
Abstract
The synaptic cleft of the neuromuscular junction (NMJ) consists of a highly specialized extracellular matrix (ECM) involved in synapse maturation, in the juxtaposition of pre- to post-synaptic areas, and in ensuring proper synaptic transmission. Key components of synaptic ECM, such as collagen IV, perlecan and biglycan, are binding partners of one of the most abundant ECM protein of skeletal muscle, collagen VI (ColVI), previously never linked to NMJ. Here, we demonstrate that ColVI is itself a component of this specialized ECM and that it is required for the structural and functional integrity of NMJs. In vivo, ColVI deficiency causes fragmentation of acetylcholine receptor (AChR) clusters, with abnormal expression of NMJ-enriched proteins and re-expression of fetal AChRγ subunit, both in Col6a1 null mice and in patients affected by Ullrich congenital muscular dystrophy (UCMD), the most severe form of ColVI-related myopathies. Ex vivo muscle preparations from ColVI null mice revealed altered neuromuscular transmission, with electrophysiological defects and decreased safety factor (i.e., the excess current generated in response to a nerve impulse over that required to reach the action potential threshold). Moreover, in vitro studies in differentiated C2C12 myotubes showed the ability of ColVI to induce AChR clustering and synaptic gene expression. These findings reveal a novel role for ColVI at the NMJ and point to the involvement of NMJ defects in the etiopathology of ColVI-related myopathies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The synaptic cleft of the neuromuscular junction (NMJ) is made of a functionalized extracellular matrix (ECM), facing on the one side the presynaptic motor nerve terminal, and on the other side the muscular postsynaptic compartment [63]. Synaptic ECM cues are essential for the establishment of functional NMJ, being actively involved in regulating synaptic terminal development and maturation [24, 41, 42], in defining the precise juxtaposition of pre- to post-synaptic areas [1, 6, 44], and in guaranteeing a permissive environment for synaptic transmission [4, 35, 48]. The synaptic basal lamina contains agrin (the main player driving acetylcholine receptor aggregation and stabilization of acetylcholine receptor (AChR) aggregates at the postsynaptic membrane), laminin, nidogen, collagen IV, perlecan, and heparan sulfate glycosaminoglycans [56]. Some of these molecules, such as agrin, specific laminin isoforms and collagen IV variants, are only present at the synaptic cleft. Other molecules, such as perlecan and nidogen, display a broader deposition, being found also in the extrasynaptic muscle basement membrane, where they can differentially partner with other proteins [20, 45]. A number of these molecules, such as collagen IV, perlecan and biglycan, are actually interacting molecules of one of the most abundant ECM proteins of skeletal muscle, ColVI [11, 31, 69].
Collagen VI (ColVI) is a protein of the basement membrane of skeletal muscle [31]. It is composed of three main subunits encoded by distinct genes, where the most abundant form is made by the stoichiometric association of the α1(VI), α2(VI) and α3(VI) chains, while other three minor chains can substitute for α3(VI) in specific tissues [11, 23, 52]. Mutations of ColVI genes are causative for a broad group of inherited muscle diseases, including Ullrich congenital muscular dystrophy (UCMD), Bethlem myopathy (BM) and myosclerosis myopathy [8]. Studies in Col6a1 null mice and in BM/UCMD patients showed that ColVI plays a key role in muscle homeostasis and regeneration [7, 27, 67]. Interestingly, our recent work in Col6a1 null mice revealed that ColVI ablation affects peripheral nerve structure and function [13]. However, the potential role of ColVI at the NMJ and whether its deficiency affected neuromuscular transmission remained unknown.
Here we demonstrate that ColVI is found within the synaptic cleft of NMJs and is required for proper maintenance of the postsynaptic apparatus, as well as for preserving in vivo neuromuscular transmission in mice. Furthermore, we show that ColVI is able to modulate synaptic gene transcription, promoting AChR clustering and mimicking agrin effects. These findings highlight a novel role for ColVI at the NMJ, and point at novel and previously unforeseen features of BM/UCMD pathology.
Materials and methods
Animals
Wild-type and Col6a1−/− mice in the C57BL/6N background were used in this study [30]. All studies were carried out in 5- to 6-month-old mice. Animal procedures were approved by the Ethics Committee of the University of Padova and authorized by the Italian Ministry of Health.
Immunofluorescence on tissues
TA muscles were dissected and rapidly frozen in liquid nitrogen. Cross (7 μm) and longitudinal (40 μm) sections were collected on glass slides and stored at − 20 °C until use. Samples were fixed and permeabilized with cold 50% methanol/50% acetone at − 20 °C for 10 min. After drying, samples were washed in phosphate buffered saline (PBS) solution and incubated for 1 h with 10% goat serum (Sigma) in PBS. Alternatively, when mouse-derived primary antibodies were used, samples were incubated for 2.5 h with 4% IgG free bovine serum albumin (BSA; Sigma) in PBS at room temperature and blocked for 30 min with anti-mouse IgG Fab fragment (5 mg/ml; Jackson Immunoresearch). Samples were incubated overnight at 4 °C with the primary antibodies listed in Suppl. Table S1. After washing in PBS, samples were incubated with fluorescent secondary antibodies (Suppl. Table S2). AChR was detected with tetramethylrhodamine-conjugated α-bungarotoxin (1:200; αBT, Invitrogen) and nuclei were stained with Hoechst 33528 (Sigma). Glass slides were mounted in 80% glycerol and analyzed by confocal fluorescence microscopy. Denervated NMJ quantification was performed as described in Supp. Material.
Electron microscopy
EDL muscles were dissected from mice and fixed with 4% paraformaldehyde (PFA) in PBS for 30 min, washed and labeled with αBT. Tissue areas containing NMJs were dissected and embedded in London Resin White (London Resin Company) for immunoelectron microscopy [51] or fixed with 2.5% glutaraldehyde in cacodylate buffer and embedded in Epon812 for ultrastructural analysis. Immunoelectron microscopy analysis of ColVI was performed on London White resin ultrathin sections with a rabbit polyclonal anti-ColVI antibody (1:50; 70-XR95, Fitzgerald) and revealed with a 5-nm colloidal gold conjugated goat anti-rabbit antibody (Sigma). Sections were stained with uranyl acetate and lead citrate and observed with a Jeol JEM-1011 electron microscope operating at 100 kV.
Whole-mount staining on diaphragms
Diaphragms were dissected and cut in order to divide the left half of the tissue from the right one. Samples were permeabilized at 36 °C for 15 min in a solution containing trypsin–EDTA (50 mM; Gibco) and collagenase I (1 mg/ml; Worthington) and fixed with 4% PFA in PBS for 30 min. Then, diaphragms were soaked in a blocking/permeabilizing solution (3% BSA, 10% goat serum, 0.1% Triton X-100) at room temperature for 2 h, washed in PBS and incubated overnight at 4 °C with primary antibodies (see Suppl. Table S1) in a PBS solution containing 3% BSA and 0.5% Triton X-100 in PBS. After PBS washes, samples were incubated with secondary antibodies (see Suppl. Table S2) and αBT (1:1000) in a 3% BSA/PBS solution. Samples were finally washed in PBS and mounted in 80% glycerol before analysis by confocal fluorescence microscopy. Denervated NMJ quantification was performed as described in Supp. Material.
3D imaging and analysis
Soleus and EDL muscles were dissected and fixed with 2% PFA in PBS for 2 h at 4 °C. Muscle bundles containing 5–10 fibers were prepared and stained with αBT (1:2500) for 1 h at room temperature. Stained bundles were washed in PBS and embedded in Mowiol (Sigma). Additional staining with neurofilament and synaptophysin was performed as described in Cheusova et al. [14]. Thin bundles of teased muscles were blocked in 100 mM glycine for 15 min; permeabilized in 0.5% Triton X-100, 5% BSA, and 1% FCS for 1 h; incubated with both, rabbit anti-neurofilament antibody (1:500; Millipore) and rabbit anti-synaptophysin antibody (1:500; DAKO). Secondary antibodies conjugated to Alexa-488 (Molecular Probes) were applied together with αBT. Diaphragms were dissected and fixed with 4% PFA in PBS for in 30 min at 4 °C, washed in Tris-buffered saline (TBS), incubated in blocking solution (4% BSA, 0.1% Triton X-100 in TBS) overnight at 4 °C, and stained with αBT (1:1000) overnight at 4 °C. After rinsing again in TBS, diaphragms were mounted in 80% glycerol. Muscles were analyzed by confocal fluorescence microscopy, and 3D images of NMJs were taken with 40× oil objective (Zeiss Examiner E1) at 55 ms exposure time. Images were deconvoluted and analyzed using different modules in AxioVision software. The following parameters were determined for each NMJ: volume, surface, sum fluorescence, mean fluorescence, number of fragments.
