
Abstract
Neuronal ceroid lipofuscinoses (NCL) are a group of inherited neurodegenerative disorders with lysosomal pathology (CLN1-14). Recently, mutations in the DNAJC5/CLN4 gene, which encodes the presynaptic co-chaperone CSPα were shown to cause autosomal-dominant NCL. Although 14 NCL genes have been identified, it is unknown if they act in common disease pathways. Here we show that two disease-associated proteins, CSPα and the depalmitoylating enzyme palmitoyl-protein thioesterase 1 (PPT1/CLN1) are biochemically linked. We find that in DNAJC5/CLN4 patient brains, PPT1 is massively increased and mis-localized. Surprisingly, the specific enzymatic activity of PPT1 is dramatically reduced. Notably, we demonstrate that CSPα is depalmitoylated by PPT1 and hence its substrate. To determine the consequences of PPT1 accumulation, we compared the palmitomes from control and DNAJC5/CLN4 patient brains by quantitative proteomics. We discovered global changes in protein palmitoylation, mainly involving lysosomal and synaptic proteins. Our findings establish a functional link between two forms of NCL and serve as a springboard for investigations of NCL disease pathways.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
NCLs are a group of inherited progressive neurodegenerative diseases, which mainly affect children, but also occasionally adults [10, 24, 38]. These devastating diseases cause blindness, seizures, motor symptoms, and early death. A hallmark of NCLs is the aberrant accumulation of autofluorescent pigment in the lysosomal compartment in neuronal cell bodies. Fourteen disease-causing genes have been identified to date. All but one of the disease-causing genes harbor recessive mutations. Identification of these genes has revolutionized our understanding of the etiology of NCLs. Consistent with the known function of NCL genes, lysosomal dysfunction, dysregulated ER-lysosomal trafficking and aberrant lipid modifications are thought to be the basis for these diseases [10, 24, 38]. Several of these gene (PPT1/CLN1, CLN3, DNAJC5/CLN4) products are localized to presynaptic termini [17, 18, 20], suggesting that synaptic dysfunction may also play a role in the pathogenesis of NCLs [17, 20, 36]. A lacuna in the field is that unlike other neurodegenerative diseases, notably Parkinson’s disease [25], it is not known if NCL genes function in common pathways. However, there is mounting evidence that these genes may interact, providing valuable insight into disease mechanisms [21, 28].
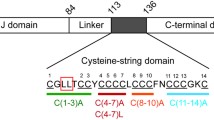
Recently, the locus for the only autosomal-dominant form of NCL was identified. Two mutations, a L115R substitution and an in-frame deletion L116Δ in DNAJC5 (Fig. 1a) cause adult NCL (ANCL) [4, 7, 23, 35]. DNAJC5/CLN4 patients have a disease onset of 25–45 years of age and present variably with generalized seizures, dyskinesia, psychiatric manifestations, and progressive dementia, leading to early death. CSPα is unique among the NCL proteins as it has no known function in lysosomes or ER-lysosome trafficking. CSPα, via its J and C-terminal domains (Fig. 1a), forms a chaperone complex with Hsc70 on synaptic vesicles and folds select clients in the presynaptic terminal [32, 42]. The two CLN4 mutations are not in either of these domains, but in the cysteine string domain, which is comprised of 14 cysteines that are normally all palmitoylated (Fig. 1a). Accordingly, the CLN4 mutations do not directly affect CSPα chaperone activity in vitro [41], although they promote self-assembly in vitro [13, 41]. Thus, it is not immediately evident why CSPα mutations cause a dominantly inherited lysosomal storage disease.

Characterization of human ANCL brains. a Schematic diagram of CSPα domain architecture. The cysteine string domain consists of 14 cysteines and the two mutations that lead to ANCL are within this domain. b CSPα staining of age-matched controls, L115R and L116Δ human cortical sections. Scale bar 50 μm. c Quantification of CSPα immunofluorescence in individual brains shown in b. d Western blotting of homogenates derived from age-matched controls, L115R, and L116Δ cortex. Note that CSPα in patient samples runs at 35 KDa, the expected molecular weight of fully palmitoylated CSPα. e Quantification of CSPα levels in immunoblots shown in d. f Blotting of cell lysate from CSPα KO mouse neurons lentivirally transduced to express WT, L115R, or L116Δ CSPα. Heterologous expression of mutant CSPα leads to decreased monomeric CSPα expression as well as the formation of HMW species. g Quantification of monomeric CSPα from Western blots shown in f; n = 3 independent experiments, ***p < 0.001. h Saposin A immunostaining of control and L115R cerebellum sections. Scale bar 50 μm. i LFQ of control and CLN4 brains. Double log2 plot of average fold change in CLN4 brains compared to control versus the negative (q value). Dotted vertical lines demarcate twofold changes, while the dotted horizontal line represents q value = 0.05. Each triangle represents a protein (n = 549 proteins). Green and red triangles represent significant changes between patient and control samples and the proteins are labeled. Filled triangles symbolize proteins whose levels change was replicated in both L115R and L116Δ brains. Cortical samples from one patient and age matched control each, were used when examining protein changes in L115R and L116Δ CLN4 patients (n = 4 brains, three technical replicates each). j The top diseases or functions annotation, sorted by p value, from Ingenuity’s Core Analysis of significantly changed proteins in CLN4 patient brains, identified in i
Here, we show that ANCL mutations in CSPα affect PPT1 (CLN1) expression, localization and enzymatic activity leading to changes in global protein palmitoylation, especially of lysosomal and synaptic proteins. Our data strongly suggest that CSPα and PPT1 are biochemically linked and function in a common neurodegenerative pathway.
Materials and methods
Human samples
Frozen brains from ANCL patients and age-matched controls were accessed and used under the auspices of IRB guidelines administered by the NYS Institute for Basic Research in Developmental Disabilities. One of the L116Δ patients has been described previously [31].
Mice
The generation and characterization of CSPα KO mice has been described previously [11]. All mice were kept under an approved IACUC protocol.
Immunohistochemistry
Patient and control brain sections were immunostained using standard procedures, and mounted on glass slides with Vectashield mounting medium (Vector Laboratories, Inc.). Prior to mounting, sections were incubated in 10 mM CuSO4 in ammonium acetate buffer (50 mM, pH 5.0) to reduce autofluorescence of lipofuscin. Stained sections were imaged with a point scanning laser confocal microscope (Zeiss LSM 510 MP), and analyzed with Volocity software (Perkin Elmer). The following primary antibodies were used at the given concentration: CSPα (Rabbit, 1:1000, Enzo ADI-VAP-SV003-E), LAMP-2 (Mouse, 1:100, DSHB H4B4), NeuN (Mouse, 1:500, Millipore MAB377), PPT1 (Rabbit, 1:200, GeneTex 110677), and Saposin A (Rabbit, 1:200, a gift from Dr. Gregory Grabowski, Cincinnati Children’s Hospital Research Foundation). Alexa conjugated secondary antibodies were used at a 1:500 dilution.
