
Abstract
Filamentous tau inclusions are hallmarks of Alzheimer’s disease (AD) and other neurodegenerative tauopathies. An increasing number of studies implicate the cell-to-cell propagation of tau pathology in the progression of tauopathies. We recently showed (Iba et al., J Neurosci 33:1024–1037, 2013) that inoculation of preformed synthetic tau fibrils (tau PFFs) into the hippocampus of young transgenic (Tg) mice (PS19) overexpressing human P301S mutant tau induced robust tau pathology in anatomically connected brain regions including the locus coeruleus (LC). Since Braak and colleagues hypothesized that the LC is the first brain structure to develop tau lesions and since LC has widespread connections throughout the CNS, LC neurons could be the critical initiators of the stereotypical spreading of tau pathology through connectome-dependent transmission of pathological tau in AD. Here, we report that injections of tau PFFs into the LC of PS19 mice induced propagation of tau pathology to major afferents and efferents of the LC. Notably, tau pathology propagated along LC efferent projections was localized not only to axon terminals but also to neuronal perikarya, suggesting transneuronal transfer of templated tau pathology to neurons receiving LC projections. Further, brainstem neurons giving rise to major LC afferents also developed perikaryal tau pathology. Surprisingly, while tangle-bearing neurons degenerated in the LC ipsilateral to the injection site starting 6 months post-injection, no neuron loss was seen in the contralateral LC wherein tangle-bearing neurons gradually cleared tau pathology by 6–12 months post-injection. However, the spreading pattern of tau pathology observed in our LC-injected mice is different from that in AD brains since hippocampus and entorhinal cortex, which are affected in early stages of AD, were largely spared of tau inclusions in our model. Thus, while our study tested critical aspects of the Braak hypothesis of tau pathology spread, this novel mouse model provides unique opportunities to elucidate mechanisms underlying the selective vulnerability of neurons to acquire tau pathology and succumb to or resist tau-mediated neurodegeneration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tau is a microtubule-binding protein which is mainly localized to axons of neurons and plays an important role in promoting the assembly and stabilization of microtubules [29]. However, under pathological conditions, tau becomes abnormally phosphorylated, conformationally altered, and aggregates into insoluble neurofibrillary tangles (NFTs) as well as in neuropil threads that are the signature lesions of Alzheimer’s disease (AD), progressive supranuclear palsy, corticobasal degeneration, Pick’s disease, frontotemporal lobar degeneration (FTLD) linked to tau pathology (FTLD-Tau) and other tauopathies [26]. In AD, NFTs progressively accumulate throughout increasingly widespread CNS regions in a stereotypical manner, from the transentorhinal and entorhinal cortex to the hippocampal formation and eventually the neocortex [9].
Recent studies have demonstrated that exogenously supplied preformed fibrils (PFFs) assembled from recombinant human tau can readily “seed” or template the aggregation of endogenously expressed soluble tau in cultured cells [20, 23]. While Clavaguera et al. showed induction and propagation of tau pathology in vivo by injecting brain extracts containing aggregated tau from tau transgenic (Tg) mice [16] or tauopathy patients [15] into ALZ17 tau Tg mice that overexpress human wild-type (WT) tau but do not normally develop tau pathology, we demonstrated that synthetic tau PFFs alone can induce templated transmission of pathological tau when injected into tau Tg mice overexpressing human P301S mutant tau (PS19) months before the emergence of transgene-driven tau pathology [25]. Furthermore, these and other studies suggest that tau transmission occurs over long distances, presumably along axons that are efferent and afferent to the injection site rather than as a result of the proximity of neurons to the injection site [1, 18, 25, 30]. This connectome-dependent propagation of aggregated tau has been suggested to underlie the highly predictable pattern of NFT progression in AD [12, 21].
Interestingly, tau PFF injections into either the striatum and overlying neocortex or the hippocampus of young PS19 mice consistently led to significant tau pathology in the locus coeruleus (LC), which is located in the lower part of the 4th ventricle in the brainstem and is the major noradrenergic nucleus in the CNS with widespread efferent projections and limited afferent inputs [3, 19]. Moreover, recent neuropathological studies on postmortem human brains from different age groups revealed surprisingly frequent “pretangle” tau lesions in brainstem nuclei, predominantly LC, of people under 30 years old, suggesting that the LC could be the first brain region affected by AD pathology from whence pathological tau then spreads rostrally to affect multiple telencephalic regions including transentorhinal cortex and hippocampus [10, 11]. For these reasons, we hypothesized that LC may be a critical brain structure giving rise to the stereotypical progression of NFTs in AD brains. Here, we report that unilateral injection of tau PFFs into LC of young PS19 mice induced time-dependent neuron loss from ipsilateral LC and significant tau pathology in numerous brain regions anatomically connected with LC, although the spreading pattern of tau pathology is different from that found in AD brains.
