
Abstract
Glioblastoma is the most frequent malignant primary brain tumor. In a hierarchical tumor model, glioblastoma stem-like cells (GSC) play a major role in tumor initiation and maintenance as well as in therapy resistance and recurrence. Thus, targeting this cellular subset may be key to effective immunotherapy. Here, we present a mass spectrometry-based analysis of HLA-presented peptidomes of GSC and glioblastoma patient specimens. Based on the analysis of patient samples (n = 9) and GSC (n = 3), we performed comparative HLA peptidome profiling against a dataset of normal human tissues. Using this immunopeptidome-centric approach we could clearly delineate a subset of naturally presented, GSC-associated HLA ligands, which might serve as highly specific targets for T cell-based immunotherapy. In total, we identified 17 antigens represented by 41 different HLA ligands showing natural and exclusive presentation both on GSC and patient samples. Importantly, in vitro immunogenicity and antigen-specific target cell killing assays suggest these peptides to be epitopes of functional CD8+ T cell responses, thus rendering them prime candidates for antigen-specific immunotherapy of glioblastoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma, the most frequent malignant primary brain tumor, carries a poor prognosis despite advances in surgery, radiation- and chemotherapy [37]. This dilemma calls for innovative approaches, one of the most promising of which is T cell-based immunotherapy. The impressive clinical efficacy of immune checkpoint inhibitors in a subset of patients with extracranial malignancies demonstrates the potential of T cell-based immunotherapy to induce and maintain long-lasting anti-tumor immune responses [4, 10, 29]. Since the clinical benefit of immune checkpoint inhibition is associated with high numbers of tumor-associated somatic mutations and since this mutational burden is rather low in glioblastoma [1], T cell epitopes from non-mutated sources may represent the most actionable targets in glioblastoma. Such targets may be utilized to boost or direct the specificities of anti-tumoral T cell responses, e.g., by combination therapies of checkpoint blockade and peptide vaccination or in adoptive cellular approaches. In this context, the identification of appropriate targets is crucial. A central challenge in targeting a highly heterogeneous tumor such as glioblastoma is to prevent positive clonal selection of antigen loss variants [19].
In recent years, there is growing evidence for the presence of glioma-initiating cells exhibiting stem cell properties, which can be designated as glioblastoma stem-like cells (GSC) [9, 27]. In a hierarchical tumor model, GSC are supposed to have important functions in the initiation, maintenance, and therapy resistance of glioblastoma and therefore warrant evaluation as a possible target for immunotherapy.
By mapping the HLA peptidome of glioblastoma and GSC, we identified a new panel of T cell antigens characterized by exclusive detection on malignant specimens, a high frequency of presentation as well as representation on the glioblastoma stem cell compartment. This study is based on the in-depth HLA peptidome analysis of 3 established GSC lines (GS-2, GS-5, GS-9), which have been characterized extensively for stable stemness properties [9, 40], as well as 9 freshly resected glioblastoma samples. This glioblastoma cohort was analyzed in relation to a comprehensive dataset of HLA ligands presented on various normal human tissues including normal human brain. In contrast to previous studies on the HLA peptidome of glioblastoma [7, 20] our extensive dataset allows for a purely HLA peptidome-centric analysis of the antigenic landscape of bulk patient tumors and the glioblastoma stem cell compartment as represented by GSC. Notably, cluster and network analyses revealed direct evidence for the gliogenesis and stem cell fate determination being reflected in the intersection of patient tumor and GSC immunopeptidomes. Furthermore, in vitro priming assays demonstrated the induction of specific T cell responses for a subset of these novel target candidates, underscoring their potential for T cell immunotherapy.
Materials and methods
Patients and blood samples
Primary tissue of patients with histologically confirmed glioblastoma who underwent surgery at the Department of Neurosurgery at the University Hospital Zurich was immediately snap-frozen in liquid nitrogen during surgery and stored at − 80 °C. Written informed consent was obtained from each glioblastoma patient as well as from patients and healthy donors that contributed to the normal tissue database in accordance with the local ethical requirements and the Declaration of Helsinki. Institutions that contributed samples to the normal tissue database are listed in the Supplementary Methods (Online Resource 1).
Peripheral blood mononuclear cells (PBMC) of healthy donors for functional experiments were isolated by density gradient centrifugation. HLA typing of cell lines and tumor samples was carried out at the Department of Hematology and Oncology, University of Tübingen, Germany. PBMC of HLA-typed healthy donors were provided by the Department of Transfusion Medicine, University of Tübingen, Germany, and by the Swiss Red Cross Blood Transfusion Services Zurich and Chur, Switzerland.
All analyses were performed according to the guidelines of the local ethics committees (KEK-ZH-Nr. 2015-0163).
Cell lines and GSC culture expansion
We used established GSC lines GS-2, GS-5, GS-9, which have been characterized extensively for stable stemness properties [9, 40]. GSC were provided by the Department of Neurosurgery at the University Hospital Hamburg-Eppendorf. Cells were tested for mycoplasma contamination every 3 months using a PCR-based test kit. Cell line authentication and exclusion of cross-contamination was performed showing stable HLA typing (PCR with sequence-specific primers) from original cell lines and cell pellets used for experiments. GSC were thawed around passages 15–20 and before using the cells for experiments they were passaged 6–8 times. GSC were maintained in neurobasal medium with B-27 supplement (20 µl/ml) and glutamax (10 µl/ml) (all Invitrogen, Karlsruhe, Germany), as well as fibroblast growth factor (FGF)-2, epidermal growth factor (EGF) (20 ng/ml each; Peprotech, Rocky Hill, PA, USA) and heparin (32 IE/ml; Ratiopharm, Ulm, Germany). Stem cell factors were supplemented twice a week, the medium was changed once a week. Cells were passaged when spheres reached an estimated diameter of 500 μm or an estimated density of 5 × 104 cells/cm2. Spheres were dissociated mechanically and enzymatically (accutase; PAA, Vienna, Austria). After culture expansion, GSC line cell pellets (1 ml each) were harvested and snap-frozen. Three experimental replicates per GSC line were produced and each experimental replicate was used for the isolation of HLA ligands with subsequent analysis by LC–MS/MS in 5 technical replicates (as described below).