C2C12 culture and differentiation
C2C12 cells were seeded on 12-well plates or glass coverslips and cultured in DMEM (Gibco) supplemented with 20% fetal bovine serum (FBS, Gibco). Myotube differentiation was initiated by switching to DMEM supplemented with 2% horse serum (Gibco). After 7 days myotubes were left untreated or treated for 20 h with agrin (10 ng/ml; R&D), or with native murine ColVI (1 μg/ml) [30] purified by gel filtration chromatography [51] in the absence or presence of anti-ColVI antibody (40 μg/ml; sc-47712, Santa Cruz). After treatments, myotubes were lysed with either Trizol (Life Technologies), protein lysis buffer, or fixed with 4% PFA.
Real-time qRT-PCR in murine muscles and myotubes
For RNA extraction, muscles and myotube cultures were lysed in Trizol reagent (Invitrogen) and processed according to manufacturer instructions. Reverse transcription was performed using M-MLV reverse transcriptase, and cDNA products were analyzed by real-time PCR with the Rotor Gene SYBR PCR kit (Quiagen). The primers used are listed in Suppl. Table S3. Data were normalized to Gapdh expression.
Western blotting
Diaphragms were dissected, stained with αBT and the NMJ-rich band was cut from the rest of the tissue under a fluorescence stereomicroscope. Samples were homogenized with a pestle in a lysis buffer (50 mM Tris HCl, pH 7.5, 150 mM, NaCl, 1 mM EDTA, 10% glycerol) containing 0.5 mM DTT, 2% SDS, 1% Triton X-100, phosphatase inhibitors (Cocktail II, Sigma) and protease inhibitors (complete EDTA free, Roche). Myotubes were lysed in a lysis solution (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 20 mM EDTA, 0.5% NP-40) supplemented with phosphatase inhibitors (Cocktail II, Sigma) and protease inhibitors (complete EDTA free, Roche). Proteins were quantified with BCA Protein assay kit (Pierce), and separated by SDS-PAGE. Quaternary structure of ColVI used for myotube treatment was assessed running it in a 2.5% acrylamide/0.5% agarose SDS-PAGE in non-reducing conditions [38], and contamination by other ECM proteins was excluded using laminin (L2020, Sigma) and fibronectin (F1141, Sigma) as positive controls. Proteins were then blotted onto PVDF membranes (Millipore) at 20 or 30 V (for non-reduced ColVI) for 2 h. Membranes were incubated overnight at 4 °C with primary antibodies (see Suppl. Table S1). After washing in TBS-T, membranes were incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies (see Suppl. Table S2). Detection was performed by chemiluminescence (Thermo-Scientific).
Nerve–muscle preparations for electrophysiology
Isolated diaphragm–phrenic nerve and soleus–sciatic nerve preparations were pinned to the bottom of a Sylgard (Sigma)-coated petri dish and maintained in Liley’s solution gassed with 95% O2–5% CO2 at room temperature [33]. The recording chamber was perfused at a rate of 1 ml/min. Nerve stump was stimulated with suction electrode connected to a stimulator (Grass S88, Grass) through a stimulus isolation unit (SIU5, Grass). Single supramaximal stimulation (0.1 ms duration) was delivered at 0.5 Hz in order to prevent synaptic fatigue. To block muscle action potentials, so that EPP and EPCs could be recorded [47, 49], µ-conotoxin GIIIB (µ-CTX, 2 µM; Alomone Labs) was added to Liley’s solution. In some experiments, the effect of the toxin wore off after 1–2 h and contractions resumed in response to nerve stimulation. These preparations were then exposed for a second time to the toxin. At the same time, AChRs were labeled by adding rhodamine-conjugated αBT (5 nM; Life Technologies) to the bath, as described before [17, 70].
Intracellular recordings in isolated diaphragm–phrenic nerve preparation
Two intracellular electrodes (resistance 10–15 MΩ) were inserted within 50 µm of the NMJs under visual inspection. Current was passed through one electrode to maintain the membrane potential within 2 mV of − 75 mV, while voltage transients were recorded with the other electrode. Signals were amplified by an Axoclamp 900A and digitized at 40 kHz by a Digidata 1440A under the control of pCLAMP 10 (Molecular Devices). Voltage records were filtered at 3 kHz and current recorded at 1 kHz (8-pole Bessel filter). Current transients were recorded using the two-electrode voltage-clamp facility of the Axoclamp 900A. 50–100 spontaneous quantal events were recorded during a period of 1 min. Records were analyzed using pCLAMP 10. Events recorded from each NMJ were averaged and the amplitude, rise time and single exponential decay time constant determined.
Extracellular recordings in isolated diaphragm–phrenic nerve preparation
The preparation was placed on the stage of a Zeiss Axio Examiner Z1 microscope fitted with incident light fluorescence illumination with filters for red (Zeiss filter set 20) fluorescing fluorophore (Carl Zeiss MicroImaging). At the beginning of the experiment, CMAP was recorded using a micropipette with a tip diameter of about 10 µm, filled with bathing solution. The electrode was positioned so that the latency of the major negative peak was minimized. The electrode was then positioned 100 µm above the surface of the muscle and CMAP was recorded. For curare experiments in ex vivo diaphragm preparations, the experimental conditions were established as previously reported [55]. The recording chamber was filled with 2 ml (500 or 800 nM) of d-tubocurarine chloride (Sigma). During curare treatment, trains of 25 repetitive nerve stimulations (5 Hz) were performed at 2 min intervals, and the ratio of CMAP amplitudes (mean (20th − 25th)/2nd) was calculated.
Intracellular recordings in soleus nerve–muscle preparation
Spontaneous and evoked excitatory postsynaptic potentials were intracellularly recorded from single muscle fibers using borosilicate glass microelectrodes (inner diameter 0.86 mm, outer diameter 1.5 mm; 15 MW resistance) (Science Products). Microelectrodes were inserted within 1 mm of the point of entrance of the nerve in the soleus muscle. For each recording, resting membrane potential was measured and set at − 70 mV for the entire duration of the recording. Spontaneous neurotransmitter release (miniature end plate potentials, mEPP) was initially recorded for 120 s. Subsequently, evoked neurotransmitter release (excitatory postsynaptic potentials, EPP) was recorded following nerve stimulation as described above for 30 s. For repetitive nerve stimulation, soleus nerve was stimulated at 10 Hz. Intracellularly recorded signals were amplified using an intracellular amplifier (SEC NPI Electronic) in current-clamp condition. Amplified signals were then sent to an A/D converter (National Instruments) and fed to a personal computer. Digitized recordings were analyzed offline using the WinEDR and Pclamp software for electrophysiology (Strathclyde and Pclamp6, Axon). mEPP amplitudes were computed by analyzing the first 120 s of each recording. EPP amplitudes were computed by analyzing the first 10 evoked responses stimulating the nerve at 0.5 Hz, in order to prevent the onset of synaptic fatigue. For tetanic nerve stimulations at 10 Hz, the ratio between the amplitude of each evoked response and the amplitude of the first one was calculated.
Human samples
All the human muscle biopsies used in these studies were provided by the Neuromuscular Bank of Tissues and DNA samples of the Telethon Network of Genetic BioBanks. Six subjects were selected from a cohort of BM and UCMD patients followed at the Neuromuscular Center of the University of Padova. All patients carried previously characterized mutations on ColVI genes. Relevant data concerning genetic analysis, age at the biopsy, age at the last clinical evaluation, and disease progression are reported in Fig. 5a. Control and patient samples were obtained by open muscle biopsy from vastus lateralis, with the exception of P4, whose biopsy derived from the femoral quadriceps. Four other dystrophic patients were included in the study as pathological controls: two affected by Duchenne muscular dystrophy (DMD1, DMD2) and two by polymyositis (PM1, PM2). Both DMD patients were 1 year old at the age of biopsy, carried characterized dystrophin mutations and both biopsies derived from the femoral quadriceps. PM1 biopsy was collected when the patient was 80 years old, and was derived from femoral quadriceps; PM2 biopsy was collected at the age of 69 years old, and was derived from vastus lateralis. Both PM biopsies were collected before glucocorticoid treatment.