Immunocytochemistry
Primary hippocampal cultures, prepared from P0-P1 WT mouse brains, were immunostained using standard procedures, and mounted on glass slides with Vectashield mounting medium (Vector Laboratories, Inc.). Cells were imaged with an UltraVIEW VoX (Perkin Elmer) spinning disc confocal microscope, and analyzed with the accompanying Volocity quantification software (Perkin Elmer). The following primary antibodies were used at the given concentration: CSPα (Rabbit, 1:1000, Enzo ADI-VAP-SV003-E), Synaptophysin (Mouse, 1:500, Synaptic Systems SYS-101-011), MAP2 (Chicken, 1:5000, AB5543), PPT1 (Rabbit, 1:500, GeneTex 110677), GM130 (Mouse, 1:1000, BD 610822). Alexa conjugated secondary antibodies were used at a 1:500 dilution.
Quantitative immunoblotting
Patient and control brain samples were homogenized using a Dounce homogenizer in Buffer A (50 mM HEPES, pH 7.4, 100 mM NaCl, 4 mM EGTA, 2 mM MgCl2) with 1 mM PMSF, pepstatin (1 µg/mL), aprotinin (2 µg/mL), leupeptin (1 µg/mL), Complete (Roche 11873580001), 300 U/mL DNase, and with 0.5 % SDS added following homogenization. Protein concentrations were determined using a Pierce BCA Protein Assay Kit (Thermo 23225) and equal protein amounts were loaded. Total protein was isolated from cultured cells using Buffer A with 1 mM PMSF, pepstatin (1 µg/mL), aprotinin (2 µg/mL), leupeptin (1 µg/mL), 300 U/mL DNase, and 1 % TritonX-100. For cell lysates, equal volumes were loaded. Bands were quantified using IRDye secondary antibodies and a Li-COR Odyssey infrared imaging system, with actin as an internal control. The following primary antibodies were used at the given concentration: Acid Ceramidase (Mouse, 1:100, BD 612302), Actin (Mouse, 1:5000, ImmunO 691001), AKAP-5 (Mouse, 1:500, BD 610314), ATP6V1A (Rabbit, 1:1000, GeneTex 110815), Cathepsin D (Mouse, 1:1000, Abcam ab6313), CSPα (Rabbit, 1:3000, Enzo ADI-VAP-SV003-E), GFP (Rabbit, 1:1000, T3743, gift from Thomas Südhof. Stanford University), HIP14 (Rabbit, 1:100, Sigma H7414), Hsc70 (Rabbit, 1:1000, 903A), LC3B-I/II (Rabbit, 1:1000, Sigma L7543), PPT1 (Rabbit, 1:500, GeneTex 110677), PPT1 (Rabbit anti-rat PPT1, a gift from Sandra Hofmann, University of Texas Southwestern), Syntaxin-8 (Rabbit, 1:240, Synaptic Systems 110083), TPP1 (Mouse, 1:50, 8C4, gift of Adam Golabek, NYS Institute for Basic Research in Developmental Disabilities).
Lentivirus preparation
Lentivirus was prepared via transfection of packaging (pMDLg/pRRE, pRSV/REV, VsVg) and protein-encoding vectors (PPT1, CSPα WT, L115R and L116Δ) into HEK293T cells as described previously [31]. Lentivirus was resuspended in neuron growth media supplemented with 4 µg/mL polybrene. Cortical neurons were infected on DIV 5–6.
Neuronal cultures
Primary cortical cultures were prepared from P0-P1 CSPα KO mice brains and suspended in plating medium (Gibco MEM supplemented with 5 mg/mL glucose, 0.2 mg/mL NaHCO3, 10 % fetal bovine serum, 2 mM l-glutamine, and 8 µg/mL insulin). Cells were plated at a density of 50,000 cells/cm2 on Matrigel treated coverslips or 100,000 cells/cm2 on poly-d-lysine plates (BD Biocoat). Neurons were transferred to growth medium (5 mg/mL glucose, 0.2 mg/mL NaHCO3, 5 % fetal bovine serum, 0.5 mM l-glutamine, B-27 supplement) after 18 h, then supplemented with 2 µM cytosine arabinoside after 24 h to prevent glial growth. Half media replaced every 7 days.
Reverse transcription (RT)-PCR
Total RNA were extracted from control and patient (L116Δ) brain using TRIZOL Reagent (Invitrogen) and the first strand of the cDNA was synthesized using Super Script III First-Strand Kit (Invitrogen). The cDNA derived from 50 ng of total RNA was employed as the template to perform PCR for β-actin, CSPα and PPT1 by the following PCR protocol: 1 cycle of 94 °C for 4 min; 32 cycles of 94 °C for 30 s, 56 °C for 30 s and 72 °C for 25 s; 1 cycle of 72 °C for 7 min. The intensity of PCR bands were quantified using Image J with β-actin as internal control. The primers are as follows:
-
β-actin: Sense 5′-GAGCACAGAGCCTCGCCTTTG
Antisense 5′-CCTCGTCGCCCACATAGGAATC
-
PPT1: Sense 5′-ACACTGAATGCTGGGGCGTAC
Antisense 5′-ACTCCGAATCTACAGGGTCCAC
-
CSPα: Sense 5′-ACAAGTACGGCTCGCTGGGTC
Antisense 5′-AGGCGCCTTGGGCTTACACTTC
PPT1 activity quantification assay
PPT1 activity was quantified as previously described [33]. Briefly, palmitate linked to 4-methylumbelliferyl-6-thio-β-d-gluco-pyranoside (MU-6S-Palm-βGlc) was used as a synthetic substrate. Thioester bond cleavage by PPT1 released palmitate and the intermediate 4-methylumbelliferyl-6-thio-β-d-gluco-pyranoside. This intermediate was further hydrolyzed to 4-methylumbelliferone, via exogenous almond β-glucosidase, and its fluorescence measured as a means of quantifying PPT1 cleavage of palmitate groups. Aliquots of samples used in the activity quantification assay were subjected to SDS-PAGE and Western blotting. Equal protein amounts were loaded and the relative PPT1 band intensities used for normalization of the activity data i.e. determining specific activity.
In vitro CSPα depalmitoylation PPT1 assay
HEK293T cell cultures were transfected with either PPT1 or with GFP as a negative control, using GenePORTER 3000 lipid reagent (Genlantis T203015), following the provided protocol. 48 h after transfection, the cell media was collected and passed through a 0.22 µm filter. The media was then concentrated 20-fold using centrifugal filters with a 10-kDa molecular weight cut off (Centriprep 4304). The concentrated media was supplemented with 5 mM MgCl2, 10 mM Tris, pH 7.2, pepstatin (1 µg/mL), aprotinin (2 µg/mL), and leupeptin (1 µg/mL). Concentrated media was used to incubate with CSPα immunoprecipitates (see below).