Immunofluorescence
5 × 105 cells were cytospun onto a glass slide, dried for 30 min and fixed in 4% formaldehyde for 10 min. Subsequently, the cells were stained using mouse monoclonal primary antibody to HLA-A, -B, -C (EMR8-5; 1:50; BD Pharmingen, Franklin Lakes, NJ, USA) or the mouse monoclonal primary antibody to HLA class II (ab55152; 1:50; Abcam, Cambridge, UK) followed by permeabilization with 0.5% Triton X100 and incubation with goat anti-rabbit IgG-Alexa Fluor 488-coupled secondary antibody (Invitrogen) (diluted 1:100) and nuclear DNA labeling with DAPI (Invitrogen) for visualization. Images were acquired using a Leica SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany).
Immunohistochemistry
Immunohistochemistry was carried out on cytospins (prepared as described above) or paraformaldehyde-fixed paraffin-embedded glioma sections. Cytospins or sections were stained using the mouse monoclonal primary antibody to HLA-A, -B, -C (1:50) (BD Pharmingen) or the mouse monoclonal primary antibody to HLA class II (1:50) (Abcam) and visualized using anti-mouse secondary antibody with the ImmPACT DAB kit (Vector, Burlingame, CA, USA). Binding specificity was controlled by an isotype control. Images were acquired using a Zeiss Scope.A1 microscope (Carl Zeiss Microscopy, Jena, Germany).
Isolation of HLA ligands
HLA class I molecules were isolated using standard immunoaffinity purification [8, 16]. In brief, snap-frozen GSC pellets or snap-frozen primary glioblastoma tissue samples were lysed in 10 mM CHAPS/PBS (AppliChem, St. Louis, MO, USA/Gibco, Carlsbad, CA, USA) containing 1× protease inhibitor (Complete; Roche, Basel, Switzerland). HLA molecules were single-step purified using the pan-HLA class I-specific mAb W6/32 (produced in-house) covalently linked to CNBr-activated Sepharose (GE Healthcare, Chalfont St Giles, UK). For the immunoprecipitation of HLA class II–peptide complexes, we used equal amounts of the HLA-DR-specific antibody L243 as well as the pan-HLA class II-specific antibody Tü39 (both produced in-house). HLA-DR1 is known to be expressed at higher levels than the other class II allotypes, therefore the specific L243 mAb at the given stoichiometry was used. Tü39 is utilized complementarily to pull-down the remaining class II complexes. HLA–peptide complexes were eluted by repeated addition of 0.2% TFA (trifluoroacetic acid, Merck, Whitehouse Station, NJ, USA). Eluted HLA ligands were purified by ultrafiltration using centrifugal filter units (Amicon; Millipore, Billerica, MA, USA). HLA ligands were desalted using ZipTip C18 pipette tips (Millipore). Extracted peptides were eluted in 35 μl 80% acetonitrile (Merck)/0.2% TFA, vacuum-centrifuged to complete dryness and resuspended in 25 μl of 1% acetonitrile/0.05% TFA. Samples were stored at − 20 °C until analysis by LC–MS/MS.
Analysis of HLA ligands by LC–MS/MS
Peptide samples were separated by reversed-phase liquid chromatography (nano-UHPLC, UltiMate 3000 RSLCnano; Thermo Fisher, Waltham, MA, USA) and analyzed in online-coupled LTQ Orbitrap XL or Orbitrap Fusion Lumos mass spectrometers (both Thermo Fisher). Samples were analyzed in five (Orbitrap XL) or three (Fusion Lumos) technical replicates. Sample shares of 20% (Orbitrap XL) or 10% (Fusion Lumos) were trapped on a 75 μm × 2 cm trapping column (Acclaim PepMap RSLC; Thermo Fisher) at 4 μl/min for 5.75 min. Peptide separation was performed at 50 °C and a flow rate of 175 nl/min on a 50 μm × 25 cm separation column (Acclaim PepMap RSLC; Thermo Fisher) applying a gradient ranging from 2.4 to 32.0% of acetonitrile over the course of 90 min. The majority of samples were analyzed on an LTQ Orbitrap XL using a top five CID (collision-induced dissociation) method with survey scans at 60k resolution and fragment ion detection in the ion trap (ITMS2) operated at normal scan speed. Seven normal tissue specimens from the central nervous system (brain04–brain07 and cerebellum04, cerebellum06, cerebellum07) were analyzed on the Orbitrap Fusion Lumos for tumor-exclusivity validation of targets implementing a top-speed CID method with survey scans at 120k resolution and fragment detection in the Orbitrap (OTMS2) at 60 k resolution. On both instruments, the mass range was limited to 400–650 m/z with precursors of charge states 2+ and 3+ eligible for fragmentation. For HLA class II, a mass range of 300–1500 m/z was analyzed with charge states ≥ 2 selected for fragmentation.
Database search and spectral annotation
The software Proteome Discoverer (v.1.3; Thermo Fisher) was used to perform database search using the Mascot search engine (Mascot 2.2.04; Matrix Science, London, UK) for ITMS2 data and Sequest (Thermo Fisher) for OTMS2 data. The human proteome as comprised in the Swiss-Prot database (http://www.uniprot.org, status September 2013, 20,225 reviewed sequences contained) was used as reference database. The search combined data of technical replicates and was not restricted by enzymatic specificity. Precursor mass tolerance was set to 5 ppm, and fragment mass tolerance to 0.5 Da for ITMS2 data and 0.02 Da for OTMS2 data, respectively. Oxidation of methionine residues was allowed as a dynamic modification. False discovery rate (FDR) was estimated using the Percolator node [14] and was limited to 5%. ITMS2 peptide IDs were further filtered for Mascot Ion scores ≥ 20. For HLA class I ligands, peptide lengths were limited to 8–12 amino acids (AA), for HLA class II, peptides were limited to 12–25 AA of length. Protein grouping was disabled, allowing for multiple annotations of peptides, e.g., for conserved sequences mapping into multiple proteins. As a final step of quality control, thresholds of ≥ 500 (HLA class I, primary glioblastoma tissue), ≥ 200 (HLA class I, GSC), unique ligand identifications per sample were applied. HLA annotation was performed using the prediction tool SYFPEITHI (http://www.syfpeithi.de) or an extended in-house database [22]. Experimental validation of the predicted HLA restriction was obtained by peptide synthesis and functional characterization for a subset of ligands.