Materials and methods
Tau Tg mice
We used PS19 tau Tg mice of both genders (n = 49) which express human T34 tau (1N4R) with P301S mutation found in patients with familial FTLD-Tau [43]. These mice were maintained on a B6C3H background. All animal protocols were approved by The University of Pennsylvania’s Institutional Animal Care and Use Committee (IACUC).
Generation of synthetic tau PFFs
A cDNA encoding the longest human tau isoform (2N4R) containing the P301S mutation with a 3′Myc tag (3′Myc-T40/PS) was cloned into the pRK172 bacterial expression vector using the Ndel/EcoRI restriction sites. The protein was then expressed in BL21 (DE3) E. coli cells. The recombinant tau protein was purified and allowed to fibrillize in vitro in the presence of heparin with constant agitation at 1000 rpm at 37 °C for 1 day as previously described [22]. Fibrillization was confirmed by Thioflavin T fluorescence assay, sedimentation assay, and electron microscopy. Following ultracentrifugation at 100,000g for 30 min at 22 °C, the pellet of tau PFFs was resuspended in heparin-free buffer, aliquoted and stored at −80 °C until ready to use. Prior to injection, PFFs were sonicated with 40 brief pulses while on ice as described [25].
Stereotaxic surgery
Animals were aseptically injected with 3′Myc-T40/PS PFFs (referred to subsequently as PFFs for simplicity) or PBS as a control between 2 and 3 months of age following animal care and use protocols of the University of Pennsylvania as described [25], except that the target here was the LC. Animals were anesthetized using ketamine/xylazine/acepromazine, placed in a stereotaxic frame (David Kopf Instruments) and injections were made into the LC of the right hemisphere (bregma −5.45 mm, lateral +1.28 mm, depth −3.65 mm) [13] using a Hamilton syringe. A total of 4 µg of tau PFFs in a total volume of 1 µl were injected at a rate of 0.1 µl/min. Mice were monitored during the surgery by observing their respiration and monitoring toe reflex. Following the surgery, mice were monitored until they recovered from anesthesia, at which point they were given the analgesic buprenorphine in compliance with animal care and use protocols of the University of Pennsylvania.
Histology and immunohistochemistry (IHC)
Mice were sacrificed at 2 weeks (n = 3 for PFF injection and n = 2 for PBS injection), 1 month (n = 10 but only 8 were used for analysis since the other 2 missed the LC), 3 months (n = 8), 6 months (n = 10), and 12 months (n = 10) post-surgery by ip injection with a ketamine/xylazine mixture and transcardial perfusion with 30 ml of phosphate-buffered saline (PBS) as described [25]. The brains and spinal cords were removed and placed in neutral-buffered formalin (NBF) overnight at 4 °C. Additional 6 mice were sacrificed at 2 days and 2 weeks post-injection period (n = 3 each), and they were fixated in 70 % Ethanol overnight at 4 °C. The tissue was then thoroughly washed before being embedded in paraffin. Paraffin blocks were sectioned using a microtome to produce 6-µm-thick tissue sections.
Exactly as described earlier [25], tissue sections were deparaffinized in xylene and rehydrated through a series of increasingly dilute ethanol solutions. Endogenous peroxidase activity was quenched by treating sections with 5 % hydrogen peroxide in methanol for 30 min. Slides were incubated with primary antibody overnight at 4 °C. Following incubation with horseradish peroxidase (HRP)-conjugated secondary antibody, slides were developed using a non-biotin polymer HRP detection system (Biogenex) and counterstained with Mayer’s hematoxylin unless otherwise noted.
Pathological tau was visualized by staining every 20th slide with a series of antibodies previously used and described [25] including: AT8, a mouse monoclonal antibody (MAb) specific for tau phosphorylated at Ser202/Thr205 (1:10,000; Pierce) and MC1, a mouse MAb to a pathological conformation of tau (1:7000; a generous gift of Dr. Peter Davies). We also used the following additional antibodies: T14 (a mouse MAb that recognizes phospho-independent human tau, residues 141–178; 1:5000), T49 (a mouse MAb specific for mouse tau; 1:2000; detected using the Vector Mouse on Mouse Kit, BMK-2202), TG3, a mouse MAb that recognizes a conformation-dependent epitope around phosphorylated Thr231 (1:250; a generous gift of Dr. Peter Davies), ac-K280, a rabbit polyclonal antibody that recognizes tau acetylated at Lys280 (1:1000 with citrate buffer pre-treatment), a rabbit polyclonal antibody against tyrosine hydoxylase (TH; 1:10,000), a rat MAb to glial fibrillary acidic protein (GFAP; 1:3000), and 9E10 a mouse MAb that recognizes c-myc (1:5000; Santa Cruz). Thioflavin S staining was also performed to visualize NFTs and neuropil threads. In addition, a subset of slides were double labeled with AT8 (1:10,000) and an anti-TH antibody (1:5000) followed by detection using Alexa Fluor-labeled secondary antibodies (Invitrogen) to demonstrate accumulation of pathological tau within the noradrenergic TH-positive LC neurons. To determine if tau lesions are ubiquitinated, tissue sections were double labeled with AT8 (1:5000) and anti-ubiquitin antibody (Ubi-1, 1:500; rabbit monoclonal, Millipore, with citrate buffer treatment), and several sections were also double stained with AT8 (1:5000) and anti-tau oligomer antibody (TOC-1, 1:1000; mouse MAb with citrate buffer treatment, a generous gift of Dr. Lester Binder) to test for the presence of tau oligomers.