Analysis of gene expression in human samples
Total RNA was isolated from muscle biopsies using Trizol Reagent (Life Technologies). First-strand cDNA synthesis was performed using High-Capacity cDNA Reverse Transcription Kit (Life Technologies) and transcript levels were quantified by SYBR Green Real-Time PCR (Life Technologies) using the ABI PRISM 7000 sequence detection system. The primers used are listed in Suppl. Table S4.
Immunofluorescence on human samples
8-µm-muscle cryosections were collected on Superfrost slides, fixed with 4% PFA, treated with 0.5% Triton X-100, blocked with 10% FBS in PBS for 30 min and then incubated overnight at 4 °C with primary antibodies (see Suppl. Table S1) diluted in blocking solution. After washing, samples were incubated with secondary fluorescent antibodies (see Suppl. Table S2) for 1 h at room temperature together with αBT (1:200). Slides were mounted using Vectashield medium with DAPI stain (Vector) and examined on a Leica TCS SP5 confocal microscope. Muscle specimens were stained with hematoxylin and eosin to check histopathology integrity, and myofibrillar ATPase with preincubation at pH 4.3 to assess muscle fiber type composition. Slides were examined on upright microscope (BX60, Olympus) equipped with a CCD camera (DP70, Olympus). Images were post-processed using ImageJ suite. Type I and type II muscle fibers were counted. On average, 1248 (range 524–4082) myofibers per patient were evaluated.
Statistical analysis
Data are presented as mean ± s.e.m., except where indicated. The statistical significance was determined by unpaired two-tailed Student’s t test or by unpaired one-tailed Mann–Whitney test as indicated. A P value < 0.05 was considered as a significant difference.
Results
ColVI is associated to the neuromuscular synaptic ECM
To investigate the presence of ColVI at the NMJ, we performed immunostaining for ColVI together with fluorophore-coupled α-bungarotoxin (αBT), which specifically binds AChRs. Immunolabeling of both 7-μm-thick cross sections (Fig. 1a) and 40-μm-thick longitudinal sections (Fig. 1b) of mouse tibialis anterior (TA) muscle showed a close localization of ColVI with AChR clusters. Single stack confocal planes helped in analyzing at higher resolution the continuity of ColVI with laminin and AChR clusters (Fig. 1c). It is well known that ColVI is composed of three different α chains, and that the most frequent α3(VI) can be alternatively substituted by α4(VI), α5(VI) or α6(VI) in its association to α1(VI) and α2(VI) chains in specific tissues [11]. Interestingly, α5(VI) was previously suggested to be present at the NMJ [22]. Therefore, we compared α3(VI) and α5(VI) immunostaining on longitudinal muscle sections, detecting only partial colocalization of these two ColVI chains (Suppl. Fig. S1a). While immunolabeling data suggested that both ColVI chains are found in association with NMJs, comparison between the two staining patterns in single stack confocal planes indicated that α3(VI) is closer to the AChR clusters than α5(VI) (Suppl. Fig. S1b).

ColVI is present in the NMJ synaptic cleft. a Immunofluorescence for ColVI (green) in 7-μm-thick cross sections of wild-type and Col6a1−/− TA muscles, showing ColVI colocalization with AChRs in wild-type animals. AChRs were stained with αBT (red), and nuclei were stained with Hoechst (blue). Arrowheads point at some NMJs, and the inset shows higher magnification of one NMJ in wild-type muscle. Scale bar, 100 μm. b Confocal analysis of ColVI immunostaining (green) in 40-μm-thick longitudinal sections of wild-type TA, revealing that ColVI staining encases αBT-labeled AChR clusters (red) and is in close contact with them. Laminin immunostaining (grey) is provided as marker of basal membrane. Scale bar, 40 μm. c Serial stacks, corresponding to single focal planes, from the immunostaining analysis in b are provided, showing at higher resolution the tight contact between ColVI and AChR clusters (red). Scale bar, 40 μm. d Immunogold labeling for ColVI in wild-type EDL, showing the presence of ColVI, identified by gold particles (arrowheads), in the ECM associated with the postsynaptic membrane within the synaptic cleft. Presynaptic compartment is indicated in transparent blue. The right panel shows a higher magnification of the squared area. Scale bar, 300 nm
Ultrastructural analysis by immunoelectron microscopy confirmed ColVI deposition in the ECM associated with the synaptic cleft (Fig. 1d), thus identifying ColVI as a novel constituent of the synaptic basal lamina.
Motor endplates are altered in the absence of ColVI in vivo
To explore the in vivo role of ColVI at the NMJ, we performed whole-mount αBT labeling of adult wild-type and Col6a1−/− mouse muscles. Postsynaptic boutons of Col6a1−/− muscles appeared less regular in shape, being frequently fragmented (Fig. 2a). Moreover, parts of presynaptic regions were not covered by αBT-positive clusters in Col6a1−/− muscles, in correspondence with areas of NMJ fragmentation, when stained with neurofilament and synaptophysin (Fig. 2b). Fragmentation was confirmed by morphometric analysis of extensor digitorum longus (EDL), soleus and diaphragm muscles, revealing a marked fragmentation of NMJs in Col6a1−/− muscles when compared to wild-type muscles (Fig. 2c and Suppl. Fig. S2). In soleus and EDL, NMJ fragmentation was accompanied by changes in three-dimensional (3D) morphometric parameters, with an increase of the surface area, volume and fluorescence intensity of NMJs (Suppl. Fig. S3).

ColVI ablation induces AChR clusters fragmentation. a Representative micrographs of wild-type and Col6a1−/− EDL and soleus endplates, obtained by whole-mount staining with αBT (red). Arrowheads indicate the presence of AChR cluster fragmentation. b Representative images of wild-type and Col6a1−/− EDL and soleus endplates, stained for AChR clusters with αBT (red) and immunostained for neurofilament and synaptophysin (green), showing regions of nerve terminals not completely covered by subsynaptic AChR aggregates (arrowheads), corresponding to areas of cluster fragmentation. Images in a and b are deconvoluted Z-stack micrographs presented as 2D. c Morphometric analysis of whole-mount αBT staining of EDL (left panel) and soleus (right panel) from wild-type and Col6a1−/− mice. Muscles lacking ColVI display a significantly higher fragmentation of NMJs, when compared to wild-type muscles (***P < 0.001; unpaired two-tailed Student’s t test; n = 115 NMJs, wild-type EDL; n = 106 NMJs, Col6a1−/− EDL; n = 69 NMJs, wild-type soleus; n = 84 NMJs, Col6a1−/− soleus). NMJs were sampled from three mice per genotype. Error bars indicate s.e.m
Endplate fragmentation is often due to aging or muscle denervation [3, 50]. Although we did not detect denervation defects in the absence of ColVI (Suppl. Fig. S4a,b), Col6a1−/− muscles displayed a remarkable upregulation of AChRγ and AChRα, but not AChRε, mRNA transcripts (Fig. 3a), thus confirming NMJ alterations and pointing at an abnormal neuromuscular transmission [71]. On the other hand, the presynaptic compartment did not display any overt alteration in Col6a1−/− muscles (Suppl. Fig. S4c). Interestingly, some ColVI interactors, such as perlecan, collagen IV and biglycan [11], were reported to be critical in stabilizing the postsynaptic apparatus [2, 4, 21]. In order to evaluate the impact of ColVI ablation on the extracellular and cell surface NMJ protein network, we performed immunofluorescence for a panel of NMJ-enriched proteins (Fig. 2d). This analysis revealed that perlecan, known to engage the dystrophin–glycoprotein complex (DGC) [4, 46], is abnormally increased in the NMJs of Col6a1−/− muscles, and a similar increased labeling was displayed by other DGC components, such as utrophin, dystrophin and NOS1, but not δ-sarcoglycan (Fig. 3b and Suppl. Fig. S5a). Collagen type IV, an ECM protein involved in NMJ maturation [21], was also upregulated in the NMJs of Col6a1−/− muscles (Fig. 3b). The MuSK/LRP4 heterodimer acts as an agrin receptor and was shown to be contacted by biglycan, thus playing a key role on synapse stability [2]. Although MuSK immunolabeling appeared similar between wild-type and Col6a1−/− NMJs, LRP4 displayed increased labeling in Col6a1−/− NMJs (Fig. 3b and Suppl. Fig. S5a). In agreement with the increased immunolabeling of NMJ-associated proteins in ColVI-deficient muscles (semi-quantitative analysis is provided in Fig. 3c), real-time PCR analysis of Col6a1−/− diaphragms showed altered expression of postsynaptic genes, with significant upregulation of MuSK, LRP4 and utrophin mRNA transcripts (Fig. 3d). In addition, MuSK, NOS1 and AChRγ protein levels resulted upregulated in Col6a1−/− isolated synaptic diaphragms, as revealed by semi-quantitative western blot analysis (Suppl. Fig. S5b, c).