Peptide synthesis
Synthetic peptides for the validation of mass spectrometric identifications as well as for functional experiments were synthesized using the automated peptide synthesizer EPS221 (Abimed, Langenfeld, Germany) and the 9-fluorenylmethyloxycarbonyl/tert-butyl (Fmoc/tBu) strategy [28].
CD8+ T cell priming using aAPC
Priming of CD8+ T cells was performed using artificial antigen-presenting cells (aAPC) according to a standard protocol [35]. PBMC were isolated from fresh buffy coats of healthy volunteers using standard density gradient centrifugation. CD8+ T cells were MACS-enriched by positive selection (Miltenyi Biotec, Bergisch Gladbach, Germany).
In vitro stimulations were initiated in 24-well plates with 5 × 106 responder cells plus 106 beads per well in 1.5 ml of T cell medium consisting of RPMI 1640 containing 25 mM HEPES (Life Technologies/Invitrogen) supplemented with 10% heat-inactivated human AB serum (CC Pro, Neustadt/Weinstraße, Germany), 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, and 20 μg/ml gentamicin (all BioWhittaker/Cambrex, Verviers, Belgium). Five nanograms per milliliter human IL-12 p70 (R&D Systems) was added with aAPC. After 4 days of co-incubation at 37 °C, fresh medium and 20 U/ml human IL-2 (R&D Systems) were added, and cells were incubated for 3 days. This stimulation cycle was repeated twice and tetramer staining was performed on day 21.
Intracellular cytokine and tetramer staining
The frequency and functionality of peptide-specific CD8+ T cells was analyzed by intracellular IFN-γ and TNF-α staining as described previously [21, 39]. PBMC were pulsed with 10 μg/ml of individual peptide and incubated in the presence of 10 μg/ml Brefeldin A (Sigma Aldrich) and 10 μg/ml GolgiStop (BD Biosciences) for 6–8 h. Cells were labeled using Cytofix/Cytoperm (BD Biosciences), CD8-PECy7 (Beckman Coulter, Fullerton, CA, USA), CD4-APC (BD Biosciences), TNFα-PE (Beckman Coulter), and IFNγ-FITC (BD Biosciences). Samples were analyzed on a FACS Canto II (BD, Franklin Lakes, NJ, USA). The frequency of peptide-specific CD8+ T cells was determined by staining with anti-CD8 and HLA:peptide-tetramer-PE [25]. Cultures were considered positive when > 1% of tetramer-positive cells among CD8 + cells were detected.
Cytotoxicity assay
Tetramer-positive cells were enriched by FACS cell sorting (BD FACSAria III, BD Biosciences) and subsequent expansion using irradiated PBMC as feeder cells and 3000 U/ml IL-2 (PeproTech, London, UK) and 30 ng/ml OKT3 (eBioscience, Thermo Fisher). Medium supplemented with IL-2 and OKT3 was changed twice a week. Tetramer-negative cells were expanded in parallel as controls. T cell-mediated GSC lysis was determined using a flow cytometry-based cytotoxicity assay. A single cell suspension of target cells was stained with PKH-26 (Sigma-Aldrich, St. Louis, MO, USA) for 3 min and then co-incubated with T effector cells at different effector to target (E:T) ratios as indicated for 20 h. Subsequently, live/dead staining was performed with Zombie NIR Fixable Viability Kit (BioLegend, San Diego, CA, USA) followed by assessment of target cell lysis by flow cytometry. Specific cell lysis was expressed as percentage of dead cells within the PKH-26-positive target cells, corrected for spontaneous background lysis.
Software, statistical analysis and online tools
Flow cytometric data analysis was performed using FlowJo 7.2 (Tree Star Inc., Ashland, OR, USA). In-house R scripts (https://www.r-project.org) were used for the calculation of TAA false discovery rates and unsupervised hierarchical clustering of HLA ligand source proteins. Overlap analyses were done using VENNY (Oliveros, J.C. (2007–2015) Venny. An interactive tool for comparing lists with Venn’s diagrams (http://bioinfogp.cnb.csic.es/tools/venny/index.html) as well as the online tool BioVenn [13]. Functional Annotation charts were created using DAVID (https://david-d.ncifcrf.gov) [11, 12]. The network analysis of GSC- and patient-specific source proteins was performed using GeneMania (http://www.genemania.org; default settings) [36]. Bar charts were created using Microsoft Excel 2015 (Redmond, WA, USA), box plots and line graphs were done using Prism 5 (GraphPad Software, San Diego, CA). Kaplan–Meier analysis of survival probability and gene expression analysis for the Glioma–French–284–MAS5.0–u133p2 dataset (n = 284, glioblastoma n = 156) in the database of The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov) was done using glioblastoma database entries only (n = 156) and using the R2 microarray analysis and visualization platform (http://r2.amc.nl) using a cut off for high expression (top 25%) versus the rest.
Survival was analyzed separately for glioblastoma patients regardless of IDH1 mutation status (n = 156) and for patients with known IDH1 mutation status harboring no IDH1 mutation (IDH1 wt, n = 92).