Mouse brains were homogenized using an IKA disperser in Buffer A with 0.5 % TritonX-100 added following homogenization. CSPα was immunoprecipitated using standard procedures, using CSPα antibodies (Rabbit, 1:100, Millipore AB1576) with Protein A beads (Pierce Thermo). Protein A bead-bound immunoprecipitated CSPα was rotated at 37 °C in concentrated HEK293T cell media, containing recombinant PPT1 or GFP, or in PBS for 30 min. The beads were washed and the bound protein subjected to SDS-PAGE and Western blotting. When appropriate, a 50 % hydroxylamine (HA) solution (Sigma) was added to 1.6 M, following the washing of the beads.
Sample preparation for label-free quantitation
Protein preparation was carried out on ~370 µg or ~600 µg of total protein or palmitoylated enriched samples, respectively, that were dissolved in 75-µl 8 M urea, 0.4 M ammonium bicarbonate. The pH was verified to be within the 7.5–8.5 range. 50 µg aliquots of the samples were used for a dual (Lys-C and Trypsin) digest at an enzyme:protein ratio of 1:10. Digestion by Lys-C was carried out at 37 °C for 4 h, then trypsin was added to the mixture and sample incubation was continued overnight (~16 h). Digestion was quenched during the de-salting step with C18 UltraMicroSpin columns. The effluents from the de-salting step were dried and re-dissolved in 5 µl 70 % FA and 35 µl 0.1 % TFA. An aliquot was taken to obtain total digested protein amount. A 1:10 dilution of Pierce Retention Time Calibration Mixture (Cat# 88321) was added to each sample prior to injecting on the UPLC LTQ Orbitrap ELITE mass spectrometer for normalization of LFQ data.
Label-free quantitation (LFQ)
Label-Free Quantitation (LFQ) was performed on a Thermo Scientific LTQ Orbitrap Elite connected to a Waters nanoACQUITY UPLC system equipped with a Waters Symmetry® C18 180 μm × 20 mm trap column and a 1.7-μm, 75 μm × 250 mm nanoACQUITY UPLC column (35 °C). The digests were diluted to 0.05 µg/µl with 0.1 % TFA prior to injecting 5 µl of each duplicate analysis in block randomized order. To ensure a high level of identification and quantitation integrity, a resolution of 30,000 was utilized for MS and 15 MS/MS spectra were acquired per MS scan using CID in the linear ion trap. All MS (Profile) and MS/MS (centroid) peaks were detected in the Orbitrap. Trapping was carried out for 3 min at 5 µl/min in 99 % Buffer A (0.1 % FA in water) and 1 % Buffer B [(0.075 % FA in acetonitrile (ACN)] prior to eluting with linear gradients that reach 30 % B at 140 min, 40 % B at 155 min, and 85 % B at 160 min. Two blanks (1st 100 % ACN, 2nd Buffer A) followed each injection to ensure there was no sample carry over.
The LC–MS/MS data was processed with Progenesis LC–MS software (Nonlinear Dynamics, version 3.4) with protein identification carried out using the Mascot search algorithm. The Progenesis LC–MS software performs feature/peptide extraction, chromatographic/spectral alignment (one run was chosen as a reference for alignment), data filtering, and quantitation of peptides and proteins. A normalization factor for each run was calculated to account for differences in sample load between injections as well as differences in ionization. The normalization factor was determined by comparing the abundance of the spike in Pierce Retention Time Calibration mixture among all the samples. The experimental design was set up to group multiple injections from each run. The algorithm then tabulated raw and normalized abundances, and maximum fold change for each feature in the data set. The MS/MS spectra were exported as .mgf (Mascot generic files) for database searching. The Mascot search results were exported as .xml files using a significance cutoff of p < 0.05 and FDR of 1 % and then imported into the Progenesis LC–MS software, where search hits were assigned to corresponding peptides. Relative protein-level fold changes were calculated from the sum of all unique, normalized peptide ion abundances for each protein on each run.
Acyl-resin-assisted capture (AcylRAC)
A modified acylRAC protocol [12] was performed, as previously described [37]. For this procedure, we used the same brains as the total homogenate LFQ analysis. Briefly, N-ethylmaleimide was used to block free thiol groups followed by addition of hydroxylamine, which cleaves the thioester bonds of palmitoylated proteins, releasing the palmitate and exposing new free thiol groups. Thiopropyl-sepharose was used to bind these nascent thiol groups and capture these palmitoylated proteins allowing for the enrichment of endogenously palmitoylated proteins. Through this procedure we identified >850 distinct proteins, identified from >30,000 peptides as defined by at least two peptides.
Differential detergent extraction
Brain tissue was serially extracted with detergents following previously published protocols. Patient and control brain samples were homogenized using a Dounce homogenizer in Homogenization Buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 4 mM EGTA, 2 mM MgCl2) with 1 mM PMSF, pepstatin (1 µg/mL), aprotinin (2 µg/mL), leupeptin (1 µg/mL), Complete (Roche 11873580001), and 300 U/mL DNase. The homogenate was mixed at 1:1 with buffer A (Homogenization Buffer; 1 % NP40) and then sonicated for 30 s using a Branson 2510 Ultrasonic Cleaner. The sonicated lysate was centrifuged for 5 min at 54,000g, using an Optima TLX Ultracentrifuge, to separate supernatant S1 and pellet P1. The P1 pellet was washed with Buffer B (Homogenization Buffer; 0.5 % NP40) followed by sonication for 30 s and the lysates were centrifuged for 5 min at 54,000 g to obtain pellet P2. P2 was suspended in Buffer C (Homogenization Buffer; 0.5 % NP40; 0.5 % deoxycholic acid; 0.25 % SDS) and then extracted by sonication for 30 s, followed by centrifuging for 5 min at 54,000 rpm to sediment P3. P3 was resuspended in buffer D (Homogenization Buffer; 0.5 % NP40; 0.5 % deoxycholic acid; 2 % SDS) and sonicated as above. Supernatants S1, S3 and Pellet P3 were immunoblotted for PPT1, Actin, and Hypoxanthine–guanine phosphoribosyltransferase (HPRT), which is a soluble protein used to assess purity of fractions.
Statistical analysis
The unequal variance Welch t test was used for the differential protein expression analyses. The Fisher’s combined probability test was used to combine the p values from two independent expression profile sets of patient sample and age-matched control data. To correct for multiple comparisons, we applied the false discovery rate (FDR) approach [5] to adjust for the raw Fisher’s combined p values, with a q value of 0.05 chosen to indicate statistical significance. The expression change of a protein was quantified by the effect measure of fold change. Volcano scatter plots of −log2(q value) on the y-axis by log2(fold change) on the x-axis were displayed to visualize the differentially expressed proteins.