Absence of ColVI causes NMJ defects and affects the postsynaptic compartment. a qRT-PCR analysis for the Chrng, Chrna and Chrne genes in diaphragm (top panel) and TA (lower panel) of wild-type and Col6a1−/− mice, showing a significant increase of Chrng and Chrna transcripts in Col6a1−/− muscles (***P < 0.001; **P < 0.01; unpaired two-tailed Student’s t test; diaphragm, n = 6 mice, each genotype; TA, n = 8 mice, each genotype). Error bars indicate s.e.m. b Representative immunolabeling for NMJ-associated proteins (green) in cross sections of wild-type and Col6a1−/− TA muscles stained with αBT staining (red). The localization at the NMJ of collagen IV, perlecan, utrophin, NOS1 and LRP4 was analyzed in at least three animals per genotype. Scale bar, 20 μm. c Quantification of immunofluorescence intensity from assays in b, showing altered expression of the proteins at the Col6a1−/− NMJ. a.u.: arbitrary units (*P < 0.05; unpaired one-tailed Student’s t test; n = 3 mice, each genotype; a minimum of 20 NMJs per genotype was analyzed). Error bars indicate s.e.m. d qRT-PCR analysis for postsynaptic genes in diaphragms (DIA) and TA muscles of wild-type and Col6a1−/− mice (***P < 0.001; *P < 0.05; unpaired two-tailed Student’s t test; diaphragm, n = 6 mice, each genotype; TA, n = 8 mice, each genotype). Error bars indicate s.e.m
Impaired NMJ function in the absence of ColVI
Previous studies demonstrated decreased muscle strength in Col6a1−/− mice, with lower locomotor activity during running wheel or treadmill exercise [28] and normal activity on rotarod [13]. However, NMJ-related functional aspects were never evaluated in ColVI-deficient mice. When measuring the latency to fall from an upside-down rotated horizontal grid, we found that Col6a1−/− mice hung for significantly shorter times than wild-type animals (Fig. 4a). Ex vivo electrophysiological recordings showed that both nerve-evoked endplate potentials (EPP) and nerve-independent miniature endplate potentials (mEPP) had significantly reduced amplitudes in soleus (a muscle with predominance of slow twitch fibers) [31] and diaphragm (a muscle with predominance of mixed fast twitch fibers) [60] of Col6a1−/− mice when compared to wild-type animals (Fig. 4b, c). The reduced EPP and mEPP amplitudes could be due either to decreased quantal size or to a reduced postsynaptic sensitivity to individual quanta. Notably, both the above-mentioned neuromuscular transmission defects were observed, as the mean quanta were significantly reduced in Col6a1−/− muscles, together with decreased endplate currents (Suppl. Table S5) and lower input resistance (Fig. 4c).

ColVI ablation alters neuromuscular synapse function and affects safety factor. a The time for which wild-type and Col6a1−/− mice clung upside down to a horizontal grid was measured, showing impaired performance in Col6a1−/− animals (***P < 0.001; unpaired two-tailed Student’s t test; n = 10 mice, each genotype). Error bars are s.e.m. b, c Measurement of the EPP and mEPP amplitudes in wild-type and Col6a1−/− soleus (b) (n = 126 EPP, wild-type, n = 238 EPP, Col6a1−/−; n = 362 mEPP, wild-type; n = 416 mEPP, Col6a1−/−) and diaphragm (c) (n = 58 EPP, wild-type; n = 63 EPP, Col6a1−/−; n = 78 mEPP, wild-type; n = 104 mEPP, Col6a1−/−), revealing alterations of the electrophysiological features in Col6a1−/− muscles. The input resistance (c, left panel) (n = 76, wild-type; n = 82, Col6a1−/−) was also significantly lower in Col6a1−/− diaphragms (***P < 0.001; **P < 0.01; *P < 0.05; unpaired two-tailed Student’s t test). All measurements were obtained from 4 mice per genotype. Error bars indicate s.d. (b) or s.e.m. (c). d EPP amplitude after repetitive stimulation at 5 or 10 Hz for 25 s in wild-type and Col6a1−/− diaphragms, showing enhanced reduction of postsynaptic stimulation after stress in Col6a1−/− animals (***P < 0.001; unpaired two-tailed Student’s t test; n = 123 EPP, 5 Hz wild-type, n = 152 EPP, 5 Hz Col6a1−/−; n = 116, 10 Hz wild-type; n = 168, 10 Hz Col6a1−/−). Measurements were obtained from 8 mice per genotype. Error bars indicate s.d. e EPP amplitude intracellularly recorded after tetanic stimulation (10 Hz) for 25 s in wild-type and Col6a1−/− soleus fibers (n = 8 fibers, each genotype). Measurements were obtained from 2 mice per genotype. Data are percentage to baseline EPPs evoked at 0.5 Hz before tetanic stimulation. Error bars indicate s.d. f CMAP measurements in wild-type and Col6a1−/− diaphragms under untreated conditions and in the presence of increasing concentrations (500 and 800 nM) of d-tubocurarine. Col6a1−/− animals show a significantly higher decrease of CMAP, which is already displayed at 500 nM d-tubocurarine, thus unmasking a reduced safety factor in the absence of ColVI (***P < 0.001; **P < 0.01; *P < 0.05; unpaired two-tailed Student’s t test; n = 6–11 mice, each genotype). Error bars indicate s.e.m
We next assessed neuromuscular transmission by repetitive stimulation of the phrenic nerve for 25 pulses with 10 Hz (Fig. 4d). When analyzing the EPP amplitudes after 10 Hz stimulation, the decrement in the diaphragm of Col6a1−/− mice was higher than that of wild-type littermates, and a similar difference was found in soleus (Fig. 4e). We finally evaluated the safety factor, which reflects the fact that the threshold required to generate a muscle action potential is exceeded by the excitatory effect generated by nerve stimulation [72]. Towards this aim, we carried out compound muscle action potential (CMAP) measurements on wild-type and Col6a1−/− diaphragms in the presence of increasing concentrations of d-tubocurarine, in order to monitor the effect of a partial block of AChRs. Treatment with d-tubocurarine led to a strong decrease of CMAP amplitudes in response to repetitive stimuli in Col6a1−/− muscles but not in wild-type muscles (Fig. 4f), pointing at a reduced safety factor in ColVI-deficient animals. These results indicate that lack of ColVI impairs neuromuscular synaptic transmission, by affecting both sustained neurotransmitter release and safety factor.