Results
Mapping the antigenic landscape of naturally presented HLA class I and II ligands on primary glioblastoma and GSC
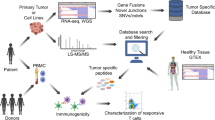
As a prerequisite for HLA ligandome profiling we analyzed HLA class I and class II expression on GSC (Fig. 1a, b) and patient samples (Fig. 1c), showing robust membrane staining for both GSC and patient samples. The workflow of our antigen discovery approach is summarized in Fig. 2a, b. Three different GSC lines (GS-2, GS-5, GS-9) were culture-expanded and cell pellets (1 ml each) as well as 9 freshly resected glioblastoma samples were snap-frozen. Tumor samples and three experimental replicates per GSC line were used for the isolation of HLA ligands with subsequent analysis by LC–MS/MS in 5 technical replicates. HLA ligandome mapping was done in comparison with an extensive HLA peptidome database. For HLA class I, this database comprised 160 different human normal tissues including central nervous system samples (n = 13) with a total of 46,429 peptides derived from 12,245 source proteins. A list of normal tissues included in this database is provided in Supplementary Table 1 (Online Resource 2). Comparative profiling of HLA ligand source proteins from tumor samples and benign tissue samples was performed separately for HLA class I and II based on the frequency of HLA-restricted representation (Fig. 2c). The overlap between GSC, patient samples and normal tissues is depicted in Fig. 2d–g for both, source protein and HLA ligand level. HLA class I peptidome analysis of the GSC lines and patient samples yielded a total of 9770 peptide identifications, representing 5703 source proteins. Mapping of the HLA class II ligandome identified 2942 different peptides derived from 1121 distinct source proteins. Basic patient characteristics including HLA typing and peptide yields are provided in Table 1. The 3 cell lines and 9 patient samples included a total of 23 different HLA-A and -B allotypes; 99.3% of individuals of the Caucasian population have at least one HLA-A or -B allotype that is represented by this tumor dataset (calculated by the IEDB population coverage tool www.iedb.org). The most frequent allotypes were HLA-A*02 (n = 7), -A*03 (n = 6), -B*44 (n = 5), and -B*18 (n = 3). The HLA allotype-dependent population coverage is depicted in Supplementary Fig. 1 (Online Resource 3). Peptide yields per sample and the length distribution of peptides are shown in Fig. 3a, b. A list of all peptides and corresponding source proteins from tumor samples and normal tissue is provided in Supplementary Table 2 (Online Resource 4).

HLA class I and II molecules are expressed on GS cells and patient samples. a, b GS-2, GS-5 and GS-9 cells were analyzed for HLA class I and II cell surface levels by immunocytochemistry (a) or immunofluorescence (b) and nuclei were counterstained with hematoxylin or DAPI, respectively (scale bar, 50 µm). c HLA class I and II protein levels were assessed on glioblastoma specimens by immunohistochemistry and nuclei were counterstained with hematoxylin (scale bar, 50 µm for ×20 or ×40 magnification)

Experimental workflow and overview of HLA ligandome mapping results. a Snap-frozen GSC pellets or snap-frozen glioblastoma patient samples were lysed and HLA molecules were single-step immunopurified using the pan-HLA class I-specific mAb W6/32 covalently linked to CNBr-activated Sepharose. For HLA class II, we used equal amounts of the HLA-DR-specific antibody L243 as well as the pan-HLA class II-specific antibody Tü39. Eluted HLA–peptide ligands were purified by ultrafiltration, desalted and subjected to analysis by LC–MS/MS. b Candidate peptides for immunotherapy were defined by tumor exclusivity, shared presentation on both GSC and patient samples and a sufficient frequency on tumor samples in order to pass false discovery rate criteria for antigens. Peptides mapping into multiple proteins that do not fulfill these criteria were excluded. Candidates fulfilling the criteria mentioned above underwent functional immunogenicity testing. c Comparative profiling of HLA class I and class II ligand source proteins based on the frequency of HLA-restricted representation in malignant tissues (red bars, GSC and patient samples) versus normal tissues (blue bars). Frequencies (%) for HLA-restricted presentation of the respective source protein (x axis) are indicated on the y axis for tumor-derived samples and normal controls separately. d–g VENN diagrams depicting the overlap between GSC, glioblastoma patient samples and an HLA ligandome normal tissue database (including normal brain) on source protein level for HLA class I (e) and HLA class II (g) as well as on peptide level [HLA class I (d); HLA class II (f)]. The red circle in e depicts the 50 proteins, on which the network diagram shown in h is based on. h GeneMANIA network analysis of HLA class I-presented tumor-exclusive source proteins derived from the overlap of GSC and patient samples (n = 50). Nodes of involved tumor-exclusive source proteins (n = 50) are striped (grey), nodes of supplemented genes for network analysis are completely filled. Edges represent physical interactions (red), co-expression (violet), genetic interactions (green), shared protein domains (yellow) and co-localization (blue). Selected enriched functions are depicted as colored pie charts of nodes. All enriched pathways are listed in Supplementary Table 6 (Online Resource 8)

Characterization of HLA ligands. a Peptide yields per sample are shown for HLA class I (blue bars) and HLA class II (red bars). b Length distribution of HLA class I (blue bars) peptides and HLA class II (red bars) peptides. c Venn diagrams depicting the overlap between the 3 GSC lines (GS-2, GS-5, GS-9) on source protein level as well as on peptide level for HLA class I and HLA class II. d Unsupervised heatmap analysis of core dataset samples that were acquired with the Orbitrap XL mass spectrometer based on source proteins represented by HLA ligands of GSC (yellow; n = 3), patient samples (red; n = 9) and normal brain samples [green; brain (n = 2), cerebellum (n = 2), amygdalohippocampectomy (AHE, n = 1)]. Hierarchical clustering is based on qualitative data and color coding is based on semi-quantitative data (area of precursor ions). Only unique protein IDs (proteins represented by peptides, which match to a single source protein) represented by peptides with predicted HLA binding (NetMHC 3.4 percentile rank < 2%) for at least one of the HLA-A and -B allotypes expressed by the sample were included. The number of protein entries is determined by the size of the smallest sample (Cerebellum-02; n = 135). e The overlap between HLA class I and HLA class II regarding all tumor source proteins (GSC and patient samples combined) as well as the overlap of tumor-exclusive proteins is depicted. In addition, HLA class I ligand sequences that were embedded in HLA class II sequences were compared to HLA class I tumor-exclusive peptide sequences
Gene ontology and network analysis shows enrichment for stem cell- and development-associated functions of GSC-associated glioblastoma antigens
Functional annotation based on gene ontology (GO) terms revealed distinct cellular compartments for HLA ligand source proteins presented on HLA class I compared to HLA class II [Supplementary Table 3 (Online Resource 5)]. The most frequent compartment identifiers were “cytoplasm” for HLA class I and “extracellular/exosome” for HLA class II. Interestingly, plasma membrane components were overrepresented in HLA class I tumor-exclusive source proteins. In line with that finding, GO term analysis for “biological process” detected “homophilic cell adhesion molecules” to be enriched in HLA class I and II tumor-exclusive source proteins (Supplementary Table 4 (Online Resource 6), hinting at an important role for membrane proteins as a source for glioblastoma-specific HLA ligands. Molecular function GO annotation did not reveal any major differences between the two HLA classes [Supplementary Table 5 (Online Resource 7)]. Notably, network analysis of the 44 class I antigens shared by GSC and patient samples showed involvement of stem cell- and development-related functions [Fig. 2h, Supplementary Table 6 (Online Resource 8)] with “gliogenesis” and “stem cell development” among the two top-ranking terms.
