levels and positive regulation by autophosphorylation [17]. Finally, recent work suggests a tight interplay between initiation and elongation control, as activated mTORC1 blocks eEF2K via S6 kinase (S6K) mediated eEF2K inhibitory phosphorylation, thus enhancing elongation rates during anabolic growth [48]. In addition, ribosome stalling during translation promotes eIF2α phosphorylation to inhibit translation initiation [82]. While a recent review has extensively summarized the role of mTORC–AMPK signalling in brain [52], mechanisms of translation elongation control unique to neuronal cells are discussed in “Translational control in normal brain function”.
Translational control through RNA granules
In addition to control of initiation and elongation, eukaryotic cells have evolved other sophisticated mechanisms to regulate translation. An important example is the formation RNA granules, including stress granules (SGs) and processing bodies (P-bodies) in somatic cells, neuronal granules in neurons, and germinal granules in germ cells [2]. Due to their relevance to neuronal function and CNS pathology, this review will mainly focus on SGs (discussed below) and neuronal granules (discussed in “Translational control in normal brain function”).
Diverse cellular stresses such as heat shock, oxidative stress, hypoxia, nutrient deprivation, or viral infections lead to a rapid translational inhibition and disassembly of translationally active polysomes [168]. Under these conditions, translationally stalled complexes consisting of the 40S ribosome, silenced polyadenylated mRNAs, translation initiation factors, and distinct RNA-binding proteins (RBPs) aggregate into membrane-less organelles called SGs [2]. Previous studies indicate that eIF2α phosphorylation is necessary for SG formation [93], suggesting that, along with the above-mentioned ISR, SG formation might contribute to stress-induced global translation inhibition. However, other studies have brought this notion into dispute. First, SGs are reported to form in the absence of eIF2α phosphorylation [123]. Second, a detailed analysis of SG-associated mRNAs suggests that only ~ 10% of cellular mRNAs reside in SGs under stress [98]. In addition, no specific functional mRNA classes are significantly over- or under-represented in SGs, and the only consistent features of these mRNAs are longer lengths and higher translational inefficiency scores [98]. This calls into question whether SGs actually function to reduce global translation. Additional functional studies are necessary to uncover the role of these perplexing structures in translation regulation.
Translational control in normal brain function
Neurons are morphologically unique, highly polarized cells, with the ability to integrate excitatory and inhibitory stimuli from hundreds to thousands of synaptic inputs. Changes in protein synthesis necessary for synaptic activity must occur very rapidly, which is biologically challenging for neurons, given the distance (in the range of hundreds of micrometers or even millimeters) between cell body and axonal or dendritic terminals. Although de novo protein synthesis in neuronal cell bodies is important, emerging evidence suggests that hundreds of mRNAs are localized at synaptic terminals [23, 198], and that local translation at the dendritic terminal is key to synaptic functions. Furthermore, localized mRNA translation appears critical for processes such as synaptic development, synaptic plasticity, and memory formation [19, 33, 166].
The strengthening of a synapse, a process also called long-term potentiation (LTP), and the long-lasting decrease in synaptic strength, long-term depression (LTD), are thought to be key mechanisms mediating a number of critical brain functions, including learning and memory formation [30, 135]. Strong evidence supporting a role for localized mRNA translation at the dendritic terminal relies on seminal neurobiology work, indicating that LTP and LTD occur even when the neuronal body is physically disconnected from the synapse [80, 91].
Translation initiation control
Translation initiation control by the above-mentioned eIF2α pathway, acting as a negative regulator, is critical for memory function. Indeed, genetically engineered mice deficient in eIF2α phosphorylation (+/S51A) display increased memory capacity [31]. In contrast, increased eIF2α phosphorylation in mouse hippocampus reduces long-term memory [89]. Mechanistically, eIF2α-mediated translational control appears to inhibit the progression from transient to persistent LTP [142] and to induce LTD [45], both phenomena requiring de novo protein synthesis. Thus, it is likely that these processes are mediated by eIF2α-induced decreases in translation of a subset of mRNAs, a few of which (e.g., oligophrenin-1) have been identified [45].
The highly conserved mTOR enzyme is ubiquitously expressed in neuronal cells, where its activity is also linked to synaptic function and cognition [107]. The activity of mTOR and its downstream targets is also critical for memory formation, as extensively reviewed [32]. This is supported by the finding that mTORC1 inhibition negatively impacts LTP and memory formation [169]. Hippocampal LTP induces the expression of eukaryotic elongation factor 1A (eEF1A), the mRNA of which contains a 5′TOP motif, within five minutes after stimulation [179]. This is preserved in dendrites disconnected from their cellular bodies, but is impaired by the mTOR inhibitor rapamycin, suggesting that the mTOR pathway controls the rapid synthesis of proteins during LTP formation at the dendritic terminals. However, these studies did not address whether changes in dendritic protein expression induced by rapamycin are required for LTP. The process of LTD also requires selective mRNA translation and localized dendritic protein synthesis, which appears to be mostly mediated by activation of the PI3K–Akt–mTOR pathway at dendritic terminals [197].
Translation elongation control
Translation control at the level of elongation is also critical for synaptic plasticity. In addition to the regulation of elongation by the AMPK–eEF2K–eEF2 pathway under nutrient deprivation, ample evidence implicates additional mechanisms regulating neuronal eEF2K activation in response to synaptic activity (Fig. 3b) [69]. For instance, chemical stimulation of metabotropic glutamate receptors mGluR5 using (S)-3,5-dihydroxyphenylglycine (DHPG), depolarization by potassium chloride bath application, and treatment with the gamma-aminobutyric acid (GABAA) antagonist bicuculline increase eEF2K activity (i.e., p-eEF2 Thr56) in cultured neurons (Fig. 3b) [69, 97]. Instead, ketamine-mediated suppression of NMDA receptors (NMDAR) activity suppresses eEF2K and induces eEF2 dephosphorylation [128]. The significance of eEF2K activity and eEF2 phosphorylation in synaptic plasticity, such as LTP and LTD, has been reviewed elsewhere [69]. In brief, eEF2K mediates activity-dependent changes in dendritic mRNA translation, particularly postsynaptic changes in protein synthesis in response to acute or prolonged NMDAR and mGluR signaling [69]. Although eEF2K activation inhibits general mRNA translation in dendrites, some studies suggest that translation of messages encoding brain-derived neurotrophic factor (BDNF), activity-regulated cytoskeletal-associated protein (Arc), the alpha subunit of calcium/CaM-dependent kinase II (CaMKII), microtubule-associated protein 1B (MAP1B), and other microtubule-associated proteins is actually increased in response to eEF2K activation [95, 184]. In addition, translational control of Arc synthesis by the eEF2K-eEF2 axis is lost in Fragile X disease mice, suggesting an unexpected coordinated control of Arc translation by eEF2K and the Fragile X mental retardation protein FMRP [138]. These processes are linked to diverse forms of synaptic plasticity including recycling and internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [69, 138]. Finally, eEF2K activation inhibits GABAergic synaptic transmission, thus interfering with the GABAA activation/inhibition balance and decreasing resistance to epileptic seizures [70].
Translation control by neuronal RNA granules
Neuronal granules constitute the major contributors to the spatio-temporal regulation of mRNA translation and protein synthesis in neurons. Serving as motile macromolecular entities that translocate mRNA from the nucleus to synaptic terminals within neurons, neuronal granules are composed of mRNA, translation initiation factors, small and large ribosomal subunits, and RBPs [2, 102]. While neuronal granules are mostly considered to package and transport silenced mRNA, recent literature has suggested that some components of neuronal granules might also promote translation of subsets of mRNAs [191]. At the dendritic terminal, membrane depolarization leads to a redistribution of mRNAs from neuronal granules to actively translating polysomes [62, 102]. Accordingly, neuronal RNA granules appear to directly contribute to localized dendritic mRNA translation. Recent evidence also suggests that local translation of specific mRNAs, such as β-actin, might contribute to synaptic plasticity upon granule disassembly [21]. For additional discussion on translational control in memory and normal brain function, we refer the readers to recent review works [56, 166, 187].
Translational control in brain tumors
Since the discovery that proliferating cells rely on increased translation rates for enhanced protein synthesis, it has become clear that mRNA translation plays a critical role in cell transformation and tumor progression [177]. However, translation control in cancer has been historically overlooked in favor of the other steps regulating protein expression, such as control of transcription. A growing body of literature indicates that transcript abundance does not predict protein level for a wide variety of genes in different tumors [193], including brain cancer [49, 149]. For comprehensive reviews on translation control in cancer, we refer the reader to other specific work [178]. In the present review, we will focus exclusively on work that has shed light on translational control in brain tumors. The two most common and most aggressive primary brain tumors, which have been extensively dissected through genomics, epigenomics, and transcriptomic studies over the last decade, are glioblastoma (GBM, [189]) and, in childhood populations, medulloblastoma (MB, [175]). While the most important alterations of mRNA translation in brain tumors are discussed below, a summary is also presented in Table 1.