To quantify tau pathology in LC neurons, every 5th section through the entire rostral to caudal extent of the LC was stained with MC1 and positively stained cells were counted in the LC ipsilateral and contralateral to the injection site. Similarly, neuron loss in the LC was quantified by staining every 5th section through the LC with TH antibody and counting TH-positive LC neurons in each hemisphere.
To quantify tau pathology in amygdala and bed nucleus of the stria terminalis (BNST), every 5th section containing those brain regions was immunostained with MC1 and positively stained cells were counted for the side ipsilateral to the injection site.
For semi-quantitative analysis of AT8-positive neurons in the entire CNS, we selected nine different coronal CNS levels (bregma 2.10, −0.22, −1.22, −2.18, −3.52, −4.48, −5.52, −6.36, and −6.84 mm), marked brain regions based on anatomical/cytoarchitectural patterns [36] and used a previously published grading system adapted from the Braak staging system for AD to score the extent of tau pathology in PFF-injected PS19 mice [24, 44]. After grading individual brain regions in each mouse at 1 (n = 8), 3 (n = 8) and 6 (n = 10) months post-injection, we imported averaged values of these scores into custom-designed heat map software to create pathology distribution maps for the telencephalon and brainstem [8, 25].
Statistical analysis
Quantification results were analyzed using one-way ANOVA followed by Tukey’s post hoc test using Graph-Pad Prism 4.0 software (GraphPad Software, San Diego, CA). Data were expressed as mean ± SEM, and differences with P values less than 0.05 were considered significant.
Results
Induction of tangle pathology in LC
To test whether tau PFF injections into LC would lead to progressive development of tau pathology in a pattern similar to that observed in AD brains [10], we unilaterally injected Myc-T40/PS PFFs into a region 300 µm lateral to the LC so that the injection itself would not damage LC neurons [13] (Fig. 1a). Most of the injections were successfully placed (n = 47/49, 96 %) and immunoreactivity for GFAP was observed only ipsilaterally near the injection site but outside the main body of the LC at 2 weeks post-injection (Fig. 1b). Immunostaining using an anti-Myc antibody showed abundant tau PFFs around the injection site at 2 days post-injection, which appeared to be largely extracellular, but the Myc immunoreactivity was completely undetectable at 2 weeks post-injection, suggesting rapid clearance of exogenous tau PFFs (Supplementary Figure 1a, c). To confirm that the injected tau PFFs recruited endogenous and transgene expressed tau into pathological aggregates, we performed double-labeling immunofluorescence with AT8 (recognizing hyperphosphorylated Ser202/Thr205 tau) and antibody against TH. Since tau PFFs assembled from recombinant tau are not phosphorylated and since normal endogenous tau in the PS19 mice are not phosphorylated at the AT8 site, the presence of AT8 immunoreactivity marks the presence of tau pathology. As expected, a very high proportion of TH-positive LC neurons were filled with hyperphosphorylated tau inclusions, some of which were Thioflavin S positive as early as 2 weeks post-injection, suggesting rapid formation of mature tangles (Fig. 1c). We also immunostained sections throughout the LC region with different anti-tau antibodies at 3 months post-injection and found that PFF-induced tau pathology in LC neurons was conformationally changed (MC1), phosphorylated and conformationally changed (TG3), acetylated (ac-K280), and contained both human tau (T14) and mouse tau (T49) (Fig. 1d). To confirm that the development of tau pathology is not due to nonspecific effects of injection, mice were injected with PBS and immunostained with AT8 2 weeks post-injection. No AT8-positive neurons were detected in the ipsilateral LC and therefore provide support that all the tau pathology detected was due specifically to PFF-induced templated recruitment of endogenous tau followed by spreading and not due to other non-specific responses such as stress (Supplementary Figure 1d).