HLA class I ligand source proteins from GSC cluster with patient-derived tumors, but both are distinct from normal brain samples
Regarding GSC, a total of 4863 peptides derived from 3698 different source proteins were identified. The overlap of HLA class I source proteins and peptide ligands of the 3 GSC lines is depicted in Fig. 3c. As expected, the overlap was much smaller on the HLA peptidome level compared to the source protein level, mainly due to expression of different HLA allotypes between cell lines. Regarding primary patient samples, a total of 6459 peptides representing 4198 source proteins were identified. Unsupervised hierarchical clustering of HLA ligand source proteins represented on GSC, patient samples or normal brain specimens delineated a distinct cluster for normal brain specimens, which was well separated from the mixed cluster of primary glioblastoma and GSC samples (Fig. 3d). Notably, normal brain specimens showed a considerable degree of inter-sample homogeneity with a broad set of common antigens represented in their HLA peptidomes. In contrast, primary glioblastoma and GSC lines showed high levels of inter-patient/tumor heterogeneity with large blocks of unique antigens represented in their HLA peptidomes indicative of unique tumor biology and/or disruptive effects of malignant transformation on HLA-restricted antigen presentation. Nevertheless, sets of tumor-associated common antigens were identified leading to the distinct clustering of patient tumors and GSC samples. The similarity of individual GSC and patient samples based on the calculation of the Jaccard coefficient is depicted in Supplementary Fig. 2 (Online Resource 9). The underlying raw data of the clustering is provided in Supplementary Table 7 (Online Resource 10).
Comparative profiling of HLA class I peptidomes of primary samples and GSC versus normal tissues identifies a subset of GSC-associated glioblastoma antigens
When compared to normal tissues, 224 source proteins were exclusively represented in the HLA peptidome of GSC, 44 proteins were commonly represented on both GSC and solid glioblastoma, and 219 source proteins were specific for primary bulk glioblastoma (Fig. 2e). The FDR of antigen discovery at different thresholds of tumor-exclusive frequency were estimated based on dataset randomization and are shown for HLA class I in Fig. 4a. Aiming for an antigen-discovery-specific FDR of < 5%, antigens were required to be exclusively presented on at least 3 of 12 (25%) malignant samples leading to an estimated FDR of 3.15% for the identified tumor antigens. Using the criteria of (1) tumor-exclusive representation on the HLA peptidome level; (2) common representation on both GSC and primary patient samples, and (3) a frequency of at least 3/12 malignant samples to yield < 5% false-positive antigen discovery, we identified 14 HLA class I antigens represented by 28 HLA ligands [Supplementary Table 8 (Online Resource 11)]. Peptides mapping into multiple proteins that do not fulfill these criteria were excluded. Based on microarray data from TCGA, these candidates are expressed in both glioblastoma samples (Fig. 4c) and normal brain tissue [Supplementary Fig. 3 (Online Resource 12)]. However, differential gene expression was deliberately not a criterion of this antigen discovery approach. As a rough estimate for the relative abundance of candidate peptides within each sample, we plotted the precursor intensity distributions and highlighted candidate peptides in red for GSC (Fig. 4d–f) and for primary patient specimens [Supplementary Fig. 4 (Online Resource 13)], revealing a broad range of presentation levels for these candidate targets. Kaplan–Meier survival analysis of candidates based on TCGA data revealed prolonged survival in a cohort of glioblastoma patients with high expression of RFTN2. The same analysis in this cohort considering only patients with known IDH1 mutation status and IDH1 wild-type tumors did not show a significant survival difference. No survival effects were observed for the other candidates [Supplementary Fig. 5 (Online Resource 14)].

Characterization of candidate antigens. a, b Statistical analysis of the likelihood of false-positive tumor-associated antigen (TAA) identifications at different representation frequencies for HLA class I TAA (a) and HLA class II TAA (b). The numbers of original TAA identified based on the analysis of GSC and patient samples were compared with random virtual TAA. Virtual samples were generated in silico based on random weighted sampling from the entirety of protein identifications in both original cohorts. Virtual ligandomes were generated using a defined size of the mean number of protein identifications in all analyzed samples. The process of protein randomization, cohort assembly and TAA identification was repeated 1000 times and the mean value of resulting virtual TAAs was calculated and plotted for the different threshold values. Experimental and virtual TAA are shown as bars and corresponding false discovery rates for any chosen TAA threshold are shown as a line diagram. c TCGA microarray data were used to screen for gene expression of candidate antigens. d, e The intensity rank of candidates within the precursor area distribution in GSC samples is shown for d GS-2, e GS-5, and f GS-9. The area under the curve (AUC) of the MS1 precursor ion of a given peptide is used as semi-quantitative marker for peptide abundance
A minority of HLA class I ligands derived from cancer testis antigens is tumor-exclusive and most of the tumor-exclusive CTA-derived ligands are presented on GSC
Cancer testis genes code for a group of immunogenic proteins (CTA, cancer testis antigens) expressed in theory almost exclusively in germ cells located in immunoprivileged sites, but notably also in various human cancer types. This expression pattern led to CTA being considered prime targets for antigen-specific immunotherapy and CTA are targets of ongoing clinical trials in glioblastoma (e.g., NCT02546102 or NCT02208362; http://www.clinicaltrials.gov). Overlap analysis of established CTA (as comprised in CTDatabase; http://www.cta.lncc.br/) and tumor-exclusive source proteins from GSC and patient samples revealed the presentation of 6 CTA represented by 6 corresponding peptides, shown in Fig. 5a, b. The majority of proteins listed in the CTDatabase (133/206; 65%) was neither represented in the HLA ligandome of normal tissues nor on tumor samples. A total of 23/206 (11%) proteins were represented in the HLA ligandome overlap between malignant and benign tissue, 44/206 (21%) were exclusively represented on normal tissues and only a minority of CTA was tumor-exclusive (6/206; 3%). Interestingly, 4/6 (67%) CTA were only identified on GSC although GSC made up only 3/12 (25%) of the samples, indicating aberrant expression or HLA-restricted presentation of these targets selectively on glioblastoma stem cells. Regarding survival analysis, no effects were seen for high expression of the CTA except for an unfavorable prognosis of high interleukin-13 receptor subunit alpha-2 (IL13RA2) expression, which was not the case in the subgroup of IDH1 wild-type tumors [Supplementary Fig. 5 (Online Resource 14)].