Translation initiation control
A limited number of studies of the translational state of brain tumors have been reported, and largely in the context of GBM. To investigate in vivo translation regulation in brain tumors, Helmy et al. applied a method of ribosome-bound mRNA immunoprecipitation followed by deep sequencing to a genetically engineered glioma mouse model [72]. By comparing ribosome-bound mRNA to total mRNA levels, the authors measured the translation efficiency of single mRNAs in vivo. They found that > 60% of all genes were differentially represented in the ribosome-bound and non-bound mRNA sets, suggesting a high degree of protein biosynthesis regulation at the level of translation. Compared to non-neoplastic cells, the most efficiently translated genes belonged to the broad class of cell division and biosynthetic pathways. In addition, deletion of Phosphatase and Tensin homolog (PTEN, mutated or deleted in 36% of GBM, [24]) was found to translationally downregulate a set of genes involved in cellular respiration, consistently with the switch towards anaerobic glycolysis observed in PTEN-deleted tumors [13]. Combining a RiboTag system with ribosome profiling, Gonzalez et al. measured genome-wide ribosomal mRNA occupancy and translation rates in an in vivo glioma model [60]. They found that translation efficiency is highly cell-dependent, with remarkable differences between tumor and non-tumor cells. As opposed to non-transformed cells, there were high ribosome densities at the 5′-UTRs of mRNAs in glioma cells, suggesting that these cells have higher translation rates for the existing mRNAs. Among the genes highly translated in tumor cells, many were associated with cell motility, cell adhesion, and regulation of angiogenesis. These studies highlight translational control as an important phenomenon which should not be overlooked in the study of brain cancer.
The PI3K-Akt pathway is frequently hyperactive in GBM [189]. Oncogenic transformation in mouse derived GBM cells by Ras and activation of Akt signaling was shown to increase the translation of existing mRNAs, rather than altering transcript expression [144]. In particular, pharmacological inhibition of Akt leads to a decrease in the polysome-bound mRNA fraction, as opposed to the levels of total mRNA, of several target genes. Some of these genes, such as ATM (Ataxia-Telangectasia Mutated), Miz1, VEGF, and Notch1, are well known to be involved in GBM pathogenesis. The PI3K–Akt pathway is also known to stimulate cap-dependent protein synthesis, via activation of mTORC1, and to inhibit IRES-dependent translation. This effect is mediated by direct phosphorylation of the multifunctional RNA-binding heterogeneous nuclear ribonucleoprotein (hnRNP) A1 by Akt [121]. As a consequence, inhibition of mTORC1 by rapamycin in GBM is ineffective in inhibiting the translation of IRES mRNAs critical for cell cycle progression, such as Cyclin D1 and c-Myc [159]. However, co-targeting of IRES- and cap-dependent translation (i.e., by IRES-J007 and PP242, respectively) reduces tumor growth in a preclinical model of GBM [77]. IRES-dependent translation in GBM occurs also for c-Jun, a transcription factor that stimulates cell cycle progression [12], and accumulation of the c-Jun protein results from increased translatability of the transcript, rather than from upregulated mRNA levels. Additional links between GBM pathogenesis and translation control are suggested by the fact that isocitrate dehydrogenases 1 and 2 (IDH1/2) mutations, which are frequent in WHO grade 2 and 3 astrocytomas, oligodendrogliomas and secondary GBM [189], have been reported to modulate mTOR signaling [25]. Mechanistically, the oncometabolite R-2-hydroxyglutarate (2HG), produced by IDH1/2 gain of function mutations, inhibits the activity of the α-ketoglutarate-dependent enzyme Lysine-specific demethylase 4A (KDM4A). This enzyme normally promotes the demethylation of di- and tri-methylated H3K9 and H3K36. Inhibition of KDM4A results in a drastic decrease in levels of DEPTOR, an mTOR inhibitor; this stimulates mTOR signalling, in turn, increasing cap-dependent translation rates and protein synthesis [25]. This suggests that the potential therapeutic benefit for mTOR inhibition specifically in IDH-mutant glioma should be examined. Collectively, these results underscore deregulation of mRNA translation in glioma as a critical component of the disease. Further mechanistic studies are required to fully elucidate how translation control contributes to the aggressive behavior of GBM, and how targeting of this process might result in effective therapeutic strategies.
Translation elongation control
As discussed above, cellular stress reduces translation rates and protein synthesis [110]. The classic view is that acute cellular stress impairs the survival of normal cells, but adaptive mechanisms in cancer cells help them to survive in harsh milieus, such as under O2 and nutrient deprivation. Our recent data support the notion that processes regulating elongation are altered in tumor cells, and that such adaptation confers a survival advantage. We reported that the AMPK–eEF2K–eEF2 axis is part of a highly conserved pathway hijacked by cancer cells to overcome nutrient deprivation [109], a stress form typical of the tumor microenvironment. As stated earlier, under acute cellular stress, such as nutrient deprivation, phosphorylation of eEF2 (Thr56) by eEF2K reduces translation elongation rates and protein synthesis [152], a highly energy demanding cellular process. Indeed, mouse embryonic fibroblasts lacking eEF2K show lower polysome-associated mRNA levels under acute nutrient deprivation, suggesting that eEF2K stalls polyribosomes on mRNAs to help cells preserve energy and survive under nutrient deprivation. This mechanism seems particularly relevant for tumors driven by c-MYC or MYCN, such as pediatric MB and neuroblastoma, as transformation by these oncogenes renders cells hypersensitive to nutrient starvation [41, 42]. Indeed, in these tumors, as well as in GBM, eEF2K is upregulated, hyperactive, and linked to worse clinical outcome [42, 109]. Consistent with this, recent phospho-proteomic data reveal that in Group 3 MB, typically characterized by c-MYC overexpression, eEF2K is one of the most active kinases [5]. While detailed mechanistic studies are currently lacking, it is tempting to speculate that tight translation elongation control by eEF2K is critical for MYC-driven tumors, and that the effects of a loss of this process become phenotypically evident under conditions of cellular stress. Although the effects on translation were not examined, making it difficult to understand the significance, upregulation of translation elongation regulatory proteins such as eEF2 and eukaryotic translation elongation factor 1α (EEF1A2) is also linked to oncogenic transformation in genetically engineered mouse models of MB [54]. Collectively, these observations suggest that, by altering the rate of translation elongation, the AMPK–eEF2K–eEF2 pathway is critical for the survival and growth of brain tumors [42, 109]. While the role of this pathway in inhibiting bulk translation is well established, it remains to be deciphered if the same pathway is used by cancer cells for preferential translation of mRNAs required for survival and adaptation in their microenvironment.
Additional mechanisms of translation control
SG formation might influence tumor biology by altering the cellular translation landscape under stress, through silencing of SG-sequestered mRNA molecules [143]. In this context, a recent study reported increased SG formation in MB, leading to impaired global translation, due to frequent mutations of the DEAD-box RNA helicase DDX3X gene [54]. In addition, phospho-proteomics data suggest important differences in the activity of pathways regulating mRNA translation across distinct MB subgroups, including differential expression of RBPs, SG proteins, and mTOR pathway components [49]. While these results are preliminary, further mechanistic studies should help to further elucidate the role of SGs formation in brain tumors. A better understanding of how tumor cells exploit these mechanisms to survive and progress in their microenvironment will likely uncover new therapeutic opportunities.
Translational control in neurodevelopment and mental retardation disorders
Given the role of dendritic mRNA translation in synaptic function, it stands to reason that a number of neurological disorders result from altered mRNA translation. Fragile X syndrome is the most prominent monogenic cause of autism spectrum disorders and is probably the first neurological disease linked to alterations in translation [183]. Napoli et al. reported that FMRP, an RBP, inhibits translation initiation at the dendritic terminal through its binding partner, CYFIP1, which acts as a 4E-BP to block eIF4E activity [133]. An additional role for FMRP in this disease is suggested by the finding that mutations in the RNA-binding domain of FMRP impair its ability to bind to polyribosomes to alter translation elongation, underlying the Fragile X phenotype in mice and humans [192]. High-throughput sequencing of RNAs bound to FMRP revealed that FMRP reversibly stalls polyribosomes specifically on its target mRNAs, thus inhibiting their translation [39]. There was strong overlap between autism candidate genes, such as NLGN3, NRXN1, SHANK3, and FMRP target mRNAs, suggesting that loss of translation inhibition of specific mRNAs by FMRP in neurons may be the etiologic basis for the disease. It will be important to uncover whether these mechanisms are complimentary or mutually exclusive, and what their exact functional significance is in Fragile X phenotype. Alterations of other components of the translation initiation machinery, such as hyperphosphorylation of eIF4E, also contribute to the pathogenesis of Fragile X syndrome [57]. In addition, knockout of eIF4E-binding protein 2 (4EBP2) induces autistic-like behavior in mice, due to increased translation of neuroligins, which have been previously linked to this disease [58]. As mentioned, recent studies have implicated m6A modifications in cap-independent translation [124], and loss-of-function mutations of the FTO gene are described as leading to growth delay and mental retardation [14]. Additional mechanisms of RNA-mediated neurotoxicity are beyond the scope of this article and have been extensively reviewed elsewhere [10].
Although mTOR activity is essential for synaptic plasticity, overactive mTOR signaling can also hamper synaptic function and cognition, as can be witnessed in tuberous sclerosis complex (TSC) disease [35]. This rare disorder is caused by mutations in tumor suppressors TSC1 or TSC2, encoding hamartin and tuberin, respectively [37]. These proteins constitute a complex downstream of the PI3K-Akt pathway to stimulate the GTPase activity of the RAS family member RHEB (Ras Homolog Enriched in Brain), inducing GTP hydrolysis and conversion of RHEB into its inactive GDP-bound form [81]. Active RHEB localizes at the lysosome and induces phosphorylation and activation of mTOR [65]. Therefore, hyperactive mTOR signaling induced by genetic mutations in either TSC1 or TSC2 likely represents a crucial pathogenic mechanism for neuronal dysfunction in the TSC syndrome.