Pathway analysis
Protein interactions and functions of significant proteomic hits (Tables 1, 2) were analyzed using the QIAGEN Ingenuity Pathway Analysis software, which utilizes the curated Ingenuity Pathway Knowledge Base. Using a list of relevant protein accession numbers, a Core Analysis was performed, the output of which included statistically significant Canonical Pathways, Diseases, and Functions associated with the analyzed proteins. The software used right-tailed Fisher’s Exact Test to calculate p values, reflecting the likelihood of associations being due to random chance. No cutoff was included in the Core Analysis, as only proteins found to be statistically relevant were imported into the software. Only molecules and relationships where species = human were included in the analysis.
Subcellular localization analysis
CELLO2GO [40] was used to obtain the pie charts reflecting the predicted subcellular distribution of sets of identified proteins. The amino acid sequence from the relevant list of proteins was obtained in FASTA format from the Uniprot KnowledgeBase database. These sequences were then uploaded to the online CELLO2GO platform, which uses BLAST to parse out sequences homologous to those of proteins within the UniProtKB/SwissProt database. Identified proteins were assigned one or more subcellular localizations via CELLO predictions (http://cello.life.nctu.edu.tw/) and the distribution of localizations within the data set were presented in pie chart format. BLAST searches through the CELLO2GO platform were performed with an E value of 0.001, without TrEMBL usage.
Results
Identification of molecular changes in DNAJC5/CLN4 brains reveals a new classification of NCLs
To aid our understanding of the molecular pathogenesis of ANCL, we obtained age-matched control and DNAJC5/CLN4 patient brains and analyzed tissue sections. As lipofuscin displays broad-spectrum autofluorescence, we imaged brain sections without staining. We observed autofluorescent lipofuscin accumulations in neuronal soma of both L115R and L116Δ patient brains (Fig. S1a). After quenching autofluorescence with CuSO4 treatment, we examined the human brain sections for CSPα by immunostaining. We observed that L115R sections show little CSPα staining, and L116Δ sections have low immunofluorescence levels (Fig. 1b), consistent with previous findings [23]. Quantification indicated that L115R and L116Δ have 17 % and 50 % CSPα immunofluorescence compared to controls, respectively (Fig. 1c). We confirmed these results by western blotting (Fig. 1d, e), and found 7 and 49 % CSPα in L115R and L116Δ brains compared to controls (Fig. 1e, S1b). The strong decrease in CSPα occurs post-transcriptionally as the levels of CSPα mRNA are equivalent in control and CLN4 patient brains (Fig. S2). Diminished levels of CSPα L115R and L116Δ appear to be a consequence of the two CLN4 mutations in neurons as similar decreases were observed in CSPα mouse knockout (KO) neurons lentivirally transduced with the CLN4 mutants (Fig. 1f, g; WT 1.0 ± 0.003, L115R 0.2 ± 0.002, L116Δ 0.22 ± 0.015, p < 0.001). Higher molecular weight (HMW) CSPα species are seen both in patient brains upon higher exposure [13] (not shown) and in mouse neurons transduced with CLN4 mutants (Fig. 1f, g).
We determined the nature of the storage material by immunostaining for the two major proteins known to accumulate in lipofuscin: saposin A and the c-subunit of the mitochondrial ATPase. Neuronal cell bodies in L115R sections strongly stained for saposin A (Fig. 1h), but not for the c-subunit (not shown), consistent with earlier histopathological analysis of ANCL patients showing accumulation of saposin D [22, 30]. Intriguingly, similar pathology is seen in CLN1 (PPT1 deficiency) and to a lesser degree in CLN10 patients (deficiency of cathepsin D, a lysosomal aspartyl protease) [26]. As all other forms of NCLs accumulate exclusively the c-subunit of the mitochondrial ATP synthase, we can classify CLN1 and CLN4, and possibly CLN10, as a distinct subgroup of NCLs that accrete saposins. This classification suggests a common pathogenesis for these forms of NCL.
DNAJC5/CLN4 patient brains accumulate PPT1 enzyme with low specific activity
To identify all the major protein changes in ANCL brains, we performed high resolution LC–MS/MS based label-free quantification (LFQ) [6] on human control and CLN4 patient cortical brain samples. One L115R and one L116Δ patient sample each with age-matched controls were used to examine the protein changes in CLN4 brains. Through this unbiased and systematic analysis, we analyzed over 500 proteins and 20,000 peptides and used stringent criteria (±2 fold; false discovery rate (FDR) [5] of 5 %) to determine protein changes in CLN4 brains (Fig. 1i; Table 1). We identified 14 proteins whose levels were increased above threshold and 3 proteins whose levels were decreased in both L115R and L116Δ CLN4 brains compared to matched controls (Fig. 1i; Table 1). The most striking changes were for PPT1 and saposin. PPT1 peptide levels were increased on average 21-fold in CLN4 patient brains. This finding was intriguing as PPT1 deficiency, i.e. loss of its depalmitoylating activity causes both infantile and adult forms of NCL (CLN1). As confirmed by the immunohistochemistry (Fig. 1b, c, h), saposin levels were also dramatically increased (21-fold), while CSPα levels were decreased (0.66-fold). Consistent with our classification based on saposin accretion, Cathepsin D was increased (2.6 fold). We analyzed the significant proteomic hits (Table 1) using the QIAGEN Ingenuity Pathway Analysis (IPA) software, which identifies functions and pathways based on curated protein–protein relationships. Of the functions and diseases that were associated with our list of proteins (Table 1), the top hits were ‘Neuronal Ceroid Lipofuscinosis’ (p = 1.7e−7), and ‘Lysosomal Storage Disease (p = 4.4e−7)’ (Fig. 1j). The prominent functions altered accurately describe ANCL (Fig. 1j), suggesting that our proteomic hits merited further investigation. The top transcription factor associated with our list of proteins was TFEB, the master lysosomal transcription factor (p = 5.37e−4) [27].
We validated the top LFQ hits by performing quantitative immunoblotting (Fig. 2a). This analysis confirmed that PPT1 protein levels were indeed increased dramatically in CLN4 patient brains, averaging 90-fold (Fig. 2a, S1b), while cathepsin D was increased an average of 10-fold (Fig. 2a, S1b). Interestingly, proteins that interact with CSPα, such as Hsc70 [32] and the enzyme that palmitoylates CSPα, HIP14/DHHC17, were largely unaltered in CLN4 brain samples, though L115R brains show a tendency to decreased levels of Hsc70 (Fig. 2a, S1b). We confirmed the lack of changes in other lysosomal enzymes (TPP1/CLN2; ATP6V1A; LC3B II), indicating that the massive induction in PPT1 protein levels does not merely indicate enlarged or increased number of lysosomes. Next, we tested if the induction of PPT1 occurred transcriptionally. PPT1 mRNA levels are increased in ANCL brains compared to controls (Fig. S2), suggesting DNAJC5/CLN4 mutations lead to increased transcription of the PPT1 gene most likely though the transcription factor TFEB [27].