HLA ligandome representation of cancer testis antigens. a Overlap between CTA according to the CTDatabase (http://www.cta.lncc.br), tumor source proteins and normal tissue source proteins. b 7 peptides derived from 6 CTA that were exclusively presented on tumor samples
Mapping the HLA class II ligandome of glioblastoma and GSC extends the options for synergistic immunotherapy
Although the numbers of peptide identifications were lower than on HLA class I, class II peptidome analysis resulted in the identification of 1321 peptides representing 623 source proteins from GSC as well as in 1877 peptides representing 814 source proteins derived from patient samples. Comparative HLA peptidome analysis revealed 137 source proteins and 1260 corresponding peptides to be identified exclusively on malignant samples and 6 source proteins (represented by 26 peptides) to be shared between GSC and patient samples (Fig. 2f, g). For HLA class II antigen discovery, the FDR threshold was set to a frequency of presentation on at least 3/12 malignant samples resulting in an estimated FDR of 3.33% (Fig. 4b). Three out of 6 tumor-exclusive proteins shared between GSC and patient samples (SERPINE1, FABP7, PTGFRN) represented by 13 HLA-bound peptides passed this threshold [Supplementary Table 8 (Online Resource 11)]. Comparing HLA class I and class II peptidomes, we identified 765 dually represented source proteins, 12 of which were tumor-exclusive (Fig. 3e). We found 263 HLA class I peptide sequences that were embedded within longer HLA class II peptides. Notably, of these, 59 overlapped with HLA class I tumor-exclusive peptide sequences pointing towards an attractive option for synergistic immunotherapy. No differences in survival was seen for the three HLA class II candidate antigens [Supplementary Fig. 5 (Online Resource 14)].
Functional characterization of GSC-associated HLA class I peptides reveals specific immune recognition, degranulation and antigen-specific cytotoxicity of T cells
The top 3 (based on frequency) shared glioblastoma/GSC peptides (all A*02-restricted) ALFPERITV (ATAT1), RLAPFVYLL (HEPACAM), and SILDIVTKV (RFTN2) as well as 2 CTA-derived peptides LLDTNYNLFY (IL13Rα2; A*01-restricted) and FVYGEPREL (MAGEC2; A*02-restricted) were synthesized and subjected to immunological characterization. Peptide-specific immunogenicity was verified by priming of naïve T cells from healthy donors followed by HLA multimer staining (Fig. 6a). In addition, for ATAT1 and RFTN2 T cells were functionally characterized after priming by intracellular cytokine staining (ICS) detecting the induction of the master pro-inflammatory cytokines interferon gamma (IFNγ), tumor necrosis factor alpha (TNFα) and the degranulation marker CD107a (Fig. 6b). Supplementary Table 9 (Online Resource 15) summarizes the T cell priming results. Antigen-specific cytotoxicity of T cells after sorting of tetramer-positive CD8+ T cells and subsequent expansion was shown for ALFPERITV (ATAT1), RLAPFVYLL (HEPACAM), and SILDIVTKV (RFTN2) (Fig. 6c). Gene expression in GSC was shown for these three candidates. However, RNA-mediated gene silencing did not show a consistent effect on the GSC phenotype regarding proliferation, migration, and self-renewal [Supplementary Fig. 6 (Online Resource 16), Supplementary Methods (Online Resource 1)].

T cell priming of top-ranking candidates. Immunogenicity of TAA-derived peptides was assessed by priming of CD8+ T cells with artificial antigen-presenting cells (aAPC; polystyrene beads coated with HLA/peptide complex monomers and anti-CD28 antibodies). T cell responses were detected by staining with phycoerythrin-labeled HLA–peptide multimer complexes and a response was considered positive when > 1% of tetramer-positive cells among CD8+ cells were observed. a HLA–peptide multimer + populations following priming are shown for SILDIVKTV compared to a negative control (HIV peptide). b FACS-based intracellular cytokine staining of CD8+ T cells after in vitro priming. Following re-stimulation with the peptide SILDIVKTV (RFTN2), T cells produce the pro-inflammatory cytokines interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα) as well as the degranulation marker CD107a. YLLPAIHVI (RNA helicase p68) was used as a negative control. c Tetramer-positive cells were enriched by flow cytometry-based cell sorting and subsequent expansion. Tetramer-negative cells were expanded in parallel as controls. Expanded T cells were used as effectors in a 20-h cell lysis assay with GS-9 target cells, peptide-pulsed (1 µM) or unpulsed, at various effector: target (E:T) ratios as indicated. The percentage of target cell lysis corrected for spontaneous background lysis is shown (++p < 0.001, relative to corresponding unpulsed tetramer-negative T cells; *< 0.05, relative to pulsed tetramer-negative T cells; **< 0.01, relative to pulsed tetramer-negative T cells)
Discussion
By (1) mapping the natural immunopeptidome of the glioblastoma stem cell compartment, (2) a first description of the HLA class II ligandome in glioblastoma (3) the definition of a novel subset of GSC-associated antigens in glioblastoma, which were defined based on direct evidence on the immunologically pivotal level of HLA-restricted antigen presentation, rather than based on gene expression and/or in silico predictions, our study may help guiding the informed design of immunotherapeutic interventions targeting the glioblastoma stem cell compartment.
Notably, the potential of targeting HLA class II-restricted antigens was recently underscored by clinical data on the crucial role of HLA class II and CD4+ T cells in immunological tumor rejection [17, 26, 30]. Furthermore, detailed knowledge on natural HLA class II antigen presentation patterns will allow for tailoring multi-epitope peptide vaccines containing both HLA class I- and HLA class II-restricted targets, which may induce and boost synergistic CD8+ and CD4+ anti-tumor responses.