Translational control in neurodegenerative diseases
Neurodegenerative diseases represent a significant burden on the healthcare system. Alzheimer disease (AD) is the most common cause of dementia worldwide [78], while Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington disease (HD) represent leading neurodegenerative causes of motor disability [46]. Given that mRNA translation and protein synthesis are vital in the maintenance of synaptic structure and function, it is not surprising that defects in translation control are increasingly linked to these diseases [92]. Accordingly, a number of reports have shown that aberrations of translation regulation, including ribosomal recruitment, ribosomal components, RBPs, and metabolic pathways are common in neurodegenerative diseases [113].
Translation initiation control
As mentioned, eIF2α phosphorylation inhibits protein synthesis and is cytoprotective under acute stress, as part of the ISR [137]. Several reports have shown that eIF2α phosphorylation is elevated in AD and PD brains, pointing to persistent defects in protein synthesis and pathologically overactive stress responses in these diseases [117, 170]. Further evidence supporting defective mRNA translation in neurodegenerative diseases, possibly due to amyloidogenic protein aggregates, comes from studies on misfolded prion proteins [67, 130]. These works report that prion protein self-assembly induces translational repression through eIF2α phosphorylation, and restoration of mRNA translation efficiency in mouse hippocampi rescues neuronal loss and symptoms. One of the proposed mechanisms of pathology involves chronic eIF2α phosphorylation and consequent induction of SG formation, as discussed further below.
In regard to translation initiation control, mTOR and its downstream signaling targets have widely been studied in AD and PD pathology [1, 36, 136, 141], and in models of ALS [154] and HD [145]. These studies indicate that mTOR activity is increased in AD and PD brains, as reviewed elsewhere [36, 141]. In brief, phosphorylation of mTOR (Ser2481), 70S6K (Thr389 or Thr421/Ser424), eIF4E (Ser209), and 4EBP1/2 (Thr70 and Ser65) are elevated in postmortem AD medial temporal cortex [1, 111]. Similar observations of aberrant mTOR and eIF2/eIF4 signaling have also been reported in PD midbrain substantia nigra [51]. Cell culture studies and transgenic animal models suggest that mTOR activity may also be involved in pathological processes triggered by aggregation of disease-associated proteins, such as amyloid-β (Aβ) and tau in AD [1, 22, 196], and alpha-synuclein in PD [167]. This is not surprising, since mTOR acts as a negative regulator of autophagy, a known factor in neurodegenerative diseases, and possibly affects the clearance of aggregated amyloidogenic proteins [141].
Selective mRNA translation affecting neuronal survival in neurodegenerative diseases is also supported by studies showing decreased expression of the tumor suppressor HACE1 in HD. Expression of this protein, an HECT family E3 ligase that controls cellular oxidative stress [40], is decreased in HD striatal neurons [151]. HACE1 protects neurons from mutant huntingtin toxicity by increasing translation and activity of the nuclear factor erythroid 2-related (NRF2), an antioxidant master regulator [151]. Additional evidence for translation initiation alterations in neurodegenerative disease comes from the observation that some allelic variants of FTO demethylase that normally affect m6A modifications and, hence, cap-independent translation are associated with increased risk of developing dementia/AD [94]. However, detailed mechanistic studies on this topic are currently lacking.
Translation elongation control
Defective translation elongation control is also emerging as a crucial factor in the pathogenesis of many neurological disorders. In this regard, altered AMPK–eEF2K signaling is increasingly recognized in AD, PD, and HD (Fig. 4). AMPK signaling has been shown to decline with age, and may contribute to metabolic dysfunction and altered energy homeostasis in aging or age-related diseases [148, 153]. In AD and other tauopathies, phosphorylation of AMPK at Thr172, indicating increased activity, is found in cerebral neurons and to a lesser extent in astrocytes [186]. In these pathologies, p-AMPK immunohistochemical (IHC) localization is particularly abundant in neurons bearing pretangles and tangles, and in close proximity to dystrophic neurites surrounding amyloid plaques [186]. Similarly, increased p-AMPK is reported in the midbrain of chemical (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, MPTP) induced parkinsonism [27], and in the striatum of mutant huntingtin expressing R6/2 transgenic mice [126]. Interestingly, AMPK also stimulates autophagy by direct phosphorylation of ULK1, and there is evidence that AMPK negatively regulates mTOR activity and vice versa [158]. Altered translation elongation in neurodegenerative diseases is also highlighted by the observation that, in a subset of the autosomal dominant spinocerebellar ataxias, a single-nucleotide variant results in a P596H amino acid substitution in eEF2 [71]. This mutation impairs the ability of eEF2 to maintain binding to the mRNA reading frame and results in impaired ribosome translocation during elongation. Moreover, Ishimura et al. identified a single nuclear polymorphism in the T loop stem of n-Tr20, a tRNA isodecoder, as inducing neurodegeneration in mice, when combined with loss Gtpbp2, part of the ribosome stalling detection system [83]. Ribosome profiling experiments in mutant mice revealed that the n-Tr20 mutation stalls ribosomes specifically on AGA codons of mRNA. The lack of other AGA isodecoders in the brain suggests that the expression of the n-Tr20 tRNA isodecoder is required for correct translation elongation in mammals.

Activity of eEF2 kinase is increased in Alzheimer’s (AD) and Parkinson’s (PD) disease brains. a Immunohistochemical detection of phospho-eEF2 (Thr56), as a readout of eEF2K activity, and phospho-TAU was performed on postmortem serial sections of the CA1 hippocampal region in control and AD brains, as previously described [86]. b Immunohistochemistry for phospho-eEF2 (Thr56) was performed on sections of the substantia nigra and Hippocampus in control and PD brains, as previously described [85]. Scale bar = 80 μm
Recent reports, including our own data, show that the expression and/or activity of eEF2K are increased in AD brain. Earlier studies by Li et al. reported that eEF2K activity, as assessed by IHC detection of eEF2 (Thr56) phosphorylation or by Western blots, is increased in postmortem AD mesial temporal cortex [111]. They also demonstrated decreased eEF2 levels in AD brain, particularly in AD cases neuropathologically diagnosed as stages IV–VI of Braak neurofibrillary tangle pathology [111]. Ma et al. confirmed these observations and found increased eEF2 (Thr56) phosphorylation in homogenates of postmortem AD mesial temporal cortex [116]. Expanding these studies to gain further mechanistic insights, we found that eEF2 (Thr56) phosphorylation is particularly increased in the hippocampal fields, cornu ammonis 1–3 (CA1–CA3), and in cortical layers III–V in postmortem AD brains [86] (Fig. 4a). Furthermore, eEF2 phosphorylation is significantly increased in the hippocampus of aged transgenic AD mice, although with conflicting evidence that this was due to changes in AMPK activity [86, 116]. In a follow-up report, we found, by IHC analysis, that eEF2 phosphorylation was markedly increased in postmortem PD midbrain (neurons of substantia nigra and periaqueductal gray), and to a lesser extent in hippocampus sections (CA1 and CA2) [85] (Fig. 4b). It remains to be determined whether aberrant AMPK–eEF2K signaling is also involved in other neurodegenerative diseases such as HD and ALS.
Overall, the anticipated effect of these alterations is the global shutdown of protein synthesis, except for mRNAs required for stress adaptation and survival, and suppression of long-term plasticity [69]. It is possible that, in the short term, these changes represent adaptive responses of neurons and may have some survival advantages in the face of disease processes such as protein aggregation (e.g., tau in AD and alpha-synuclein in PD). However, prolonged and/or chronic reduction in translation and protein synthesis may seriously hamper synaptic function and lead to synaptic and neuronal loss. Taken together, alterations in translation elongation control appear to contribute to the pathogenesis of several neurodegenerative diseases. Nevertheless, further studies are required to determine the exact molecular determinants of these aberrations, how they contribute to symptomatology and whether therapeutic targeting will have any beneficial effect on the disease course.
Repeat-associated non-AUG (RAN) translation in neurodegeneration
Nucleotide repeat expansions have been extensively studied in relation to their potential to produce insoluble protein aggregates toxic to neurons, which is the mechanistic basis of several neurodegenerative diseases, as extensively reviewed elsewhere [105]. Until recently, it was considered that this class of mutations encodes proteins only when retained as an open-reading frame within an annotated protein coding region. The recent discovery of repeat-associated non-AUG (RAN) translation has shed further light on the pathogenesis of microsatellite repeat diseases [199].
RAN translation occurs across expanded repetitive RNA repeats even in the absence of an AUG start codon, and was initially described in association with CAG repeats in spinocerebellar ataxia type 8 (SCA8) [199]. Loss of the canonical requirements for translation initiation results in six possible reading frames: CAG repeats can be read as CAG, AGC, GCA, and the antisense CUG, UGC, GCU [140], which can be translated into potentially toxic proteins containing homopolymeric polypeptides of polyglutamine, polyserine, polyalanine, polyleucine, polycysteine, and polyalanine. Expressed proteins resulting from RAN translation, characterized by dipeptide repeats, display an increased tendency to self-aggregate and induce neurodegeneration [140, 157]. This form of aberrant translation has been described for multiple-nucleotide repeat sequences, including but not limited to CGG repeats in Fragile X Tremor Ataxia Syndrome (FXTAS) [176], GGGGCC and GGCCCC repeats in C9orf72 ALS and frontotemporal dementia (FTD) [53, 122, 131, 132, 155, 195, 200], and CAG repeats in HD [8], as extensively reviewed [29].