Accumulation of PPT1 with low specific activity in ANCL brains. a Validation of select proteomic hits by immunoblotting of homogenates derived from age-matched controls, L115R and L116Δ cortex. Actin is a loading control. b Probing PPT1 levels in control and patient brains with other neurodegenerative diseases that accumulate lipofuscin. NCL neuronal ceroid lipofuscinosis, MPS mucopolysaccharidoses, AD Alzheimer’s Disease. The brain used for the NCL sample was donated prior to the advent of genetic testing for diagnosis of CLN1. Based on age of death and decrement in PPT1 it is presumed that the brain used is from a CLN1 patient explaining the decrease in PPT1. c Confocal microscopy of cortical sections of control and ANCL brains stained for PPT1 and NeuN. Panels show that PPT1 staining is largely diffuse throughout the section in control brains, but highly localized to cell bodies in L116Δ tissue. Boxed area is shown in higher magnification pictures in panels in right columns. Scale bar 50 μm for both panels. d PPT1 enzymatic activity in individual human control, L116Δ, and L115R brain tissue normalized to total protein levels. e PPT1 levels in samples used for assaying PPT1 activity in d. f PPT1 enzymatic activity in human control, L116Δ, and L115R brain tissue normalized to PPT1 levels quantified in e. i.e. specific activity
To test if the increase in PPT1 levels was specific for ANCL, we surveyed other neurodegenerative diseases that exhibit lipofuscin accumulations. Human cortex samples for a variety of lysosomal storage diseases and Alzheimer’s disease were immunoblotted for PPT1 (Fig. 2b). We observed that PPT1 protein levels in this disease cohort was not increased and did not differ significantly from human controls. Thus, the increase in PPT1 levels is not just reflecting increased lipofuscin accumulation and may be selective for DNAJC5/CLN4.
CLN1 occurs due to a loss of PPT1 activity. We therefore investigated PPT1 localization and enzymatic activity in control and CLN4 brains. The normal subcellular distribution of PPT1 in neurons is broad [3, 17, 18]. In control human brains PPT1 staining was mainly diffuse throughout the neuropil, however in marked contrast, in CLN4 patient brains we observed PPT1 staining concentrated in the soma and surrounding nuclear NeuN staining (Fig. 2c, S1c). Further histological evaluation showed that PPT1 is accumulating at lysosomes (Fig. S1d). These experiments demonstrate an altered subcellular distribution of PPT1 in CLN4 brains.
Next, we measured PPT1 activity in human brain homogenates, with an assay that is used to determine PPT1 activity in CLN1 patients [33]. We tested whether the dramatic increase in PPT1 protein levels (Figs. 1i, 2a, e) was accompanied by a commensurate increase in enzymatic activity. Paradoxically, we observed that PPT1 activity is only moderately increased 2–3-fold (Fig. 2d). Hence, the specific activity of PPT1 was very strongly decreased in CLN4 brain samples compared to controls (~6 % of control; Fig. 2d–f). To test if this decrease in specific activity is due to aggregation of PPT1, we carried out sequential detergent extractions. Indeed, we find that PPT1 is aggregated in L116Δ brains (Fig. S3), supporting the hypothesis that an alteration in PPT1 function is contributing to the pathogenesis of CLN4 disease.
CSPα is a PPT1 substrate
Since mutations in both PPT1 and CSPα independently cause NCL, and the two genes appear to be in a common pathway (Fig. 2), we explored the relationship between PPT1 and CSPα. We first tested if wildtype CSPα regulates PPT1 levels and activity by examining these in CSPα KO mouse brains. We observed no change in PPT1 levels (Fig. S4a–b) but a small increase in PPT1 activity (Fig. S4c). This indicates that alterations in PPT1 are not due to loss of CSPα, but rather that CLN4 CSPα mutants have a dominant effect due to their altered cysteine string domain. Since CSPα is one of the most palmitoylated brain proteins [16] and is palmitoylated in its cysteine string domain [14], we tested if CSPα is depalmitoylated by PPT1. We set up in vitro depalmitoylation assays using immunoprecipitated CSPα and recombinantly expressed human PPT1 (Fig. 3a, b). Our results show that PPT1 can robustly depalmitoylate CSPα, suggesting that CSPα and PPT1 have a substrate-enzyme relationship (Fig. 3c–e). PPT1 can be expected to depalmitoylate CSPα, which may facilitate its degradation.

CSPα is depalmitoylated by PPT1. a Schematic of the in vitro depalmitoylation assay using CSPα immunoprecipitated from mouse brain and heterologously expressed human PPT1. HEK293T cell cultures were transfected with either PPT1 or GFP as a negative control. Culture media, containing the recombinant protein, was then incubated with bead-bound CSPα, immunoprecipitated from mouse brain. b Western blotting of culture media used in depalmitoylation assay taken from GFP or PPT1 transfected HEK293T cell cultures. c Immunoblotting of mouse brain homogenate (Start) and immunoprecipitated CSPα incubated with PBS alone or with GFP or PPT1 containing media for 30 min. An aliquot of the PBS sample was treated with hydroxylamine (HA) following incubation in order to completely depalmitoylate CSPα. A time course was also performed showing that depalmitoylation of CSPα is time-dependent (data not shown). d Quantification of fully depalmitoylated CSPα as a percentage of total CSPα within lane (n = 3). e Quantification of palmitoylated CSPα (n = 3). **p < 0.01
Global changes in palmitome in DNAJC5/CLN4 patient brains
While PPT1 is a major neuronal depalmitoylating enzyme, its precise substrates are unknown. As palmitoylation is a dynamic posttranslational modification and is linked, in yet to be identified ways, to the degradation of proteins [19], we tested if the redistribution of PPT1 to perinuclear areas and the decrease in its specific activity in CLN4 brains (Fig. 2) disrupts global neuronal palmitoylation. We purified palmitoylated proteins from the same control and CLN4 brains we analyzed in Fig. 1i using AcylRAC [12], followed by LFQ identification (Fig. 4a). We validated our approach by immunoblotting the AcylRAC purification steps for known palmitoylated proteins (Fig. S5). We purified >850 proteins and identified 28 proteins exhibiting at least 2-fold changes in CLN4 samples (Fig. 4b; Table 2). Most of these proteins have been previously identified as palmitoylated [16, 37], endorsing our purification procedure. A subset of these palmitoylated proteins is likely to be direct PPT1 substrates. By directly comparing the protein hits from the total brain homogenates (Fig. 1i) and the AcylRAC (Fig. 4b), we can distinguish between palmitome changes in DNAJC5/CLN4 (Table 2) that are due to proteins whose total levels are changed versus those only whose palmitoylation are changed (Fig. 4c; Fig. S6a, b). This analysis confirmed that CLN4 brains do indeed have altered palmitomes and suggests that these palmitoylated proteins may be important for the pathogenesis of both CLN1 and CLN4.