A major focus of this study was the comparative analysis of the ligandome of GSC and patient samples with the aim of identifying GSC-associated targets also present on primary patient tumors. Bearing in mind that GSC are culture-expanded ex vivo and therefore subject to potential cell culture artifact, we performed a comparative side-by-side analysis of GSC and patient samples. Freshly resected glioblastoma patient samples represent the natural state of the HLA peptidome of glioblastoma as present in vivo. However, freshly resected patient samples are contaminated by non-tumorous cells (i.e., endothelium, microglia, infiltrating leukocytes and leukocytes from blood contamination). In contrast, GSC are composed of tumor cells only, thus tumor-exclusive patient sample-derived ligands that can also be found on GSC are unlikely to be derived from non-tumorous cells. Vice versa, GSC-derived HLA ligands that can also be found in freshly resected patient samples are unlikely to be an artifact of cell culture. In addition, hierarchical clustering analysis of HLA class I peptidomes clearly delineated normal brain samples as a distinct cluster, whereas GSC were determined to be more similar to patient samples. Another advantage of focusing on the overlap between GSC and primary patient samples is the potential similarity between GSC and adult stem cells. As GSC and normal neuroglial stem cells might share common properties [38], a high acuity of separation between these two is needed in order to prevent collateral damage to the physiological stem cell pool. Together, this strongly suggests antigens identified by our strategy represent naturally presented and highly specific targets for the immunotherapy of the glioblastoma stem cell compartment.
Recently, an antigen discovery approach for GSC-associated T cell targets was presented by Rapp and colleagues, which is based on proteomics and subsequent immunogenicity testing [23]. Peptides used for immunogenicity assays were based on in silico predictions. Albeit the abundance and turnover of source proteins has been shown to globally correlate with HLA class I-restricted peptide presentation [3], there is still a high degree of variability in sampling for HLA-restricted presentation for individual proteins, which warrants the direct analysis of HLA peptidomes for antigen discovery. Therefore, we strictly relied on HLA peptidome level analyses to identify targets by comparative HLA ligandome profiling of GSC and patient samples in relation to a comprehensive database of various human normal tissues including human brain. Both approaches complement each other and add important targets for immunotherapy of glioblastoma including the GSC compartment.
Regarding HLA ligands derived from CTA it is worth noting that 4/6 (67%) of CTA were solely identified on GSC, although GSC only contribute 3/12 (25%) of the samples. We hypothesize that this over-representation of CTA on GSC might be due to a shift towards germ-line gene expression patterns and an undifferentiated phenotype of GSC, which is consistent with the findings of our network analysis. This is in line with previous findings of enhanced CTA expression in GSC [42]. Interestingly, the 3686 HLA-A*02-restricted peptides identified on freshly resected glioblastoma in a previous study by Dutoit et al. [7] did not include previously reported HLA-A*02-restricted glioblastoma peptides from EGFRvIII, Eph-2A, gp100, HER2, IL-13Rα2, MAGEA1, TRP-2, SOX-2, -3, -11 and WT1 [6]. Dutoit et al. identified peptides derived from EGFR and Eph-A2 other than the previously published ones. We found peptides derived from MAGEA1, SOX-2, SOX-11 und IL-13Rα2, which all were detected on GSC and never on patient samples. In addition, all peptides derived from these source proteins were restricted to HLA allotypes other than HLA-A*02. This could further highlight the importance of analyzing GSC as a source of suitable antigens and also underscores the importance of mapping non-HLA-A*02 peptidomes for antigen discovery. Of note, the HLA-A*01-restricted peptide LLDTNYNLFY is derived from the source protein IL13Rα2. Recently, a report of glioblastoma tumor regression after intracranial infusion of IL13Rα2-targeted chimeric antigen receptor (CAR) T cells was published [5], which supports the utility of IL13Rα2 as a potential target for immunotherapy.
The increasing availability of next generation sequencing was accompanied by a shifting focus towards mutation-derived neoepitopes. These neoantigens are, in theory, tumor-specific and immunogenic and recent studies showed promising clinical results in malignancies other than glioblastoma [24, 30, 32]. Our approach is not designed or able to detect neoantigens as all mass spectrometry data acquired in this study was uniformly processed against a reference proteome. However, this enables us to sensitively detect non-mutated antigens derived from “self”, which are well established in tumor immunology [31, 34] and recently have been shown to be targets of pathophysiologically relevant anti-cancer immune responses in leukemia patients [15]. Compared to other malignancies, the number of somatic mutations in glioblastoma is rather low [1]. Even in tumors with a high mutational burden, peptides derived from mutated sequences are exceedingly rare [41]. In general, the number of mutated tumor antigens that elicit frequent and effective anti-tumor immune responses appears to be quite small even in the setting of checkpoint inhibition [2, 18]. A reason for the low prevalence of neoantigens even in tumors with high mutational burden might be a clonal selection of neoantigen loss variants by immunoediting [33].
A limitation of this study is the low number of normal brain tissues and the variation of HLA allotypes on the normal tissue side. In theory, target candidates may loose their tumor exclusivity by future expansion of the normal issue database. However, the potential utility of candidate targets from our comparative approach is strengthened by the evidence of antigen-specific T cell populations in functional assays. Overall, the HLA-centric antigen discovery approach implemented in our study highlights a strategy to broaden the repertoire of targetable antigens by adding a novel class of GSC-associated antigens, which—upon further target validation—may be utilized in both patient-individualized and off-the-shelf approaches.