The potential mechanistic basis of RAN translation initiation is complex and depends on the disease, mutated gene, and nucleotide repeat, as extensively reviewed elsewhere [29, 64]. For example, some reports show that GGGGCC repeats in C9orf72 form complex RNA G-quadruplex structures [146], thus possibly indicating an effect of these structures on translation initiation efficiency. Experiments in cell free in vitro systems also show that translation of GGGGCC repeats depends on a close-cognate codon and is regulated by an uORF [172]. However, a different model, where translation starts at uncapped repeat-containing intronic regions, is supported by data in cellular systems [26]. In addition, a role for eIF2α phosphorylation under chronic cellular stress in promoting selective translation of microsatellite-containing transcripts in C9orf72 ALS/FTD has recently been proposed [26, 63]. In contrast, translation efficiency of CGG repeats in FXTAS seems to be dependent on the reading frame, with repeats resulting in the expression of polyglycine and polyalanine but not polyarginine in cell culture [176]. It is clear from different model systems that aberrant proteins resulting from RAN translation are neurotoxic and induce neurodegeneration [50, 122, 125, 182, 195]. Hence, elucidating the molecular basis of RAN translation may offer novel opportunities for its targeting, as reviewed elsewhere [29, 150].
Stress granules in neurodegenerative diseases
As discussed above, RBPs containing a prion-like low complexity domain (LCD), such as TIA1, G3BP1, TDP-43, FUS, EWRS1, and hnRNPA1, self-assemble into SGs along with mRNAs by the process of phase separation, occurring when a solution oversaturated with multiple components spontaneously separates into a solid and a liquid phase [15, 127, 139]. Self-assembly and recruitment into SGs is accelerated by pathologic mutations in the LCDs of several RBPs, and inappropriate phase separation leading to pathological self-assembly is linked to the development of neurodegenerative diseases associated with mutations in RBPs, including TDP-43 and FUS in ALS and FTD [68, 127, 139]. Consistent with this notion, an increasing number of mutations in RBPs (e.g., hnRNPA1, FUS, TPD-43, Atx2, and TIA1) that promote their self-assembly are being discovered in ALS/FTD, as recently reviewed [11].
In ALS, after the discovery that neuronal protein aggregates contain the TDP-43 protein [134], more than 30 different disease-linked mutations in the TDP-43 gene have been identified [104, 120]. Most mutations induce TDP-43 localization in the cytoplasm and its association into SGs [112]. Several other disease-causing mutations, such as C9orf72 repeat expansions [108, 118] and mutant hnRNPA2/B1 and hnRNAPA1 [99], have been described to induce SG formation. Moreover, polyGly-Arg and polyPro-Arg dipeptide repeats resulting from RAN translation of C9orf72 expansions directly interact with SGs and promote their assembly [108], and RNA self-assembly induced by pathogenic dipeptide repeats promotes SG formation [180]. In addition to SG hyperformation, alterations in SG dynamics and inhibition of their dissociation have also been proposed to induce neurodegeneration in ALS. SG clearance in mammalian cells is linked to autophagy, and is impaired by the inhibition of valosin-containing peptide (VCP) [18]. Interestingly, VCP mutations predispose to several neurodegenerative diseases, including ALS [90]. Moreover, a P362L mutation in the prion-like domain of TIA1, inducing phase separation and impaired SG disassembly, has been recently identified in ALS/FTD familial cases [75, 119]. Intermediate length polyQ expansions (27-33 glutamines) of the Ataxin-2 gene (ATX2), promoting protein self-assembly and interaction with TDP-43, are another common finding in ALS patients [47].
While multiple mechanisms of neurotoxicity have been proposed in ALS/FTD (as extensively reviewed [174, 182]), ample literature argues that mutations in RBPs also affect their RNA-binding ability, possibly altering mRNA translation. Most of this evidence comes from studies in Drosophila, where mutations that prevent the RNA-binding capacity of FUS attenuate its neurotoxicity [38]. In mice, expression of ALS-linked FUS mutations impairs local protein synthesis in neurons without inducing protein aggregates, suggesting that mutant FUS affects mRNA translation [115]. Similarly, the RNA-binding capacity of TDP-43 contributes to neurodegeneration [7], suggesting a role for RNA sequestration in the disease. Consistent with this notion, restoring translation of target mRNAs of TDP-43, such as futsch/MAP1B, decreases neurodegeneration in ALS [34]. VCP and FUS were recently shown to directly interact with ribosomes in cell culture, pointing to potential roles in controlling translation [163]. In the context of C9orf72 expansions, SG formation was reported to impair translation in both cell culture [108] and mouse models [194]. It should also be mentioned that not all the genetic mutations identified in ALS/FTD have been linked to SGs assembly [16]. Nevertheless, the existence of multiple disease-linked mutations in RBPs, promoting their self-assembly into SGs, supports a mechanistic model whereby pathological SG assembly over time impairs motor neuron function, at least partially by altering mRNA translation. Future studies are required to fully understand this process and potential targets for therapy.
In other chronic neurodegenerative diseases, SG proteins such as TIA1, eIF3, TDP-43, and polyA binding protein (PABP) have been observed to co-localize with neuropathological markers in postmortem brain tissue in AD [4, 79, 181, 190]. In cell culture, tau, huntingtin, and misfolded prion protein (PrPsc) associate with SGs and to modulate their formation [59, 188], suggesting a potential role for SGs in pathogenic mechanisms of these diseases. Studies demonstrating interactions between tau and SG proteins also suggest that SG formation increases tau phosphorylation, indicating that SGs might contribute to tau pathology [88, 181]. In PD, TDP-43 inclusions have been reported in postmortem tissue [55] and the antioxidant protein DJ-1, frequently mutated in PD, was also shown to associate with SGs during neurodegeneration [147]. It is important to distinguish between the self-assembly of SG proteins during stress, and the aggregation of amyloid-forming proteins such as tau in AD. In this context, Wolozin has proposed a model of regulated protein aggregation in neurodegenerative diseases, in which SG formation in neurons is an adaptive stress response to the toxicity of mutant proteins (i.e., TDP-43 and FUS) and/or to the pathological excess of amyloidogenic proteins such as tau [190]. Based on this model, transient formation of SGs in neurons may be protective, but unregulated and overactive SG formation may cause deleterious effects by sequestering crucial transcripts and/or proteins needed for efficient plasticity and neuronal survival. It is also plausible that transiently overactive SG assembly is triggered by aggregation of amyloidogenic proteins (e.g., tau), and SGs may help to sequester toxic amyloidogenic oligomers. However, stable (i.e., pathological) SG structures may provide a nidus for the nucleation of these amyloid-forming proteins, and, hence, represent a vicious cycle in the formation and propagation of neurotoxic oligomeric aggregates. While further studies are required to fully elucidate the role of SGs in the pathogenesis of neurodegenerative diseases other than ALS/FTD, it is reasonable to postulate a model whereby protein aggregation, chronic oxidative stress, and neuronal injury activate the cellular stress responses, inducing eIF2α phosphorylation and the formation of SGs. Supportive of this model is the common finding of phosphorylated eIF2α in neurodegenerative diseases, as well as recent studies showing that misfolded prion proteins [130] and TDP-43 [100] induce chronic eIF2α phosphorylation, and inhibition of this pathway mitigates TDP-43 toxicity [100]. Whether modulating this response might represent a therapeutic approach to these pathologies will be a relevant topic for future investigations, as discussed in the therapy section below.
Therapeutic targeting of aberrant translation in brain tumors
The following discussion will focus exclusively on emerging concepts on therapeutic targeting of brain tumors with a special emphasis on SG biology, eIF2α phosphorylation, and the AMPK–eEF2K–eEF2 axis. As mentioned, SG formation represents an important component of the adaptive response of cells to acute stress. Consistently with this notion, blocking SG formation renders cells highly sensitive to apoptosis under oxidative stress conditions [6, 173]. Furthermore, we reported that blocking SG significantly attenuates the metastatic and invasive capacity of pediatric sarcomas [164]. Although early literature suggests that SG assembly inhibition in vitro in GBM sensitizes cells to chemotherapeutic agents [185], the role of SGs in brain tumor progression, invasive capacity, and antioxidant response remains largely unexplored. Investigation of this field will be of particular interest in deciphering chemoresistance, since chemotherapeutic agents (e.g., vincristine) used in the therapy of brain tumors are reported to promote SG formation [171]. In addition, drugs inhibiting SG formation, such as nocodazole [103], should be evaluated for their capacity to inhibit brain tumor progression and invasion.
As discussed above, four different kinases (PKR, HRI, GCN2, and PERK), activated by distinct stress forms, phosphorylate eIF2α on Ser51 as part of the ISR [129]. The small molecule ISRIB, an activator of the guanine nucleotide exchange factor eIF2B, has recently been shown to render cells resistant to the effects of eIF2α phosphorylation without affecting eIF2α phosphorylation itself [160, 162]. ISRIB is a potent inhibitor of SG formation and promotes the disassembly of preformed SGs [161]. In addition, the small size of this molecule and its ability to penetrate the blood–brain barrier [66] makes it an ideal candidate for SG inhibition in brain tumors.
As also previously discussed, the AMPK–eEF2K axis is critical for adaptation of cancer cells to diverse acute microenvironmental stresses [109, 110]. In this context, we have shown that MYC overexpressing tumors are particularly sensitive to eEF2K inhibition in combination with caloric restriction diets [41, 42]. Despite the challenge of identifying and developing inhibitors for an atypical α-kinase such as eEF2K, several drugs have been described as eEF2K inhibitors, with conflicting opinions regarding their specificity [44], and this topic is reviewed elsewhere [114]. Testing the efficacy of eEF2K inhibition, particularly in combination with caloric restriction mimetics, glycolysis, or mTOR inhibitors, will be of interest in MYC-driven cancers, such as neuroblastoma, MB, and pediatric GBM.