Detection of NMJ alterations in UCMD patients
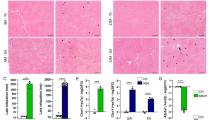
Next, we assessed whether the NMJ defects displayed by Col6a1−/− mice also occur in BM and UCMD patients. Towards this aim we investigated muscle biopsies already available in the Neuromuscular Bank of Tissues and DNA samples of the Telethon Network of Genetic BioBanks, which were isolated from six patients carrying different mutations in ColVI genes and displaying a range of clinical phenotypes, from typical BM to severe UCMD (Fig. 5a). The features of these muscle biopsies were evaluated in agreement with previous studies [59]. Histological analysis showed a prevalence of type I vs type II fibers in UCMD biopsies, when compared to BM and control biopsies (Fig. 5a and Suppl. Fig. S6). Immunofluorescence analysis for the myosin heavy chain (MHC) slow and fast isoforms largely confirmed the fiber type distribution reported in Fig. 5a for BM and UCMD patients (Suppl. Fig. S7). Interestingly, MHC immunolabeling performed in DMD and polymyositis biopsies did not show any overt disproportion in fiber type (Suppl. Fig. S7), thus sustaining the specificity of the results concerning patients affected by ColVI-related myopathies. Immunofluorescence showed close localization of ColVI labeling with αBT staining in control biopsies, whereas in BM and UCMD biopsies ColVI labeling appeared discontinuous and often detached from αBT staining (Fig. 5b). Moreover, and in keeping with the defect displayed by Col6a1−/− mice, immunostaining for collagen IV and utrophin appeared strongly upregulated at the level of NMJs in BM and UCMD patients’ biopsies, when compared to unaffected control biopsies (Fig. 5c). Finally, we evaluated the expression of genes coding for the different AChR subunits. Similarly to Col6a1−/− mice, UCMD biopsies showed increased mRNA levels of CHRNG and CHRNA genes, when compared to unaffected control biopsies (Fig. 5d). On the contrary, the mRNA levels of CHRNG and CHRNA genes in BM biopsies were, respectively, decreased or similar to those of control biopsies (Fig. 5d). This points at the presence of more overt NMJ defects in UCMD patients, compared to BM patients. Finally, we performed utrophin (Fig. 5e) and collagen IV (Fig. 5f) immunostaining also in two DMD and two polymyositis pathological controls, in order to gain further insight in the specificity of the postsynaptic defects detected in ColVI-related myopathies. This analysis did not show any major alteration in utrophin distribution in DMD or polymyositis biopsies, when compared to control biopsies (Fig. 5e). Collagen IV was abundant in DMD biopsies but not at the level of the NMJ, as instead displayed by UCMD and BM biopsies (Fig. 5f). Also compared to polymyositis biopsies, the deposition of collagen IV in UCMD/BM patients appeared higher (Fig. 5f), thus supporting the concept that the observed alterations are dependent on ColVI defects at the NMJ in BM and UCMD patients.

Hallmarks of NMJ defects in UCMD patients. a Table summarizing the clinical features, genetic characterization, and percentage of myofiber type for the BM and UCMD patients included in the study. NIMV, noninvasive mechanical ventilation; WCB, wheelchair bound. b Immunofluorescence for ColVI (green) in cross sections of muscle biopsies from unaffected control donors (CTRL), two BM patients, and two UCMD patients, stained with αBT (red). The insets show NMJ details at a higher magnification. Asterisks point at loss of contacts between ColVI and αBT labeling. Scale bar, 25 μm. c Representative immunolabeling for collagen IV and utrophin (white), performed together with αBT staining (red), in cross sections of muscle biopsies from unaffected control donor (CTRL), two BM patients, and two UCMD patients. Scale bar, 20 μm. d qRT-PCR analysis for the CHRNG and CHRNA genes in muscle biopsies from three unaffected control donors (C1-C3), three BM patients (P1-P3), and three UCMD patients (P4-P6). Dotted lines represent the mean value for each group, and the indicated P values were determined by the unpaired one-tailed Mann–Whitney test. e Representative immunolabeling for collagen IV (white) performed together with αBT staining (red), in cross sections of muscle biopsies from unaffected (CTRL) and pathological control donors, including two DMD patients (DMD1, DMD2), and two polymyositis patients (PM1, PM2). Scale bar, 25 μm. f Representative immunolabeling for utrophin (white) performed together with αBT staining (red), in cross sections of muscle biopsies from BM and UCMD patients in comparison with muscle biopsies from DMD and polymyositis patients. Arrowheads indicate NMJ, when covered by the signal of collagen IV staining. Scale bar, 25 μm
ColVI regulates AChR clustering and synaptic gene expression in vitro
To evaluate whether ColVI has a direct role at the NMJ, we tested the ability of the purified protein to affect AChR clustering in differentiated C2C12 myotube cultures, using agrin, a protein known to induce AChR clustering [12, 24], as positive control (Fig. 6a). Treatment of C2C12 myotubes with purified ColVI alone was able to induce aggregation of longer AChR clusters (Fig. 6b, c). Notably, this enhancement was completely prevented by the concurrent addition of an anti-ColVI antibody, whereas ColVI did not display any synergistic effect with agrin (Fig. 6c). In order to understand whether the effect exerted from ColVI was specific, we confirmed an extremely low endogenous expression of ColVI in differentiated myotubes, by both quantitative real-time PCR and immunofluorescence (Suppl. Fig. S8a, b). Interestingly, after ColVI was added to culture medium, the protein could be detected in close contact with AChR clusters and in tight continuity with myotube membrane (Suppl. Fig. S8b). Moreover, ColVI preparation was tested for purity, displaying almost exclusively its tetrameric form (Suppl. Fig. S8c), and showing no contamination by other ECM molecules such as laminin and fibronectin (Suppl. Fig. S8d, e).

ColVI promotes AChR clustering in differentiated myotubes. a Representative images of differentiated C2C12 myotubes under untreated conditions (Control) or treated for 20 h with purified ColVI (1 μg/ml), a mixture of purified ColVI and anti-ColVI antibodies (anti-ColVI; 40 μg/ml), or agrin (10 ng/ml). αBT staining was performed to assess AChR cluster (arrowhead) length in the different conditions. Scale bar, 50 μm. b, c Quantification of the percentage of AChR clusters belonging to different size classes (b) and of the average AChR cluster length (c) in C2C12 myotubes cultured in the different conditions as in a. ColVI promotes AChR clustering, and this effect is abolished in the presence of anti-ColVI antibodies. Data are shown as mean ± s.e.m. of three independent replicates (**P < 0.01 vs untreated condition; *P < 0.05 vs untreated condition; §P < 0.05 vs ColVI treatment; unpaired two-tailed Student’s t test; n = 15-25 AChR clusters per condition). d Western blot for MyoG in protein lysates of C2C12 myotubes cultured in the different conditions as in a. Densitometric quantification of the MyoG/GAPDH ratio, as determined by three independent experiments, is shown in the right panel. The MyoG/GAPDH ratio is expressed as fold-change relative to the untreated control conditions (**P < 0.01 vs untreated condition; §P < 0.05 vs ColVI treatment; unpaired two-tailed Student’s t test). Error bars indicate s.e.m. Ctrl, control (no treatment). e qRT-PCR analysis for genes involved in postsynaptic AChR cluster stabilization, performed in C2C12 myotubes cultured in the different conditions as in a. The mRNA levels of Lrp4, Musk, Chrna, Chrng, Utrn and Rapsn genes (coding, respectively, for LRP4, MuSK, AChRα, AChRγ, utrophin and rapsyn) were determined by three independent experiments (**P < 0.01 vs untreated condition; *P < 0.05 vs untreated condition; §§P < 0.01 vs ColVI treatment; §P < 0.05 vs ColVI treatment; unpaired two-tailed Student’s t test). Error bars indicate s.e.m
Since it was previously shown that AChR clustering depends on MyoG [34], we analyzed MyoG protein levels and found that they were significantly increased upon ColVI treatment, a response that was prevented by the addition of anti-ColVI antibodies (Fig. 6d). Agrin was shown to induce the expression of synaptic genes in vitro [32, 36]. We therefore investigated whether ColVI could, as well, modulate the expression of genes critical for postsynaptic AChR cluster stabilization. Treatment of C2C12 myotubes with native ColVI led to significant upregulation of the mRNA levels of Musk, Chrna, Chrng, Utrn and Rapsn genes, but not of the Lrp4 gene (Fig. 6e), as previously reported for agrin [25, 32, 36]. Since the presence of both N- and E-box regulatory elements was described for all the above ColVI responsive genes [16, 25, 26, 36, 43, 58, 62, 64], but not for Lpr4, these data point at the engagement of either ETS and/or bHLH transcription factor family-dependent pathways in the effects elicited by ColVI on AChR clustering.