References
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV et al (2013) Signatures of mutational processes in human cancer. Nature 500:415–421. https://doi.org/10.1038/nature12477
Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebaek E et al (2012) Dissection of T-cell antigen specificity in human melanoma. Can Res 72:1642–1650. https://doi.org/10.1158/0008-5472.CAN-11-2614
Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M (2015) Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteom 14:658–673. https://doi.org/10.1074/mcp.M114.042812
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465. https://doi.org/10.1056/NEJMoa1200694
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A et al (2016) Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 375:2561–2569. https://doi.org/10.1056/NEJMoa1610497
Dunn GP, Dunn IF, Curry WT (2007) Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human glioma. Cancer Immunity 7:12
Dutoit V, Herold-Mende C, Hilf N, Schoor O, Beckhove P, Bucher J et al (2012) Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain J Neurol 135:1042–1054. https://doi.org/10.1093/brain/aws042
Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG (1991) Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351:290–296. https://doi.org/10.1038/351290a0
Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R et al (2008) Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 27:2897–2909. https://doi.org/10.1038/sj.onc.1210949
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723. https://doi.org/10.1056/NEJMoa1003466
da Huang W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. https://doi.org/10.1093/nar/gkn923
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. https://doi.org/10.1038/nprot.2008.211
Hulsen T, de Vlieg J, Alkema W (2008) BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom 9:488. https://doi.org/10.1186/1471-2164-9-488
Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4:923–925. https://doi.org/10.1038/nmeth1113
Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L et al (2015) HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci USA 112:E166–E175. https://doi.org/10.1073/pnas.1416389112
Kowalewski DJ, Stevanovic S (2013) Biochemical large-scale identification of MHC class I ligands. Methods Mol Biol 960:145–157. https://doi.org/10.1007/978-1-62703-218-6_12
Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J et al (2015) Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520:692–696. https://doi.org/10.1038/nature14426
Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M et al (2012) TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology 1:409–418
Mohme M, Neidert MC, Regli L, Weller M, Martin R (2014) Immunological challenges for peptide-based immunotherapy in glioblastoma. Cancer Treat Rev 40:248–258. https://doi.org/10.1016/j.ctrv.2013.08.008
Neidert MC, Schoor O, Trautwein C, Trautwein N, Christ L, Melms A et al (2013) Natural HLA class I ligands from glioblastoma: extending the options for immunotherapy. J Neurooncol 111:285–294. https://doi.org/10.1007/s11060-012-1028-8
Neumann A, Horzer H, Hillen N, Klingel K, Schmid-Horch B, Buhring HJ et al (2013) Identification of HLA ligands and T-cell epitopes for immunotherapy of lung cancer. Cancer Immunol Immunother 62:1485–1497. https://doi.org/10.1007/s00262-013-1454-2
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219
Rapp C, Warta R, Stamova S, Nowrouzi A, Geisenberger C, Gal Z et al (2017) Identification of T cell target antigens in glioblastoma stem-like cells using an integrated proteomics-based approach in patient specimens. Acta Neuropathol. https://doi.org/10.1007/s00401-017-1702-1
Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J et al (2013) Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 19:747–752. https://doi.org/10.1038/nm.3161
Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D et al (2008) Potent costimulation of human CD8 T cells by anti-4-1BB and anti-CD28 on synthetic artificial antigen presenting cells. Cancer Immunol Immunother 57:175–183. https://doi.org/10.1007/s00262-007-0360-x
Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J et al (2014) A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 512:324–327. https://doi.org/10.1038/nature13387
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401. https://doi.org/10.1038/nature03128
Sturm T, Leinders-Zufall T, Macek B, Walzer M, Jung S, Pommerl B et al (2013) Mouse urinary peptides provide a molecular basis for genotype discrimination by nasal sensory neurons. Nat Commun 4:1616. https://doi.org/10.1038/ncomms2610
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454. https://doi.org/10.1056/NEJMoa1200690
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME et al (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344:641–645. https://doi.org/10.1126/science.1251102
van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B et al (1991) A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254:1643–1647
van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B et al (2013) Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol Off J Am Soc Clin Oncol 31:e439–e442. https://doi.org/10.1200/JCO.2012.47.7521
Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS et al (2016) Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. https://doi.org/10.1038/nature18945
Vigneron N, Stroobant V, Van den Eynde BJ, van der Bruggen P (2013) Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immunity 13:15
Walter S, Herrgen L, Schoor O, Jung G, Wernet D, Buhring HJ et al (2003) Cutting edge: predetermined avidity of human CD8 T cells expanded on calibrated MHC/anti-CD28-coated microspheres. J Immunol 171:4974–4978
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P et al (2010) The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38:W214–W220. https://doi.org/10.1093/nar/gkq537
Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E et al (2017) European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol 18:e315–e329. https://doi.org/10.1016/S1470-2045(17)30194-8
Westphal M, Lamszus K (2011) The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci 12:495–508. https://doi.org/10.1038/nrn3060
Widenmeyer M, Griesemann H, Stevanovic S, Feyerabend S, Klein R, Attig S et al (2012) Promiscuous survivin peptide induces robust CD4+ T-cell responses in the majority of vaccinated cancer patients. Int J Cancer 131:140–149. https://doi.org/10.1002/ijc.26365
Wolpert F, Tritschler I, Steinle A, Weller M, Eisele G (2014) A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma-initiating cells. Neuro-oncology 16:382–391. https://doi.org/10.1093/neuonc/not232
Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S et al (2014) Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515:572–576. https://doi.org/10.1038/nature14001
Yawata T, Nakai E, Park KC, Chihara T, Kumazawa A, Toyonaga S et al (2010) Enhanced expression of cancer testis antigen genes in glioma stem cells. Mol Carcinogen 49:532–544. https://doi.org/10.1002/mc.20614
Funding
This project was funded by a grant of the Canton of Zurich (HSM-2) to LR, PR and MiW. MN received financial support from the Filling-The-Gap Program (University of Zurich), the Hartmann Müller-Foundation and the EMDO Foundation. HGR received financial support from the ERC Grant 339842, MUTAEDITING.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflict of interest
DK is an employee and HGR is a shareholder of Immatics Biotechnologies GmbH. HGR is also shareholder of Curevac AG. MiW has received research grants from Acceleron, Actelion, Bayer, Isarna, MSD, Merck & Co, Novocure, Piqur and Roche and honoraria for lectures or advisory board participation or consulting from BMS, Celldex, Immunocellular Therapeutics, Isarna, Magforce, MSD, Merck & Co, Northwest Biotherapeutics, Novocure, Pfizer, Roche, Teva and Tocagen. FW has received travel support from Roche. PR has received honoraria from Roche, MSD, Novocure, Novartis and BMS for advisory board participation or lectures. The other authors declare no competing financial interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Neidert, M.C., Kowalewski, D.J., Silginer, M. et al. The natural HLA ligandome of glioblastoma stem-like cells: antigen discovery for T cell-based immunotherapy. Acta Neuropathol 135, 923–938 (2018). https://doi.org/10.1007/s00401-018-1836-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-1836-9