Purification of palmitoylated proteins in ANCL brains via AcylRAC identifies new set of proteins implicated in neurodegeneration. a Schematic of the AcylRAC procedure to purify palmitoylated proteins (NEM N-ethylmaleimide, HA hydroxylamine). b Double log2 plot of average fold change in CLN4 brains compared to control versus the negative log (q value). Dotted vertical lines represent twofold changes, while the dotted horizontal line represents q value = 0.05. Each triangle represents a protein (n = 867 proteins). Green and red triangles represent significant changes between patient and control samples and the proteins are labeled. In filled triangles the change was in the same direction for both L115R and L116Δ brains. Same cortical samples from patients and age matched controls as used in Fig. 1i were used to examine protein changes in CLN4 brains (n = 4 brains, three technical replicates). c Heat map of proteins (n = 342) identified by LFQ in both total brain homogenate (Fig. 1i) and the AcylRAC-purified samples. Shading reflects average fold change in abundance in CLN4 brains as compared to controls. Red and green shading reflect increased and decreased abundance, respectively with upper and lower limits set at twofold. d Subcellular localization of palmitoylated proteins significantly changed in CLN4 brains. CELLO was used to predict the subcellular localization of all proteins identified after AcylRAC. Percent fold changes of localization predictions by CELLO to each subcellular domain, comparing significantly changed proteins to all proteins identified by LFQ of AcylRAC-purified samples. See Fig. S7 for details
The palmitoylated proteins we identified have known functions in lysosomes/exosomes (PPT1, SAP, ISOC1), synapses (NSF, GRIN1, CPXII) and cellular signaling (DCLK1, CDK5), signifying that palmitoylation in distinct subcellular domains may be perturbed in CLN4. Indeed, by subcellular localization analysis of proteins showing significant changes in AcylRAC using CELLO [40], we find that lysosomes are the most affected organelles (Fig. 4d; Fig. S7).
Discussion
In this study, we sought to understand molecular mechanisms by which mutations in DNAJC5/CLN4 cause ANCL. Through an unbiased analysis of control and patient brains, we discovered that CLN4 and CLN1 are mechanistically related and are diseases of aberrant palmitoylation.
Combination of loss and gain-of-function mechanisms in ANCL
Many dominantly inherited neurodegenerative diseases occur through a combination of both loss- and gain-of-function mechanisms [39]. ANCL caused by DNAJC5/CLN4 mutations appears to conform to this pattern. Patient brains have lowered CSPα protein expression (7–49 % of control) indicating a partial loss-of-function or haploinsufficiency (Fig. 1b–e). This mechanism has been suggested previously by Noskova et al. [23]. While CLN4 CSPα mutants are functional as co-chaperones in vitro, they oligomerize (Fig. 1f) leading to an inhibition of co-chaperone activity [41]. We have also shown that L115R and L116Δ mutant CSPα can co-oligomerize with wildtype CSPα and reduce the overall co-chaperone activity in vitro in a dominant-negative fashion [41]. These findings are indicative of an oligomerization dependent loss-of-CSPα function. Genetically, a loss-of-function mechanism is congruent with the observation that CSPα KO mice phenocopy some clinical features seen in CLN4 patients such as seizures and early lethality [9, 11]. Whether the loss-of-CSPα function in ANCL is due to a haploinsufficiency or a dominant-negative mechanism remains to be resolved. Furthermore, how this decrement in CSPα co-chaperone capacity impact its protein clients needs to be addressed.
In addition to a loss-of-CSPα function in ANCL, the most prominent gain-of-function mechanism is the aberrant protein palmitoylation we see in CLN4 patient brains (Fig. 4). The massive increase and redistribution of PPT1 to neuronal cell bodies coupled with the reduced PPT1 specific activity (Fig. 2) is the most probable cause for the observed alterations in protein palmitoylation. The proteins which exhibit altered palmitoylation (Table 2) are expected to have changed membrane localization, signaling, and protein turnover, which is likely to account for the dominant nature of the disease and the multiplicity of clinical symptoms shown by CLN4 patients.
Previous work has suggested DHHC palmitoylation-dependent aggregation of CLN4 CSPα as the cause of the dominant nature of CLN4 disease [13]. However, we found that the fraction of aggregated CSPα is miniscule compared to that of PPT1, suggesting that the gain-of-function in ANCL is mainly due to the accumulation and mislocalization of PPT1 rather than aggregation of CSPα. Furthermore, we find that aggregation of CLN4 CSPα mutants is not dependent on palmitoylation, but the intrinsic properties of the mutant themselves [41]. It is also worthwhile to note that we did not observe a change in the levels of DHHC/palmitoylacyl transferases, though we can detect several of these enzymes in the LFQ experiments (Fig. 1i). Taken altogether, DNAJC5/CLN4 mutants act via both loss and gain-of-function mechanisms to cause neurodegeneration.
CLN1 and CLN4 are in a common disease pathway
The shared features between DNAJC5/CLN4 and PPT1/CLN1 patients from clinical to pathological to molecular aspects are so compelling that we propose these two forms of NCL are closely related and should be classified as a distinct sub-category of NCLs. At a clinical level, PPT1/CLN1 mutations that retain residual enzyme activity lead to adult NCL [34] similar to DNAJC5/CLN4 which have ~6 to 10 % PPT1 specific activity (Fig. 2f). These observations strongly suggest that PPT1 enzymatic decrement is contributing to the etiology of CLN4 disease as well. Pathologically, we find that CLN4 patients accrue saposin A and cathepsin D similar to CLN1 patients (Fig. 1h) [8, 22], but distinct from other forms of NCLs [10]. Molecularly, we indeed find changes in the palmitome of CLN4 brains compared to age-matched controls (Fig. 4). Whether these palmitome changes are shared with CLN1 patients is not known as the severe loss of neurons in CLN1 brains precludes us from ascertaining this fact. We do, however, anticipate comparing the palmitome of these CLN4 patients to mouse PPT1 KO brains in the future. Our observation that CSPα is a PPT1 substrate (Fig. 3) further cements this relationship.
Together, these facts indicate that CLN1 and CLN4 are linked by an alteration of PPT1 enzyme activity, which may lead to neurodegeneration via aberrant protein palmitoylation.
PPT1-CSPα connection
Our data strongly indicate that CSPα is a PPT1 substrate (Fig. 3). PPT1, although originally identified as a lysosomal enzyme in non-neuronal cells, has been shown in neurons to localize to axons and presynaptic termini [3, 17]. The properties of PPT1 such as its neutral pH optimum and the lack of lysosomal processing to be functional [15], probably allows it to be enzymatically active in many subcellular compartments, including presynaptic termini [17, 18]. Moreover, PPT1, like CSPα, has been shown to have a critical role in regulating the synaptic vesicle cycle, with PPT1 KO neurons exhibiting synaptic vesicle cycle deficits [1, 2, 17, 36]. Under normal conditions, PPT1 is likely to act on CSPα at the synapse.