Discussion
Previous work on ColVI in skeletal muscle, pointed at a crucial role for this protein in preserving myofiber homeostasis [7, 27], as well as in finely tuning the mechanical properties of muscle and in contributing to adult muscle satellite cell niche [67]. Here, we unravelled that ColVI is a component of the synaptic ECM and demonstrated that ColVI deficiency affects NMJ structure and function in vivo, thus highlighting an absolute novel role for ColVI in skeletal muscle. Our data indicate a major function for ColVI in promoting AChR clustering both in vivo and in vitro. Indeed, the tight contact of ColVI with AChR clusters, together with the enlargement and fragmentation of NMJs when the protein is missing, points at a role for ColVI in cluster stabilization.
While in the past only a limited number of proteins having a role within the synaptic basal lamina were described, the network of players in the last few years has become increasingly crowded. In the absence of ColVI, two of its known direct interactors [31, 66], collagen IV and perlecan, do increase at the NMJ level. Collagen IV is involved in the highly tuned organization of presynaptic terminals during development, and specific collagen IV chains are expressed at the synaptic cleft and are involved in the maintenance of NMJs [21, 39]. Perlecan was described as being involved in the stabilization of acetylcholinesterase clusters, and perlecan null mice are unable to express acetylcholinesterase at the NMJ [4]. Similarly, another interactor of ColVI, biglycan, was shown to be involved in agrin-dependent MuSK phosphorylation and in vitro AChR clustering [2]. Our data show that in the absence of ColVI both MuSK gene expression and LRP4 protein levels are altered. Altogether, these findings indicate a previously unexpected role for ColVI at the neuromuscular synaptic basal lamina, where its known binding partners are affected by its absence. Interestingly, a recent work demonstrated that Col6a3 gene expression is strongly affected in a model of congenital myasthenic syndrome, the ColQ null mouse, suggesting ColVI as a candidate partner for collagen Q [61] and opening interesting perspectives on ColVI involvement in other NMJ-specific diseases.
Lack of ColVI affects not only the expression of known interactors present in or facing the synaptic basal lamina, but also resulted in increased expression of NMJ-enriched proteins such as NOS1 and utrophin (as well as dystrophin). Of note, both proteins were reported to be modulated by electrical activity, one being overexpressed upon denervation [5], the other upon reinnervation [65], again pointing at defects in neuromuscular transmission in Col6a1−/− mice. Although nerve terminals do not seem to fail in reaching the postsynaptic boutons in Col6a1−/− mice, both NMJ fragmentation and AChRγ and AChRα mRNA upregulation were previously reported as clear signs of either defective NMJ contacts and transmission, or re-innervation defect, as it occurs in aging [9, 19, 37, 64, 71]. Indeed, our data show that ColVI deficiency affects neuromuscular transmission, as both nerve-evoked EPPs and nerve-independent mEPPs are significantly reduced in the soleus and diaphragm muscles of Col6a1−/− mice, together with a reduction of the safety factor.
Remarkably, CHRNG and CHRNA genes were also differentially upregulated in the more severe UCMD patients, compared to BM. Although limited to the analysis of muscle samples already available in a repository, due to constraints and ethical concerns in obtaining fresh nerve-guided muscle biopsies, the performed immunofluorescence data on patients’ biopsies do show alterations in ColVI deposition at the NMJ and dysregulation of NMJ-enriched proteins, such as collagen IV and utrophin, in agreement with what displayed by Col6a1−/− mice. These data suggest the presence of NMJ abnormalities not only in Col6a1−/− mice, but also in patients affected by ColVI-related diseases. In particular, the more overt NMJ defects in UCMD patients, compared to BM patients, suggest that NMJ alterations may substantially contribute to the worsening of ColVI-dependent myopathic features. In a more affected muscle, the latent safety factor defects, seen in Col6a1−/− mice, may enter into play, thus compromising neuromuscular transmission and contributing to a faster decline of muscle function in UCMD patients. On the other hand, most neuromuscular diseases ultimately impact the neuromuscular unit, leading to common clinical features including daily fluctuations of stamina, mostly related to muscle weakness [18]. Therefore, the potential implication of the nervous counterpart and of NMJ in BM and UCMD need further studies to be fully characterized. Detailed electromyographic studies of the NMJ and the demonstration of an impairment of the safety margin of neuromuscular transmission would be the proof-of-principle of the pathogenic role of NMJ in collagen VI-related myopathies [40]. However, such in-depth studies are not part of the routine assessment in either BM or UCMD and they have not been performed in our cohort of patients. Despite this, the observed defects at the NMJ in patients affected by ColVI-related myopathies, described in this work, open the field for deeper prospective studies, also in the light of recent data reporting a number of patients with both histological (fiber type grouping and fiber splitting) and electromyographic neurogenic features, suggesting the occurrence of denervation [29, 68].
Of interest, the most recent approaches for ColVI-related myopathies targeted the modulation of autophagic activity, which was found to be altered both in patients and in Col6a1−/− mice [10, 15]. Since autophagy was demonstrated to have a role in maintaining NMJ functionality [9, 50], NMJ amelioration should be taken into account as novel clinical endpoint, also prompting the search for noninvasive methods to assess it [57].
References
Ackley BD, Kang SH, Crew JR, Suh C, Jin Y, Kramer JM (2003) The basement membrane components nidogen and type XVIII collagen regulate organization of neuromuscular junctions in Caenorhabditis elegans. J Neurosci 23:3577–3587. https://doi.org/10.1523/JNEUROSCI.23-09-03577.2003
Amenta AR, Creely HE, Mercado MLT, Hagiwara H, McKechnie BA, Lechner BE, Rossi SG, Wang Q, Owens RT, Marrero E, Mei L, Hoch W, Young MF, McQuillan DJ, Rotundo RL, Fallon JR (2012) Biglycan is an extracellular musk binding protein important for synapse stability. J Neurosci 32:2324–2334. https://doi.org/10.1523/jneurosci.4610-11.2012
Andonian MH, Fahim MA (1987) Effects of endurance exercise on the morphology of mouse neuromuscular junctions during ageing. J Neurocytol 16:589–599. https://doi.org/10.1007/BF01637652
Arikawa-Hirasawa E, Rossi SG, Rotundo RL, Yamada Y (2002) Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat Neurosci 5:119–123. https://doi.org/10.1038/nn801
Biral D, Senter L, Salviati G (1996) Increased expression of dystrophin, β-dystroglycan and adhalin in denervated rat muscles. J Muscle Res Cell Motil 17:523–532. https://doi.org/10.1007/BF00124352
Blottner D, Lück G (2001) Just in time and place: NOS/NO system assembly in neuromuscular junction formation. Microsc Res Tech 55:171–180. https://doi.org/10.1002/jemt.1168
Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM (1998) Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet 7:2135–2140. https://doi.org/10.1093/hmg/7.13.2135
Bönnemann CG (2011) The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol 7:379–390. https://doi.org/10.1038/nrneurol.2011.81
Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R, Sandri M (2014) Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep 8:1509–1521. https://doi.org/10.1016/j.celrep.2014.07.061