Therapeutic targeting of aberrant translation in neurodegenerative diseases
Although many studies have explored the potential benefits of targeting translation in tumors, similar therapeutic approaches for neurodegenerative diseases remain largely unexplored. Given the complex etiology of neurodegenerative diseases discussed in this review, it may be challenging to design clinically beneficial therapeutic agents targeting more than a single mechanism in translation regulation (i.e., translation initiation or elongation). However, it is not unlikely that the neuronal dysfunction and neuronal loss induced by amyloid aggregation and/or deleterious effects of rare disease-causing mutations (e.g. APP in AD, and alpha-synuclein in PD) on neuronal protein synthesis could be mitigated. For example, clearance of SGs by autophagy has been proposed as a possible therapeutic approach in neurological diseases characterized by pathologic accumulation of SGs and RBPs [18], and it is possible that a moderate reduction in SG formation may prove beneficial in chronic pathological conditions [3]. More mechanistic studies are, however, needed to determine the therapeutic value of targeting pathways relevant to SG formation in neurological diseases. In this context, decreasing expression or activity of the eIF2α kinases GCN and PERK in transgenic AD mice (expressing the mutant human APP and presenilin 1-APP/PS1) prevented synaptic deficits and improved performance in tasks of spatial memory [117]. Consistent with the notion that eIF2α acts as an important memory repressor, preventing the effects of eIF2α phosphorylation with the small molecule ISRIB significantly improves hippocampal-dependent memory function in a mouse model of traumatic brain injury [28]. Further studies are needed to fully explore the effectiveness of ISRIB as a therapeutic strategy in neurodegenerative diseases.
With regard to the potential therapeutic utility of targeting pathological hyperactivity of the AMPK–eEF2K–eEF2 axis in neurodegenerative diseases, genetic deletion of the AMPKα2 subunit, treatment with a small molecule inhibitor of AMPK (compound C), or exposure to a small molecule inhibitor of eEF2K prevented the detrimental effects of Aβ on LTP in hippocampal slices and improved protein synthesis [116]. In line with these observations, we have explored the potential benefits of eEF2K inhibition in models of neurotoxicity induced by amyloidogenic proteins, specifically Aβ and alpha-synuclein in neuronal cultures. Indeed, eEF2K inhibition, by pharmacological and genetic approaches, prevented the toxic effects of Aβ42 oligomers on neuronal viability and dendrite maturation in neuronal cultures [86], and these types of results were recently confirmed in a transgenic mouse model of AD [9]. Furthermore, we observed that eEF2K inhibition reduced oxidative stress in cultures of dopaminergic neuroblastoma N2A cells by promoting NRF2 antioxidant response, and prevented the oxidative stress associated with application of hydrogen peroxide (H2O2) treatment [86]. Taken together, these data support the notion that targeting defective mRNA translation and protein synthesis represents a promising strategy in chronic neurodegenerative diseases.
Conclusions and perspectives
Emerging evidence suggests that loss of translation control is a common pathologic hallmark across diverse neurological disorders, including CNS tumors, neurodevelopmental and mental retardation disorders, and neurodegenerative diseases. In this article, we set out to review the role of mRNA translation in different brain diseases, and how different alterations in one pathway may contribute to very distinct pathologies. The knowledge acquired from a better understanding of the biological basis of one disease could open unexpected therapeutic opportunities for a different one. For example, unraveling the basis of RAN translation in repeat expansion diseases may suggest novel mechanisms of translation exploited by cancer cells to adapt to their microenvironment. A better understanding of how translation control impacts brain pathologies is critical for the development of novel and targeted therapies for these diseases.
References
An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K et al (2003) Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol 163:591–607. https://doi.org/10.1016/S0002-9440(10)63687-5
Anderson P, Kedersha N (2006) RNA granules. J Cell Biol 172:803–808. https://doi.org/10.1083/jcb.200512082
Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Al Abdullatif A et al (2018) Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci 21:72–80. https://doi.org/10.1038/s41593-017-0022-z
Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K et al (2009) Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 117:125–136. https://doi.org/10.1007/s00401-008-0480-1
Archer TC, Ehrenberger T, Mundt F, Gold MP, Krug K, Mah CK et al (2018) Proteomics, post-translational modifications, and integrative analyses reveal molecular heterogeneity within medulloblastoma subgroups. Cancer Cell 34(396–410):e398. https://doi.org/10.1016/j.ccell.2018.08.004
Arimoto-Matsuzaki K, Saito H, Takekawa M (2016) TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat Commun 7:10252. https://doi.org/10.1038/ncomms10252
Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E et al (2010) Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet 19:3206–3218. https://doi.org/10.1093/hmg/ddq230
Banez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK et al (2015) RAN translation in huntington disease. Neuron 88:667–677. https://doi.org/10.1016/j.neuron.2015.10.038
Beckelman BC, Yang W, Kasica NP, Zimmermann HR, Zhou X, Keene CD et al (2019) Genetic reduction of eEF2 kinase alleviates pathophysiology in Alzheimer’s disease model mice. J Clin Invest 25:25. https://doi.org/10.1172/jci122954
Belzil VV, Gendron TF, Petrucelli L (2013) RNA-mediated toxicity in neurodegenerative disease. Mol Cell Neurosci 56:406–419. https://doi.org/10.1016/j.mcn.2012.12.006
Benarroch EE (2018) Cytoplasmic RNA granules, ribostasis, and neurodegeneration. Neurology 90:553–562. https://doi.org/10.1212/WNL.0000000000005172
Blau L, Knirsh R, Ben-Dror I, Oren S, Kuphal S, Hau P et al (2012) Aberrant expression of c-Jun in glioblastoma by internal ribosome entry site (IRES)-mediated translational activation. Proc Natl Acad Sci U S A 109:E2875–E2884. https://doi.org/10.1073/pnas.1203659109
Blouin MJ, Zhao Y, Zakikhani M, Algire C, Piura E, Pollak M (2010) Loss of function of PTEN alters the relationship between glucose concentration and cell proliferation, increases glycolysis, and sensitizes cells to 2-deoxyglucose. Cancer Lett 289:246–253. https://doi.org/10.1016/j.canlet.2009.08.021
Boissel S, Reish O, Proulx K, Kawagoe-Takaki H, Sedgwick B, Yeo GS et al (2009) Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet 85:106–111. https://doi.org/10.1016/j.ajhg.2009.06.002
Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J et al (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324:1729–1732. https://doi.org/10.1126/science.1172046
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377:162–172. https://doi.org/10.1056/NEJMra1603471
Browne GJ, Finn SG, Proud CG (2004) Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem 279:12220–12231. https://doi.org/10.1074/jbc.M309773200
Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153:1461–1474. https://doi.org/10.1016/j.cell.2013.05.037
Buffington SA, Huang W, Costa-Mattioli M (2014) Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci 37:17–38. https://doi.org/10.1146/annurev-neuro-071013-014100
Burkewitz K, Zhang Y, Mair WB (2014) AMPK at the nexus of energetics and aging. Cell Metab 20:10–25. https://doi.org/10.1016/j.cmet.2014.03.002
Buxbaum AR, Wu B, Singer RH (2014) Single beta-actin mRNA detection in neurons reveals a mechanism for regulating its translatability. Science 343:419–422. https://doi.org/10.1126/science.1242939
Caccamo A, Magri A, Medina DX, Wisely EV, Lopez-Aranda MF, Silva AJ et al (2013) mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell 12:370–380. https://doi.org/10.1111/acel.12057
Cajigas IJ, Tushev G, Will TJ, tom Dieck S, Fuerst N, Schuman EM (2012) The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74:453–466. https://doi.org/10.1016/j.neuron.2012.02.036
Cancer Genome Atlas Research N (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. https://doi.org/10.1038/nature07385
Carbonneau M, Gagné LM, Lalonde ME, Germain MA, Motorina A, Guiot MC et al (2016) The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat Commun 7:12700. https://doi.org/10.1038/ncomms12700
Cheng W, Wang S, Mestre AA, Fu C, Makarem A, Xian F et al (2018) C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2alpha phosphorylation. Nat Commun 9:51. https://doi.org/10.1038/s41467-017-02495-z
Choi JS, Park C, Jeong JW (2010) AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun 391:147–151. https://doi.org/10.1016/j.bbrc.2009.11.022
Chou A, Krukowski K, Jopson T, Zhu PJ, Costa-Mattioli M, Walter P et al (2017) Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc Natl Acad Sci U S A 114:E6420–E6426. https://doi.org/10.1073/pnas.1707661114
Cleary JD, Pattamatta A, Ranum LPW (2018) Repeat-associated non-ATG (RAN) translation. J Biol Chem 293:16127–16141. https://doi.org/10.1074/jbc.R118.003237
Collingridge GL, Peineau S, Howland JG, Wang YT (2010) Long-term depression in the CNS. Nat Rev Neurosci 11:459–473. https://doi.org/10.1038/nrn2867
Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C et al (2007) eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129:195–206. https://doi.org/10.1016/j.cell.2007.01.050
Costa-Mattioli M, Monteggia LM (2013) mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci 16:1537–1543. https://doi.org/10.1038/nn.3546
Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N (2009) Translational control of long-lasting synaptic plasticity and memory. Neuron 61:10–26. https://doi.org/10.1016/j.neuron.2008.10.055
Coyne AN, Siddegowda BB, Estes PS, Johannesmeyer J, Kovalik T, Daniel SG et al (2014) Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci 34:15962–15974. https://doi.org/10.1523/JNEUROSCI.2526-14.2014