Why does inactive PPT1 accumulate in CLN4 brains? One possibility is that PPT1 and CSPα act bidirectionally and PPT1 is a substrate of CSPα, i.e. folded by the Hsc70/CSPα chaperone complex. However, we did not identify PPT1 in previous screens for CSPα substrates [42], suggesting otherwise. Further, deletion of CSPα had no effect on mouse PPT1 levels and in fact modestly increased its enzymatic activity (Fig. S4). Therefore, based on our data, we speculate that initially CLN4 mutant CSPα oligomerizes [13, 41], leading to impaired trafficking to synapses (Fig. 2c) [13]. This in turn would lead to the transcription of PPT1, through the transcription factor TFEB [27], in an effort to depalmitoylate mutant CSPα and degrade it. Over time, CLN4 mutant CSPα leads to accumulation of PPT1 and its aggregation, thus preserving the increase of PPT1 and not of other lysosomal proteins, with the exception of saposins and potential PPT1 binding partner Cathepsin D [8, 29]. This should result subsequently in altered PPT1 activity in different subcompartments (Fig. 2e–f) and changes in the palmitome (Fig. 4).
Protein palmitoylation and neurodegeneration
The network created with Ingenuity’s Pathway Explorer tool, using significantly changed palmitoylated proteins in CLN4 patient brains suggests a similarity to Huntington’s disease (Fig. S6c). Significantly, Huntington’s disease is also a dominant neurodegenerative disease linked to aberrant palmitoylation. Huntington is associated with alterations in HIP14/DHHC17, the palmitoylacyltransferase that palmitoylates proteins, including CSPα and its substrates [37]. Again affirming our hypothesis that dysregulation of palmitoylation leads to neurodegeneration.
In summary, our results highlight that CLN1 and CLN4 are related and diseases of aberrant palmitoylation. Our findings provide insight into the pathogenesis of NCLs and suggest that aberrant protein palmitoylation may be critical in the etiology of neurodegeneration.
References
Aby E, Roth A, Gumps K, Sigmon S, Jenkins SE, Kim JJ et al (2013) Mutations in palmitoyl-protein thioesterase 1 alter exocytosis and endocytosis at synapses in Drosophila larvae. Fly 7(4):267–279
Ahtiainen L, Luiro K, Kauppi M, Tyynela J, Kopra O, Jalanko A (2006) Palmitoyl protein thioesterase 1 (PPT1) deficiency causes endocytic defects connected to abnormal saposin processing. Exp Cell Res 312:1540–1553. doi:10.1016/j.yexcr.2006.01.034
Ahtiainen L, Van Diggelen OP, Jalanko A, Kopra O (2003) Palmitoyl protein thioesterase 1 is targeted to the axons in neurons. J Comp Neurol 455:368–377. doi:10.1002/cne.10492
Benitez BA, Alvarado D, Cai Y, Mayo K, Chakraverty S, Norton J et al (2011) Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS One 6:e26741. doi:10.1371/journal.pone.0026741
Benjamini YHY (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300
Bordner KA, George ED, Carlyle BC, Dugue A, Kitchen RR, Lam TT et al (2011) Functional genomic and proteomic analysis reveals disruption of myelin-related genes and translation in a mouse model of early life neglect. Front Psychiatry 2:18. doi:10.3389/fpsyt.2011.00018
Cadieux-Dion M, Andermann E, Lachance-Touchette P, Ansorge O, Meloche C, Barnabe A, Kuzniecky RI et al (2013) Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin Genet 83:571–575. doi:10.1111/cge.12020
Chandra G, Bagh MB, Peng S, Saha A, Sarkar C, Moralle M, Zhang Z, Mukherjee AB (2015) Cln1 gene disruption in mice reveals a common pathogenic link between two of the most lethal childhood neurodegenerative lysosomal storage disorders. Hum Mol Genet 24:5416–5432. doi:10.1093/hmg/ddv266
Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123:383–396. doi:10.1016/j.cell.2005.09.028
Cotman SL, Karaa A, Staropoli JF, Sims KB (2013) Neuronal ceroid lipofuscinosis: impact of recent genetic advances and expansion of the clinicopathologic spectrum. Curr Neurol Neurosci Rep 13:366. doi:10.1007/s11910-013-0366-z
Fernandez-Chacon R, Wolfel M, Nishimune H, Tabares L, Schmitz F, Castellano-Munoz M et al (2004) The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron 42:237–251
Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, Casey PH (2011) Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res 52:393–398. doi:10.1194/jlr.D011106
Greaves J, Lemonidis K, Gorleku OA, Cruchaga C, Grefen C, Chamberlain LH (2012) Palmitoylation-induced aggregation of cysteine-string protein mutants that cause neuronal ceroid lipofuscinosis. J Biol Chem 287:37330–37339. doi:10.1074/jbc.M112.389098
Gundersen CB, Mastrogiacomo A, Faull K, Umbach JA (1994) Extensive lipidation of a Torpedo cysteine string protein. J Biol Chem 269:19197–19199
Hofmann SL, Das AK, Lu JY, Soyombo AA (2001) Positional candidate gene cloning of CLN1. Adv Genet 45:69–92
Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO et al (2008) Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456:904–909. doi:10.1038/nature07605
Kim SJ, Zhang Z, Sarkar C, Tsai PC, Lee YC, Dye L, Mukherjee AB (2008) Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J Clin Invest 118:3075–3086. doi:10.1172/JCI33482
Lehtovirta M, Kyttala A, Eskelinen EL, Hess M, Heinonen O, Jalanko A (2001) Palmitoyl protein thioesterase (PPT) localizes into synaptosomes and synaptic vesicles in neurons: implications for infantile neuronal ceroid lipofuscinosis (INCL). Hum Mol Genet 10:69–75
Lu JY, Hofmann SL (2006) Thematic review series: lipid posttranslational modifications. Lysosomal metabolism of lipid-modified proteins. J Lipid Res 47:1352–1357. doi:10.1194/jlr.R600010-JLR200
Luiro K, Kopra O, Lehtovirta M, Jalanko A (2001) CLN3 protein is targeted to neuronal synapses but excluded from synaptic vesicles: new clues to Batten disease. Hum Mol Genet 10:2123–2131
Lyly A, von Schantz C, Heine C, Schmiedt ML, Sipila T, Jalanko A, Kyttala A (2009) Novel interactions of CLN5 support molecular networking between neuronal ceroid lipofuscinosis proteins. BMC Cell Biol 10:83. doi:10.1186/1471-2121-10-83
Nijssen PC, Ceuterick C, van Diggelen OP, Elleder M, Martin JJ, Teepen JL, Tyynela J, Roos RA (2003) Autosomal dominant adult neuronal ceroid lipofuscinosis: a novel form of NCL with granular osmiophilic deposits without palmitoyl protein thioesterase 1 deficiency. Brain Pathol 13:574–581