Castagnaro S, Pellegrini C, Pellegrini M, Chrisam M, Sabatelli P, Toni S, Grumati P, Ripamonti C, Pratelli L, Maraldi NM, Cocchi D, Righi V, Faldini C, Sandri M, Bonaldo P, Merlini L (2016) Autophagy activation in COL6 myopathic patients by a low-protein-diet pilot trial. Autophagy 12:2484–2495. https://doi.org/10.1080/15548627.2016.1231279
Cescon M, Gattazzo F, Chen P, Bonaldo P (2015) Collagen VI at a glance. J Cell Sci 128:3525–3531. https://doi.org/10.1242/jcs.169748
Chang Y-F, Liu T-Y, Liu S-T (2013) Arecoline inhibits and destabilizes agrin-induced acetylcholine receptor cluster formation in C2C12 myotubes. Food Chem Toxicol 60:391–396. https://doi.org/10.1016/j.fct.2013.07.079
Chen P, Cescon M, Megighian A, Bonaldo P (2014) Collagen VI regulates peripheral nerve myelination and function. FASEB J 28:1145–1156. https://doi.org/10.1096/fj.13-239533
Cheusova T, Khan MA, Schubert SW, Gavin AC, Buchou T, Jacob G, Sticht H, Allende J, Boldyreff B, Brenner HR, Hashemolhosseini S (2006) Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction. Genes Dev 20:1800–1816. https://doi.org/10.1101/gad.375206
Chrisam M, Pirozzi M, Castagnaro S, Blaauw B, Polishchuck R, Cecconi F, Grumati P, Bonaldo P (2015) Reactivation of autophagy by spermidine ameliorates the myopathic defects of collagen VI-null mice. Autophagy 11:2142–2152. https://doi.org/10.1080/15548627.2015.1108508
de Kerchove d’Exaerde A, Cartaud J, Ravel-Chapuis A, Seroz T, Pasteau F, Angus LM, Jasmin BJ, Changeux J-P, Schaeffer L (2002) Expression of mutant Ets protein at the neuromuscular synapse causes alterations in morphology and gene expression. EMBO Rep 3:1075–1081. https://doi.org/10.1093/embo-reports/kvf220
Dorninger F, Herbst R, Kravic B, Camurdanoglu BZ, Macinkovic I, Zeitler G, Forss-Petter S, Strack S, Khan MM, Waterham HR, Rudolf R, Hashemolhosseini S, Berger J (2017) Reduced muscle strength in ether lipid-deficient mice is accompanied by altered development and function of the neuromuscular junction. J Neurochem. https://doi.org/10.1111/jnc.14082
Dowling JJ, Gonorazky HD, Cohn RD, Campbell C (2017) Treating pediatric neuromuscular disorders: the future is now. Am J Med Genet Part A. https://doi.org/10.1002/ajmg.a.38418
Eftimie R, Brenner HR, Buonanno A (1991) Myogenin and MyoD join a family of skeletal muscle genes regulated by electrical activity. Proc Natl Acad Sci USA 88:1349–1353. https://doi.org/10.1073/pnas.88.4.1349
Fox MA (2008) Novel roles for collagens in wiring the vertebrate nervous system. Curr Opin Cell Biol 20:508–513. https://doi.org/10.1016/j.ceb.2008.05.003
Fox MA, Sanes JR, Borza DB, Eswarakumar VP, Fässler R, Hudson BG, John SWM, Ninomiya Y, Pedchenko V, Pfaff SL, Rheault MN, Sado Y, Segal Y, Werle MJ, Umemori H (2007) Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 129:179–193. https://doi.org/10.1016/j.cell.2007.02.035
Gara SK, Grumati P, Squarzoni S, Sabatelli P, Urciuolo A, Bonaldo P, Paulsson M, Wagener R (2011) Differential and restricted expression of novel collagen VI chains in mouse. Matrix Biol 30:248–257. https://doi.org/10.1016/j.matbio.2011.03.006
Gara SK, Grumati P, Urciuolo A, Bonaldo P, Kobbe B, Koch M, Paulsson M, Wagener R (2008) Three novel collagen VI chains with high homology to the α3 chain. J Biol Chem 283:10658–10670. https://doi.org/10.1074/jbc.M709540200
Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (1996) Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85:525–535. https://doi.org/10.1016/S0092-8674(00)81253-2
Gramolini AO, Burton EA, Tinsley JM, Ferns MJ, Cartaud A, Cartaud J, Davies KE, Lunde JA, Jasmin BJ (1998) Muscle and neural isoforms of agrin increase utrophin expression in cultured myotubes via a transcriptional regulatory mechanism. J Biol Chem 273:736–743. https://doi.org/10.1074/jbc.273.2.736
Gramolini AO, Jasmin BJ (1999) Expression of the utrophin gene during myogenic differentiation. Nucleic Acids Res 27:3603–3609. https://doi.org/10.1093/nar/27.17.3603
Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, Maraldi NM, Bernardi P, Sandri M, Bonaldo P (2010) Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16:1313–1320. https://doi.org/10.1038/nm.2247
Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M, Bonaldo P (2011) Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 7:1415–1423. https://doi.org/10.4161/auto.7.12.17877
Hunter JM, Ellen Ahearn M, Balak CD, Liang WS, Kurdoglu A, Corneveaux JJ, Russell M, Huentelman MJ, Craig DW, Carpten J, Coons SW, DeMello DE, Hall JG, Bernes SM, Baumbach-Reardon L, Jesse Hunter CM, Clow K (2015) Novel pathogenic variants and genes for myopathies identified by whole exome sequencing. Mol Genet Genom Med 3:283–301. https://doi.org/10.1002/mgg3.142
Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P (2003) Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 35:367–371. https://doi.org/10.1038/ng1270
Kuo H-J, Maslen CL, Keene DR, Glanville RW (1997) Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J Biol Chem 272:26522–26529. https://doi.org/10.1074/jbc.272.42.26522
Lacazette E, Le Calvez S, Gajendran N, Brenner HR (2003) A novel pathway for MuSK to induce key genes in neuromuscular synapse formation. J Cell Biol 161:727–736. https://doi.org/10.1083/jcb.200210156
Liley AW (1956) The effects of presynaptic polarization on the spontaneous activity at the mammalian neuromuscular junction. J Physiol 134:427–443. https://doi.org/10.1113/jphysiol.1956.sp005655
Macpherson PCD, Cieslak D, Goldman D (2006) Myogenin-dependent nAChR clustering in aneural myotubes. Mol Cell Neurosci 31:649–660. https://doi.org/10.1016/j.mcn.2005.12.005
Massoulié J, Millard CB (2009) Cholinesterases and the basal lamina at vertebrate neuromuscular junctions. Curr Opin Pharmacol 9:316–325. https://doi.org/10.1016/j.coph.2009.04.004
Meier T, Masciulli F, Moore C, Schoumacher F, Eppenberger U, Denzer AJ, Jones G, Brenner HR (1998) Agrin can mediate acetylcholine receptor gene expression in muscle by aggregation of muscle-derived neuregulins. J Cell Biol 141:715–726. https://doi.org/10.1083/jcb.141.3.715
Méjat A, Decostre V, Li J, Renou L, Kesari A, Hantaï D, Stewart CL, Xiao X, Hoffman E, Bonne G, Misteli T (2009) Lamin A/C-mediated neuromuscular junction defects in Emery–Dreifuss muscular dystrophy. J Cell Biol 184:31–44. https://doi.org/10.1083/jcb.200811035
Merlini L, Martoni E, Grumati P, Sabatelli P, Squarzoni S, Urciuolo A, Ferlini A, Gualandi F, Bonaldo P (2008) Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology 71:1245–1253. https://doi.org/10.1212/01.wnl.0000327611.01687.5e
Miner JH (1994) Collagen IV alpha 3, alpha 4, and alpha 5 chains in rodent basal laminae: sequence, distribution, association with laminins, and developmental switches. J Cell Biol 127:879–891. https://doi.org/10.1083/jcb.127.3.879
Mongiovi PC, Elsheikh B, Lawson VH, Kissel JT, Arnold WD (2014) Neuromuscular junction disorders mimicking myopathy. Muscle Nerve 50:854–856. https://doi.org/10.1002/mus.24300
Nishimune H, Valdez G, Jarad G, Moulson CL, Müller U, Miner JH, Sanes JR (2008) Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J Cell Biol 182:1201–1215. https://doi.org/10.1083/jcb.200805095
Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP (1995) Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin β2. Nature 374:258–262. https://doi.org/10.1038/374258a0
Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG (2003) E-box mutations in the RAPSN promoter regions in eight cases with congenital myasthenic syndrome. Hum Mol Genet 12:739–748. https://doi.org/10.1093/hmg/ddg089