Crino PB (2013) Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol 125:317–332. https://doi.org/10.1007/s00401-013-1085-x
Crino PB (2016) The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol 12:379–392. https://doi.org/10.1038/nrneurol.2016.81
Curatolo P, Moavero R, de Vries PJ (2015) Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol 14:733–745. https://doi.org/10.1016/S1474-4422(15)00069-1
Daigle JG, Lanson NA Jr, Smith RB, Casci I, Maltare A, Monaghan J et al (2013) RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet 22:1193–1205. https://doi.org/10.1093/hmg/dds526
Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE et al (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146:247–261. https://doi.org/10.1016/j.cell.2011.06.013
Daugaard M, Nitsch R, Razaghi B, McDonald L, Jarrar A, Torrino S et al (2013) Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat Commun 4:2180. https://doi.org/10.1038/ncomms3180
Delaidelli A, Leprivier G, Sorensen PH (2017) eEF2K protects MYCN-amplified cells from starvation. Cell Cycle 16:1633–1634. https://doi.org/10.1080/15384101.2017.1355180
Delaidelli A, Negri GL, Jan A, Jansonius B, El-Naggar A, Lim JKM et al (2017) MYCN amplified neuroblastoma requires the mRNA translation regulator eEF2 kinase to adapt to nutrient deprivation. Cell Death Differ 24:1564–1576. https://doi.org/10.1038/cdd.2017.79
Dever TE, Green R (2012) The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol 4:a013706. https://doi.org/10.1101/cshperspect.a013706
Devkota AK, Warthaka M, Edupuganti R, Tavares CD, Johnson WH, Ozpolat B et al (2014) High-throughput screens for eEF-2 kinase. J Biomol Screen 19:445–452. https://doi.org/10.1177/1087057113505204
Di Prisco GV, Huang W, Buffington SA, Hsu CC, Bonnen PE, Placzek AN et al (2014) Translational control of mGluR-dependent long-term depression and object-place learning by eIF2alpha. Nat Neurosci 17:1073–1082. https://doi.org/10.1038/nn.3754
Dugger BN, Dickson DW (2017) Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a028035
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X et al (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069–1075. https://doi.org/10.1038/nature09320
Faller WJ, Jackson TJ, Knight JR, Ridgway RA, Jamieson T, Karim SA et al (2015) mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517:497–500. https://doi.org/10.1038/nature13896
Forget A, Martignetti L, Puget S, Calzone L, Brabetz S, Picard D et al (2018) Aberrant ERBB4-SRC signaling as a hallmark of group 4 medulloblastoma revealed by integrative phosphoproteomic profiling. Cancer Cell 34(379–395):e377. https://doi.org/10.1016/j.ccell.2018.08.002
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133. https://doi.org/10.1038/nature14974
Garcia-Esparcia P, Hernandez-Ortega K, Koneti A, Gil L, Delgado-Morales R, Castano E et al (2015) Altered machinery of protein synthesis is region- and stage-dependent and is associated with alpha-synuclein oligomers in Parkinson’s disease. Acta Neuropathol Commun 3:76. https://doi.org/10.1186/s40478-015-0257-4
Garza-Lombo C, Schroder A, Reyes-Reyes EM, Franco R (2018) mTOR/AMPK signaling in the brain: cell metabolism, proteostasis and survival. Curr Opin Toxicol 8:102–110. https://doi.org/10.1016/j.cotox.2018.05.002
Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 126:829–844. https://doi.org/10.1007/s00401-013-1192-8
Genovesi LA, Ng CG, Davis MJ, Remke M, Taylor MD, Adams DJ et al (2013) Sleeping Beauty mutagenesis in a mouse medulloblastoma model defines networks that discriminate between human molecular subgroups. Proc Natl Acad Sci U S A 110:E4325–E4334. https://doi.org/10.1073/pnas.1318639110
Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ (2009) Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol 256:1205–1214. https://doi.org/10.1007/s00415-009-5069-7
Gkogkas C, Sonenberg N, Costa-Mattioli M (2010) Translational control mechanisms in long-lasting synaptic plasticity and memory. J Biol Chem 285:31913–31917. https://doi.org/10.1074/jbc.R110.154476
Gkogkas CG, Khoutorsky A, Cao R, Jafarnejad SM, Prager-Khoutorsky M, Giannakas N et al (2014) Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep 9:1742–1755. https://doi.org/10.1016/j.celrep.2014.10.064
Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB et al (2013) Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493:371–377. https://doi.org/10.1038/nature11628
Goggin K, Beaudoin S, Grenier C, Brown AA, Roucou X (2008) Prion protein aggresomes are poly(A) + ribonucleoprotein complexes that induce a PKR-mediated deficient cell stress response. Biochim Biophys Acta 1783:479–491. https://doi.org/10.1016/j.bbamcr.2007.10.008
Gonzalez C, Sims JS, Hornstein N, Mela A, Garcia F, Lei L et al (2014) Ribosome profiling reveals a cell-type-specific translational landscape in brain tumors. J Neurosci 34:10924–10936. https://doi.org/10.1523/JNEUROSCI.0084-14.2014
Gowans GJ, Hawley SA, Ross FA, Hardie DG (2013) AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18:556–566. https://doi.org/10.1016/j.cmet.2013.08.019
Graber TE, Hebert-Seropian S, Khoutorsky A, David A, Yewdell JW, Lacaille JC et al (2013) Reactivation of stalled polyribosomes in synaptic plasticity. Proc Natl Acad Sci U S A 110:16205–16210. https://doi.org/10.1073/pnas.1307747110
Green KM, Glineburg MR, Kearse MG, Flores BN, Linsalata AE, Fedak SJ et al (2017) RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun 8:2005. https://doi.org/10.1038/s41467-017-02200-0
Green KM, Linsalata AE, Todd PK (2016) RAN translation-What makes it run? Brain Res 1647:30–42. https://doi.org/10.1016/j.brainres.2016.04.003
Groenewoud MJ, Zwartkruis FJ (2013) Rheb and Rags come together at the lysosome to activate mTORC1. Biochem Soc Trans 41:951–955. https://doi.org/10.1042/BST20130037
Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J et al (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 6:e1672. https://doi.org/10.1038/cddis.2015.49
Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC et al (2017) Repurposed drugs targeting eIF2alpha-P-mediated translational repression prevent neurodegeneration in mice. Brain 140:1768–1783. https://doi.org/10.1093/brain/awx074
Harrison AF, Shorter J (2017) RNA-binding proteins with prion-like domains in health and disease. Biochem J 474:1417–1438. https://doi.org/10.1042/BCJ20160499
Heise C, Gardoni F, Culotta L, di Luca M, Verpelli C, Sala C (2014) Elongation factor-2 phosphorylation in dendrites and the regulation of dendritic mRNA translation in neurons. Front Cell Neurosci 8:35. https://doi.org/10.3389/fncel.2014.00035
Heise C, Taha E, Murru L, Ponzoni L, Cattaneo A, Guarnieri FC et al (2017) eEF2K/eEF2 pathway controls the excitation/inhibition balance and susceptibility to epileptic seizures. Cereb Cortex 27:2226–2248. https://doi.org/10.1093/cercor/bhw075
Hekman KE, Yu GY, Brown CD, Zhu H, Du X, Gervin K et al (2012) A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum Mol Genet 21:5472–5483. https://doi.org/10.1093/hmg/dds392
Helmy K, Halliday J, Fomchenko E, Setty M, Pitter K, Hafemeister C et al (2012) Identification of global alteration of translational regulation in glioma in vivo. PLoS One 7:e46965. https://doi.org/10.1371/journal.pone.0046965
Hershey JW, Sonenberg N, Mathews MB (2012) Principles of translational control: an overview. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a011528
Hinnebusch AG, Ivanov IP, Sonenberg N (2016) Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 352:1413–1416. https://doi.org/10.1126/science.aad9868
Hirsch-Reinshagen V, Pottier C, Nicholson AM, Baker M, Hsiung GR, Krieger C et al (2017) Clinical and neuropathological features of ALS/FTD with TIA1 mutations. Acta Neuropathol Commun 5:96. https://doi.org/10.1186/s40478-017-0493-x
Hoagland MB, Stephenson ML, Scott JF, Hecht LI, Zamecnik PC (1958) A soluble ribonucleic acid intermediate in protein synthesis. J Biol Chem 231:241–257
Holmes B, Lee J, Landon KA, Benavides-Serrato A, Bashir T, Jung ME et al (2016) Mechanistic target of rapamycin (mTOR) inhibition synergizes with reduced internal ribosome entry site (IRES)-mediated translation of cyclin D1 and c-MYC mRNAs to treat glioblastoma. J Biol Chem 291:14146–14159. https://doi.org/10.1074/jbc.M116.726927
Holtzman DM, Morris JC, Goate AM (2011) Alzheimer’s disease: the challenge of the second century. Sci Transl Med 3:77sr71. https://doi.org/10.1126/scitranslmed.3002369
Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC et al (2008) Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol 116:215–220. https://doi.org/10.1007/s00401-008-0400-4
Huber KM, Kayser MS, Bear MF (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288:1254–1257
Inoki K, Li Y, Xu T, Guan KL (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834. https://doi.org/10.1101/gad.1110003
Ishimura R, Nagy G, Dotu I, Chuang JH, Ackerman SL (2016) Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife 5:5. https://doi.org/10.7554/elife.14295
Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P et al (2014) RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345:455–459. https://doi.org/10.1126/science.1249749
Jackson RJ, Hellen CU, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11:113–127. https://doi.org/10.1038/nrm2838
Jan A, Jansonius B, Delaidelli A, Bhanshali F, An YA, Ferreira N et al (2018) Activity of translation regulator eukaryotic elongation factor-2 kinase is increased in Parkinson disease brain and its inhibition reduces alpha synuclein toxicity. Acta Neuropathol Commun 6:54. https://doi.org/10.1186/s40478-018-0554-9
Jan A, Jansonius B, Delaidelli A, Somasekharan SP, Bhanshali F, Vandal M et al (2017) eEF2K inhibition blocks Abeta42 neurotoxicity by promoting an NRF2 antioxidant response. Acta Neuropathol 133:101–119. https://doi.org/10.1007/s00401-016-1634-1
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y et al (2011) N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7:885–887. https://doi.org/10.1038/nchembio.687
Jiang L, Ash PEA, Maziuk BF, Ballance HI, Boudeau S, Abdullatif AA et al (2018) TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol 25:5. https://doi.org/10.1007/s00401-018-1937-5