Noskova L, Stranecky V, Hartmannova H, Pristoupilova A, Baresova V, Ivanek R et al (2011) Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am J Hum Genet 89:241–252. doi:10.1016/j.ajhg.2011.07.003
Palmer DN, Barry LA, Tyynela J, Cooper JD (2013) NCL disease mechanisms. Biochim Biophys Acta 1832:1882–1893. doi:10.1016/j.bbadis.2013.05.014
Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273. doi:10.1016/j.neuron.2014.12.007
Santavuori PGI, Haltia M, Rapola J, Lake BD, Tyynelä J, Peltonen L (1999) CLN1: infantile and other types of NCL with GROD. IOS Press, Amsterdam
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325:473–477. doi:10.1126/science.1174447
Scifo E, SzwajdaA DebskiJ, Uusi-Rauva K, Kesti T, Dadlez M, Gingras AC, Tyynela J, Baumann MH, Jalanko A, Lalowski M (2013) Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: a label-free quantitative proteomics approach. J Proteome Res 12:2101–2115. doi:10.1021/pr301125k
Scifo E, Szwajda A, Soliymani R, Pezzini F, Bianchi M, Dapkunas A, Debski J, Uusi-Rauva K, Dadlez M, Gingras AC, Tyynela J, Simonati A, Jalanko A, Baumann MH, Lalowski M (2015) Proteomic analysis of the palmitoyl protein thioesterase 1 interactome in SH-SY5Y human neuroblastoma cells. J Proteomics 123:42–53. doi:10.1016/j.jprot.2015.03.038
Siintola E, Partanen S, Stromme P, Haapanaen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynela J (2006) Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 129:1438–1445. doi:10.1093/brain/awl107
Sims KB, Cole AJ, Sherman JC, Caruso PA, Snuderl M (2011) Case records of the Massachusetts General Hospital. Case 8-2011. A 32-year-old woman with seizures and cognitive decline. N Engl J Med 364:1062–1074. doi:10.1056/NEJMcpc1013927
Tobaben S, Thakur P, Fernandez-Chacon R, Sudhof TC, Rettig J, Stahl B (2001) A trimeric protein complex functions as a synaptic chaperone machine. Neuron 31:987–999
van Diggelen OP, Keulemans JL, Winchester B, Hofman IL, Vanhanen SL, Santavuori P, Voznyi YV (1999) A rapid fluorogenic palmitoyl-protein thioesterase assay: pre- and postnatal diagnosis of INCL. Mol Genet Metab 66:240–244. doi:10.1006/mgme.1999.2809
van Diggelen OP, Thobois S, Tilikete C, Zabot MT, Keulemans JL, van Bunderen PA et al (2001) Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adult-onset patients of a childhood disease. Ann Neurol 50:269–272
Velinov M, Dolzhanskaya N, Gonzalez M, Powell E, Konidari I, Hulme W et al (2012) Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS One 7:e29729. doi:10.1371/journal.pone.0029729
Virmani T, Gupta P, Liu X, Kavalali ET, Hofmann SL (2005) Progressively reduced synaptic vesicle pool size in cultured neurons derived from neuronal ceroid lipofuscinosis-1 knockout mice. Neurobiol Dis 20:314–323. doi:10.1016/j.nbd.2005.03.012
Wan J, Savas JN, Roth AF, Sanders SS, Singaraja RR, Hayden MR, Yates JR 3rd, Davis NG (2013) Tracking brain palmitoylation change: predominance of glial change in a mouse model of Huntington’s disease. Chem Biol 20:1421–1434. doi:10.1016/j.chembiol.2013.09.018
Warrier V, Vieira M, Mole SE (2013) Genetic basis and phenotypic correlations of the neuronal ceroid lipofusinoses. Biochim Biophys Acta 1832:1827–1830. doi:10.1016/j.bbadis.2013.03.017
Winklhofer KF, Tatzelt J, Haass C (2008) The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J 27:336–349. doi:10.1038/sj.emboj.7601930
Yu CS, Cheng CW, Su WC, Chang KC, Huang SW, Hwang JK, Lu CH (2014) CELLO2GO: a web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PloS One 9:e99368
Zhang YQ, Chandra SS (2014) Oligomerization of cysteine string protein alpha mutants causing adult neuronal ceroid lipofuscinosis. Biochim Biophys Acta 1842:2136–2146. doi:10.1016/j.bbadis.2014.07.009
Zhang YQ, Henderson MX, Colangelo CM, Ginsberg SD, Bruce C, Wu T, Chandra SS (2012) Identification of CSPalpha clients reveals a role in dynamin 1 regulation. Neuron 74:136–150. doi:10.1016/j.neuron.2012.01.029
Acknowledgments
We thank Dr. Katherine Sims for contributing pathologic material from the Massachusetts General Hospital NCL Biorepository. We also thank Ted Voss, Jean Kanyo, and Kathrin Wilczak for assistance with sample prep, data collection, and LFQ analysis, respectively. We would like to thank Art Horwich, Shawn Ferguson, Pietro De Camilli, Thomas Biederer, and members of our laboratories for critical discussions related to this paper. We would also like to thank Zack Gomez for data analysis. This work was supported by the Battens Disease Research and Support Association Grant, NIH R01NS083846, R01NS064963 (to S.S.C.), NIDA Neuroproteomic Center Grant (5 P30 DA018343-07) and by a NRSA NS078861-02 (to M.X.H) as well as CTSA Grant UL1 RR024139 from the National Center for Research Resources (NCRR) and the National Center for Advancing Translational Science (NCATS).
Author contributions
M.X.H. and G.S.W. performed all immunohistochemical and immunoblot analyses; M.X.H. and G.S.W. maintained and lentivirally transduced primary mouse neuronal cultures; G.S.W. performed in vitro depalmitoylation assays, IPA and CELLO analyses of MS data; Y.Z. performed RT-PCR; A.F.R. and N.G.D. purified palmitoylated proteins for MS using AcylRAC; G.D. performed PPT1 activity assays; T.T.L. performed MS analyses; F.D. provided statistical analyses of MS data; M.V., N.D., J.F.S. and P.C.G.N. clinically characterized patients and provided control and patient tissue; S.D.G. provided additional samples; M.X.H., G.S.W., and S.S.C. were involved in study design, analyzed data, and wrote the paper. M.X.H. and G.S.W. contributed equally to this study. All authors discussed the results and commented on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Ethical standard
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Additional information
Michael X. Henderson and Gregory S. Wirak have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Henderson, M.X., Wirak, G.S., Zhang, Yq. et al. Neuronal ceroid lipofuscinosis with DNAJC5/CSPα mutation has PPT1 pathology and exhibit aberrant protein palmitoylation. Acta Neuropathol 131, 621–637 (2016). https://doi.org/10.1007/s00401-015-1512-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1512-2