Patton BL, Cunningham JM, Thyboll J, Kortesmaa J, Westerblad H, Edström L, Tryggvason K, Sanes JR (2001) Properly formed but improperly localized synaptic specializations in the absence of laminin α4. Nat Neurosci 4:597–604. https://doi.org/10.1038/88414
Patton BL, Miner JH, Chiu AY, Sanes JR (1997) Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol 139:1507–1521. https://doi.org/10.1083/jcb.139.6.1507
Peng HB, Xie H, Rossi SG, Rotundo RL (1999) Acetylcholinesterase clustering at the neuromuscular junction involves perlecan and dystroglycan. J Cell Biol 145:911–921. https://doi.org/10.1083/jcb.145.4.911
Plomp JJ, van Kempen GT, Molenaar PC (1992) Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol 458:487–499. https://doi.org/10.1113/jphysiol.1992.sp019429
Robinson KG, Mendonca JL, Militar JL, Theroux MC, Dabney KW, Shah SA, Miller F, Akins RE (2013) Disruption of basal lamina components in neuromotor synapses of children with spastic quadriplegic cerebral palsy. PLoS One 8:e70288. https://doi.org/10.1371/journal.pone.0070288
Rogozhin AA, Pang KK, Bukharaeva E, Young C, Slater CR (2008) Recovery of mouse neuromuscular junctions from single and repeated injections of botulinum neurotoxin A. J Physiol 586:3163–3182. https://doi.org/10.1113/jphysiol.2008.153569
Rudolf R, Khan MM, Labeit S, Deschenes MR (2014) Degeneration of neuromuscular junction in age and dystrophy. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2014.00099
Sabatelli P, Bonaldo P, Lattanzi G, Braghetta P, Bergamin N, Capanni C, Mattioli E, Columbaro M, Ognibene A, Pepe G, Bertini E, Merlini L, Maraldi NM, Squarzoni S (2001) Collagen VI deficiency affects the organization of fibronectin in the extracellular matrix of cultured fibroblasts. Matrix Biol 20:475–486. https://doi.org/10.1016/S0945-053X(01)00160-3
Sabatelli P, Gara SK, Grumati P, Urciuolo A, Gualandi F, Curci R, Squarzoni S, Zamparelli A, Martoni E, Merlini L, Paulsson M, Bonaldo P, Wagener R (2011) Expression of the collagen VI α5 and α6 chains in normal human skin and in skin of patients with collagen VI-related myopathies. J Invest Dermatol 131:99–107. https://doi.org/10.1038/jid.2010.284
Sabatelli P, Gualandi F, Gara SK, Grumati P, Zamparelli A, Martoni E, Pellegrini C, Merlini L, Ferlini A, Bonaldo P, Maraldi NM, Paulsson M, Squarzoni S, Wagener R (2012) Expression of collagen VI α5 and α6 chains in human muscle and in Duchenne muscular dystrophy-related muscle fibrosis. Matrix Biol 31:187–196. https://doi.org/10.1016/j.matbio.2011.12.003
Sala C, Andreose JS, Fumagalli G, Lømo T (1995) Calcitonin gene-related peptide: possible role in formation and maintenance of neuromuscular junctions. J Neurosci 15:520–528. https://doi.org/10.1523/JNEUROSCI.15-01-00520.1995
Sandrock AW Jr (1997) Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science (80-) 276:599–603. https://doi.org/10.1126/science.276.5312.599
Sanes JR (2003) The basement membrane/basal lamina of skeletal muscle. J Biol Chem 278:12601–12604. https://doi.org/10.1074/jbc.R200027200
Sarwal A, Walker FO, Cartwright MS (2013) Neuromuscular ultrasound for evaluation of the diaphragm. Muscle Nerve 47:319–329. https://doi.org/10.1002/mus.23671
Schaeffer L, de Kerchove d’Exaerde A, Duclert N, Huchet-Dymanus M, Changeux J-P J-P (1998) Implication of a multisubunit Ets related transcription factor in synaptic expression of the nicotinic acetylcholine receptor. J Physiol 92:489. https://doi.org/10.1016/S0928-4257(99)80106-5
Schessl J, Goemans NM, Magold AI, Zou Y, Hu Y, Kirschner J, Sciot R, Bönnemann CG (2008) Predominant fiber atrophy and fiber type disproportion in early Ullrich disease. Muscle Nerve 38:1184–1191. https://doi.org/10.1002/mus.21088
Schiaffino S, Reggiani C (2011) Fiber types in mammalian skeletal muscles. Physiol Rev 91:1447–1531. https://doi.org/10.1152/physrev.00031.2010
Sigoillot SM, Bourgeois F, Karmouch J, Molgó J, Dobbertin A, Chevalier C, Houlgatte R, Léger J, Legay C (2016) Neuromuscular junction immaturity and muscle atrophy are hallmarks of the ColQ-deficient mouse, a model of congenital myasthenic syndrome with acetylcholinesterase deficiency. FASEB J 30:2382–2399. https://doi.org/10.1096/fj.201500162
Simon AM, Burden SJ (1993) An E box mediates activation and repression of the acetylcholine receptor delta-subunit gene during myogenesis. Mol Cell Biol 13:5133–5140. https://doi.org/10.1128/mcb.13.9.5133
Singhal N, Martin PT (2011) Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev Neurobiol 71:982–1005. https://doi.org/10.1002/dneu.20953
Tang H, Veldman MB, Goldman D (2006) Characterization of a muscle-specific enhancer in human MuSK promoter reveals the essential role of myogenin in controlling activity-dependent gene regulation. J Biol Chem 281:3943–3953. https://doi.org/10.1074/jbc.M511317200
Tews DS, Goebel HH, Schneider I, Gunkel A, Stennert E, Neiss WF (1997) Expression of different isoforms of nitric oxide synthase in experimentally denervated and reinnervated skeletal muscle. J Neuropathol Exp Neurol 56:1283–1289. https://doi.org/10.1097/00005072-199712000-00003
Tillet E, Wiedemann H, Golbik R, Pan TC, Zhang RZ, Mann K, Chu ML, Timpl R (1994) Recombinant expression and structural and binding properties of alpha 1(VI) and alpha 2(VI) chains of human collagen type VI. Eur J Biochem 221:177–185. https://doi.org/10.1111/j.1432-1033.1994.tb18727.x
Urciuolo A, Quarta M, Morbidoni V, Gattazzo F, Molon S, Grumati P, Montemurro F, Tedesco FS, Blaauw B, Cossu G, Vozzi G, Rando TA, Bonaldo P (2013) Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat Commun 4:1964. https://doi.org/10.1038/ncomms2964
Verma S, Goyal P, Guglani L, Peinhardt C, Pelzek D, Barkhaus PE (2018) COL6A and LAMA2 mutation congenital muscular dystrophy: a clinical and electrophysiological study. J Clin Neuromuscul Dis 30324:108–116. https://doi.org/10.1097/CND.0000000000000198
Wiberg C, Hedbom E, Khairullina A, Lamandé SR, Oldberg A, Timpl R, Mörgelin M, Heinegård D (2001) Biglycan and decorin bind close to the n-terminal region of the collagen VI triple helix. J Biol Chem 276:18947–18952. https://doi.org/10.1074/jbc.m100625200
Willadt S, Nash M, Slater CR (2016) Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci Rep 6:24849. https://doi.org/10.1038/srep24849
Witzemann V, Barg B, Criado M, Stein E, Sakmann B (1989) Developmental regulation of five subunit specific mRNAs encoding acetylcholine receptor subtypes in rat muscle. FEBS Lett 242:419–424. https://doi.org/10.1016/0014-5793(89)80514-9
Wood SJ, Slater CR (1997) The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast-and slow-twitch muscles. J Physiol 500:65–176. https://doi.org/10.1113/jphysiol.1997.sp022007
Acknowledgements
We are grateful to R. Wagener for the 3(VI) antibody and Dr M. Vitadello for the neurofilament antibody. We also acknowledge support from Telethon Genetic BioBank (GTB12001D) and the Eurobiobank network. This work was supported by Italian Ministry of Education, University and Research (Grants RBAP11Z3YA_003 and 2015FBNB5Y), Telethon Foundation (Grant GGP14202) and University of Padova (to P.B.); Cariparo Foundation (Starting Grants 2015) (to M.C. and P.B.); German Research Council DFG (Grant HA3309/3-1) and Interdisciplinary Centre for Clinical Research at the University Hospital of the Friedrich-Alexander University of Erlangen-Nürnberg (Grant E17) (to S.H.). I.G. was supported by a PhD fellowship from the Cariparo Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing or financial interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cescon, M., Gregorio, I., Eiber, N. et al. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol 136, 483–499 (2018). https://doi.org/10.1007/s00401-018-1860-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-1860-9