Jiang Z, Belforte JE, Lu Y, Yabe Y, Pickel J, Smith CB et al (2010) eIF2alpha Phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J Neurosci 30:2582–2594. https://doi.org/10.1523/JNEUROSCI.3971-09.2010
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ et al (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68:857–864. https://doi.org/10.1016/j.neuron.2010.11.036
Kang H, Schuman EM (1996) A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273:1402–1406
Kapur M, Monaghan CE, Ackerman SL (2017) Regulation of mRNA translation in neurons-a matter of life and death. Neuron 96:616–637. https://doi.org/10.1016/j.neuron.2017.09.057
Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J et al (2002) Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13:195–210. https://doi.org/10.1091/mbc.01-05-0221
Keller L, Xu W, Wang HX, Winblad B, Fratiglioni L, Graff C (2011) The obesity related gene, FTO, interacts with APOE, and is associated with Alzheimer’s disease risk: a prospective cohort study. J Alzheimers Dis 23:461–469. https://doi.org/10.3233/JAD-2010-101068
Kenney JW, Genheden M, Moon KM, Wang X, Foster LJ, Proud CG (2016) Eukaryotic elongation factor 2 kinase regulates the synthesis of microtubule-related proteins in neurons. J Neurochem 136:276–284. https://doi.org/10.1111/jnc.13407
Kenney JW, Moore CE, Wang X, Proud CG (2014) Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv Biol Regul 55:15–27. https://doi.org/10.1016/j.jbior.2014.04.003
Kenney JW, Sorokina O, Genheden M, Sorokin A, Armstrong JD, Proud CG (2015) Dynamics of elongation factor 2 kinase regulation in cortical neurons in response to synaptic activity. J Neurosci 35:3034–3047. https://doi.org/10.1523/JNEUROSCI.2866-14.2015
Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R (2017) The stress granule transcriptome reveals principles of mrna accumulation in stress granules. Mol Cell 68(808–820):e805. https://doi.org/10.1016/j.molcel.2017.10.015
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z et al (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. https://doi.org/10.1038/nature11922
Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ et al (2014) Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 46:152–160. https://doi.org/10.1038/ng.2853
King HA, Cobbold LC, Willis AE (2010) The role of IRES trans-acting factors in regulating translation initiation. Biochem Soc Trans 38:1581–1586. https://doi.org/10.1042/BST0381581
Krichevsky AM, Kosik KS (2001) Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron 32:683–696
Kwon S, Zhang Y, Matthias P (2007) The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev 21:3381–3394. https://doi.org/10.1101/gad.461107
Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ (2007) TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol 114:63–70. https://doi.org/10.1007/s00401-007-0226-5
La Spada AR, Taylor JP (2010) Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 11:247–258. https://doi.org/10.1038/nrg2748
Lacerda R, Menezes J, Romao L (2017) More than just scanning: the importance of cap-independent mRNA translation initiation for cellular stress response and cancer. Cell Mol Life Sci 74:1659–1680. https://doi.org/10.1007/s00018-016-2428-2
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. https://doi.org/10.1016/j.cell.2012.03.017
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD et al (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167(774–788):e717. https://doi.org/10.1016/j.cell.2016.10.002
Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo AR, Kool M et al (2013) The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153:1064–1079. https://doi.org/10.1016/j.cell.2013.04.055
Leprivier G, Rotblat B, Khan D, Jan E, Sorensen PH (2015) Stress-mediated translational control in cancer cells. Biochim Biophys Acta 1849:845–860. https://doi.org/10.1016/j.bbagrm.2014.11.002
Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ (2005) Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J 272:4211–4220. https://doi.org/10.1111/j.1742-4658.2005.04833.x
Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T et al (2010) Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. https://doi.org/10.1371/journal.pone.0013250
Liu EY, Cali CP, Lee EB (2017) RNA metabolism in neurodegenerative disease. Dis Model Mech 10:509–518. https://doi.org/10.1242/dmm.028613
Liu R, Proud CG (2016) Eukaryotic elongation factor 2 kinase as a drug target in cancer, and in cardiovascular and neurodegenerative diseases. Acta Pharmacol Sin 37:285–294. https://doi.org/10.1038/aps.2015.123
Lopez-Erauskin J, Tadokoro T, Baughn MW, Myers B, McAlonis-Downes M, Chillon-Marinas C et al (2018) ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100(816–830):e817. https://doi.org/10.1016/j.neuron.2018.09.044
Ma T, Chen Y, Vingtdeux V, Zhao H, Viollet B, Marambaud P et al (2014) Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid beta. J Neurosci 34:12230–12238. https://doi.org/10.1523/JNEUROSCI.1694-14.2014
Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P et al (2013) Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci 16:1299–1305. https://doi.org/10.1038/nn.3486
Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K et al (2013) Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol 126:859–879. https://doi.org/10.1007/s00401-013-1181-y
Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C et al (2017) TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95(808–816):e809. https://doi.org/10.1016/j.neuron.2017.07.025
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9:995–1007. https://doi.org/10.1016/S1474-4422(10)70195-2
Martin J, Masri J, Cloninger C, Holmes B, Artinian N, Funk A et al (2011) Phosphomimetic substitution of heterogeneous nuclear ribonucleoprotein A1 at serine 199 abolishes AKT-dependent internal ribosome entry site-transacting factor (ITAF) function via effects on strand annealing and results in mammalian target of rapamycin complex 1 (mTORC1) inhibitor sensitivity. J Biol Chem 286:16402–16413. https://doi.org/10.1074/jbc.M110.205096
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM et al (2014) C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128:485–503. https://doi.org/10.1007/s00401-014-1329-4
Mazroui R, Sukarieh R, Bordeleau ME, Kaufman RJ, Northcote P, Tanaka J et al (2006) Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol Biol Cell 17:4212–4219. https://doi.org/10.1091/mbc.e06-04-0318
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O et al (2015) 5’ UTR m(6)A promotes cap-independent translation. Cell 163:999–1010. https://doi.org/10.1016/j.cell.2015.10.012
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345:1192–1194. https://doi.org/10.1126/science.1256800
Mochel F, Durant B, Meng X, O’Callaghan J, Yu H, Brouillet E et al (2012) Early alterations of brain cellular energy homeostasis in Huntington disease models. J Biol Chem 287:1361–1370. https://doi.org/10.1074/jbc.M111.309849
Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ et al (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163:123–133. https://doi.org/10.1016/j.cell.2015.09.015
Monteggia LM, Gideons E, Kavalali ET (2013) The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biol Psychiatry 73:1199–1203. https://doi.org/10.1016/j.biopsych.2012.09.006
Moon SL, Sonenberg N, Parker R (2018) Neuronal regulation of eIF2alpha function in health and neurological disorders. Trends Mol Med 24:575–589. https://doi.org/10.1016/j.molmed.2018.04.001
Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG et al (2012) Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485:507–511. https://doi.org/10.1038/nature11058
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126:881–893. https://doi.org/10.1007/s00401-013-1189-3
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339:1335–1338. https://doi.org/10.1126/science.1232927
Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S et al (2008) The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134:1042–1054. https://doi.org/10.1016/j.cell.2008.07.031
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Nicoll RA (2017) A brief history of long-term potentiation. Neuron 93:281–290. https://doi.org/10.1016/j.neuron.2016.12.015
Oddo S (2012) The role of mTOR signaling in Alzheimer disease. Front Biosci (Schol Ed) 4:941–952
Ohno M (2014) Roles of eIF2alpha kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci 7:22. https://doi.org/10.3389/fnmol.2014.00022
Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL et al (2008) Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-Ltd. Neuron 59:70–83. https://doi.org/10.1016/j.neuron.2008.05.023
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY et al (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162:1066–1077. https://doi.org/10.1016/j.cell.2015.07.047
Pearson CE (2011) Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet 7:e1002018. https://doi.org/10.1371/journal.pgen.1002018
Perluigi M, Di Domenico F, Butterfield DA (2015) mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis 84:39–49. https://doi.org/10.1016/j.nbd.2015.03.014
Placzek AN, Prisco GV, Khatiwada S, Sgritta M, Huang W et al (2016) eIF2alpha-mediated translational control regulates the persistence of cocaine-induced LTP in midbrain dopamine neurons. Elife. https://doi.org/10.7554/elife.17517
Protter DS, Parker R (2016) Principles and properties of stress granules. Trends Cell Biol 26:668–679. https://doi.org/10.1016/j.tcb.2016.05.004
Rajasekhar VK, Viale A, Socci ND, Wiedmann M, Hu X, Holland EC (2003) Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell 12:889–901
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG et al (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36:585–595. https://doi.org/10.1038/ng1362
Reddy K, Zamiri B, Stanley SY, Macgregor RB Jr, Pearson CE (2013) The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem 288:9860–9866. https://doi.org/10.1074/jbc.C113.452532
Repici M, Hassanjani M, Maddison DC, Garcao P, Cimini S, Patel B et al (2018) The Parkinson’s disease-linked protein DJ-1 associates with cytoplasmic mRNP granules during stress and neurodegeneration. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1084-y
Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ et al (2007) Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 5:151–156. https://doi.org/10.1016/j.cmet.2007.01.008
Rivero-Hinojosa S, Lau LS, Stampar M, Staal J, Zhang H, Gordish-Dressman H et al (2018) Proteomic analysis of medulloblastoma reveals functional biology with translational potential. Acta Neuropathol Commun 6:48. https://doi.org/10.1186/s40478-018-0548-7
Rohilla KJ, Gagnon KT (2017) RNA biology of disease-associated microsatellite repeat expansions. Acta Neuropathol Commun 5:63. https://doi.org/10.1186/s40478-017-0468-y
Rotblat B, Southwell AL, Ehrnhoefer DE, Skotte NH, Metzler M, Franciosi S et al (2014) HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci U S A 111:3032–3037. https://doi.org/10.1073/pnas.1314421111
Ryazanov AG, Shestakova EA, Natapov PG (1988) Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 334:170–173. https://doi.org/10.1038/334170a0
Salminen A, Kaarniranta K (2012) AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 11:230–241. https://doi.org/10.1016/j.arr.2011.12.005
Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F et al (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80–96. https://doi.org/10.1016/j.neuron.2013.07.027
Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Kupper C et al (2015) Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol 130:537–555. https://doi.org/10.1007/s00401-015-1450-z
Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J et al (2011) Global quantification of mammalian gene expression control. Nature 473:337–342. https://doi.org/10.1038/nature10098
Sellier C, Buijsen RAM, He F, Natla S, Jung L, Tropel P et al (2017) Translation of expanded CGG repeats into FMRpolyG Is pathogenic and may contribute to fragile X tremor ataxia syndrome. Neuron 93:331–347. https://doi.org/10.1016/j.neuron.2016.12.016
Shaw RJ (2009) LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 196:65–80. https://doi.org/10.1111/j.1748-1716.2009.01972.x
Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J (2005) Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J Biol Chem 280:10964–10973. https://doi.org/10.1074/jbc.M407874200
Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H et al (2013) Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2:e00498. https://doi.org/10.7554/eLife.00498
Sidrauski C, McGeachy AM, Ingolia NT, Walter P (2015) The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife. https://doi.org/10.7554/elife.05033
Sidrauski C, Tsai JC, Kampmann M, Hearn BR, Vedantham P, Jaishankar P et al (2015) Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife 4:e07314. https://doi.org/10.7554/eLife.07314
Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF et al (2017) The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 169(1051–1065):e1018. https://doi.org/10.1016/j.cell.2017.05.022
Somasekharan SP, El-Naggar A, Leprivier G, Cheng H, Hajee S, Grunewald TG et al (2015) YB-1 regulates stress granule formation and tumor progression by translationally activating G3BP1. J Cell Biol 208:913–929. https://doi.org/10.1083/jcb.201411047
Sonenberg N, Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. https://doi.org/10.1016/j.cell.2009.01.042
Sossin WS, Costa-Mattioli M (2018) Translational control in the brain in health and disease. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a032912
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R et al (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 29:13578–13588. https://doi.org/10.1523/JNEUROSCI.4390-09.2009
Spriggs KA, Bushell M, Willis AE (2010) Translational regulation of gene expression during conditions of cell stress. Mol Cell 40:228–237. https://doi.org/10.1016/j.molcel.2010.09.028
Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, Costa-Mattioli M (2011) Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc Natl Acad Sci U S A 108:3791–3796. https://doi.org/10.1073/pnas.1014715108
Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, Group PSPGS et al (2013) The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun 1:31. https://doi.org/10.1186/2051-5960-1-31
Szaflarski W, Fay MM, Kedersha N, Zabel M, Anderson P, Ivanov P (2016) Vinca alkaloid drugs promote stress-induced translational repression and stress granule formation. Oncotarget 7:30307–30322. https://doi.org/10.18632/oncotarget.8728
Tabet R, Schaeffer L, Freyermuth F, Jambeau M, Workman M, Lee CZ et al (2018) CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat Commun 9:152. https://doi.org/10.1038/s41467-017-02643-5
Takahashi M, Higuchi M, Matsuki H, Yoshita M, Ohsawa T, Oie M et al (2013) Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol Cell Biol 33:815–829. https://doi.org/10.1128/MCB.00763-12
Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539:197–206. https://doi.org/10.1038/nature20413
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC et al (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123:465–472. https://doi.org/10.1007/s00401-011-0922-z
Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M et al (2013) CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78:440–455. https://doi.org/10.1016/j.neuron.2013.03.026
Topisirovic I, Sonenberg N (2015) Translation and cancer. Biochim Biophys Acta 1849:751–752. https://doi.org/10.1016/j.bbagrm.2015.05.004
Truitt ML, Ruggero D (2017) New frontiers in translational control of the cancer genome. Nat Rev Cancer 17:332. https://doi.org/10.1038/nrc.2017.30
Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R et al (2005) Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci 25:5833–5843. https://doi.org/10.1523/JNEUROSCI.0599-05.2005
Van Treeck B, Protter DSW, Matheny T, Khong A, Link CD, Parker R (2018) RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. Proc Natl Acad Sci U S A 115:2734–2739. https://doi.org/10.1073/pnas.1800038115
Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T et al (2012) Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci 32:8270–8283. https://doi.org/10.1523/JNEUROSCI.1592-12.2012
Vatsavayai SC, Nana AL, Yokoyama JS, Seeley WW (2018) C9orf72-FTD/ALS pathogenesis: evidence from human neuropathological studies. Acta Neuropathol. https://doi.org/10.1007/s00401-018-1921-0
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A et al (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914
Verpelli C, Piccoli G, Zibetti C, Zanchi A, Gardoni F, Huang K et al (2010) Synaptic activity controls dendritic spine morphology by modulating eEF2-dependent BDNF synthesis. J Neurosci 30:5830–5842. https://doi.org/10.1523/JNEUROSCI.0119-10.2010
Vilas-Boas Fde A, da Silva AM, de Sousa LP, Lima KM, Vago JP, Bittencourt LF et al (2016) Impairment of stress granule assembly via inhibition of the eIF2alpha phosphorylation sensitizes glioma cells to chemotherapeutic agents. J Neurooncol 127:253–260. https://doi.org/10.1007/s11060-015-2043-3
Vingtdeux V, Davies P, Dickson DW, Marambaud P (2011) AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol 121:337–349. https://doi.org/10.1007/s00401-010-0759-x
Vlatkovic IS, Schuman E (2016) Local translation in dendrites. In: OS Online (ed) dendrites. Oxford Scholarship Online, Oxford
Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H et al (2001) Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12:1393–1407
Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM et al (2015) Glioma. Nat Rev Dis Primers 1:15017. https://doi.org/10.1038/nrdp.2015.17
Wolozin B (2012) Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener 7:56. https://doi.org/10.1186/1750-1326-7-56
Yasuda K, Zhang H, Loiselle D, Haystead T, Macara IG, Mili S (2013) The RNA-binding protein Fus directs translation of localized mRNAs in APC-RNP granules. J Cell Biol 203:737–746. https://doi.org/10.1083/jcb.201306058
Zang JB, Nosyreva ED, Spencer CM, Volk LJ, Musunuru K, Zhong R et al (2009) A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet 5:e1000758. https://doi.org/10.1371/journal.pgen.1000758
Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z et al (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513:382–387. https://doi.org/10.1038/nature13438
Zhang YJ, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K et al (2018) Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med 24:1136–1142. https://doi.org/10.1038/s41591-018-0071-1
Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL et al (2014) Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128:505–524. https://doi.org/10.1007/s00401-014-1336-5
Zhou XW, Tanila H, Pei JJ (2008) Parallel increase in p70 kinase activation and tau phosphorylation (S262) with Abeta overproduction. FEBS Lett 582:159–164. https://doi.org/10.1016/j.febslet.2007.11.078
Zhu PJ, Chen CJ, Mays J, Stoica L, Costa-Mattioli M (2018) mTORC2, but not mTORC1, is required for hippocampal mGluR-LTD and associated behaviors. Nat Neurosci 21:799–802. https://doi.org/10.1038/s41593-018-0156-7
Zivraj KH, Tung YC, Piper M, Gumy L, Fawcett JW, Yeo GS et al (2010) Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J Neurosci 30:15464–15478. https://doi.org/10.1523/JNEUROSCI.1800-10.2010
Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD et al (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108:260–265. https://doi.org/10.1073/pnas.1013343108
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110:E4968–E4977. https://doi.org/10.1073/pnas.1315438110
Funding
This work was partially supported by Canadian Cancer Society Research Institute (CCSRI) Impact Grant (Grant #703205; to PHS), and CIHR Foundation Grant FDN-143280 (to PHS). AD is supported by a Harry and Florence Dennison Fellowship in Medical Research, 4 Year Fellowship, and a Killam Doctoral Scholarship from the University of British Columbia, BC, Canada. Funding to AJ was in the form of an AIAS-COFUND fellowship from European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie agreement (Grant #754513) and the Lundbeckfonden, Denmark (Grant #R250-2017-1131).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Delaidelli, A., Jan, A., Herms, J. et al. Translational control in brain pathologies: biological significance and therapeutic opportunities. Acta Neuropathol 137, 535–555 (2019). https://doi.org/10.1007/s00401-019-01971-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-019-01971-8