Schematic illustration of the LC injection site and the characterization of tau tangles and dystrophic neurites induced in LC and adjacent neurons by tau PFF injections. a Shows schematic coronal (upper) and sagittal views (bottom) of the tau PFF injection site just lateral to the LC. T40/PS PFFs were injected immediately lateral to the LC (bregma −5.45 mm, lateral +1.28 mm, depth −3.65 mm). The red dots and lines indicate the injection site and needle track of the injection, respectively. b Illustrates low (left) and high (right) power microscopic images of GFAP-stained reactive astrocytes in the vicinity of the ipsilateral LC injection site. Scale bars are 1 mm (left) and 100 µm (right), respectively. c TH (red) and AT8 (green) double staining revealed that injected tau PFFs recruited endogenous tau into aggregates in the TH-positive LC neurons. Thioflavin S staining shows the formation of mature tangles in the LC within 2 weeks. Scale bar is 100 µm. d Tau tangles in LC neurons are detected by multiple anti-tau antibodies in the LC ipsilateral to the tau PFF injection site at 3 months post-injection. Scale bar is 100 µm
Spreading of tau pathology to the contralateral LC and time-dependent neuron loss from the ipsilateral LC
To investigate the spreading of tau pathology after injecting tau PFFs into the LC on one side of the brainstem, we sacrificed PS19 Tg mice at 2 weeks, 1, 3, 6 and 12 months post-injection. Figure 2a shows 4 rows of different levels of the LC and related pontine regions from the most rostral region at the top to the caudal region at the bottom immunostained with MC1. As shown, injected tau PFFs not only templated the formation of tau pathology in the ipsilateral LC throughout its rostral–caudal span, but they also induced spreading of tau pathology to a small number of neurons on the contralateral LC within 2 weeks post-injection. Tau pathology in the ipsilateral LC neurons was most abundant at 1 month post-injection and gradually decreased with longer survival periods (Fig. 2a, c), although other pontine neurons adjacent to the LC in this brainstem region, including those in the vestibular and pontine nuclei showed more MC1-positive neurons with time. A similar time-dependent reduction in tau pathology was observed for the contralateral LC (Fig. 2c).

Time-dependent progressive accumulation of tau pathology in the LC and adjacent brainstem regions. a Shown here are MC1-stained sections at four different representative levels of the LC and adjacent brainstem structures from PS19 mice at 2 weeks, 1, 3, 6, and 12 months post-injection. Scale bar is 200 µm. b TH IHC staining results from tau PFF-injected PS19 mice after different post-injection periods. Scale bar is 200 µm. c This graph shows quantitative data on MC1-positive LC neurons with contralateral on the left (white bars) and ipsilateral on the right (gray bars). d This graph shows quantitative data on TH-positive LC neurons with contralateral on the left (white bars) and ipsilateral on the right (gray bars). The number of PS19 mice used per group for MC1-positive cell count were n = 4 for 1, 3 and 12 months post-injection and n = 10 for 6 months post-injection, and n = 4 for TH-positive cell count. *P < 0.1, **P < 0.05, ***P < 0.01
In view of these findings, we next asked if this diminution of tau pathology in the LC might reflect the degeneration of tangle-bearing LC neurons. To do this, we performed IHC with the anti-TH antibody (Fig. 2b). As confirmed by quantitative analysis (Fig. 2d), TH-positive LC neurons were significantly reduced at 6 and 12 months post-injection on the ipsilateral side while they were undiminished on the contralateral side. These results suggested that LC neurons ipsilateral to the injection site first accumulate tangles as soon as 2 weeks post-injection and then began to degenerate by 6 months post-injection, whereas neurons in the contralateral LC did not degenerate following a less aggressive accumulation of tau pathology, but instead, appeared to gradually clear tau inclusions with longer post-injection survival periods. Indeed, since a lower percentage of tau inclusions in the contralateral LC were Thioflavin S positive compared to the ipsilateral LC (data not shown), this suggests that the tau pathology induced in the contralateral side was less mature than in the ipsilateral LC, and perhaps less toxic. Double labeling of AT8 with an anti-ubiquitin antibody showed little ubiquitination of tau inclusions in the contralateral LC (Supplementary Figure 1e), suggesting the ubiquitin–proteasome system is unlikely playing a major role in the clearance of tau aggregates.
Retrograde and anterograde propagation of tau pathology along the LC projections
As previously described, the rodent LC receives afferent inputs from restricted brainstem regions, namely the prepositus hypoglossi (PrH) and nucleus paragigantocellularis (PGi) [4], but LC efferents project to almost all regions of the CNS except for the striatum which receives dopaminergic terminals [6]. Figure 3a is a simplified schematic illustration of both the well-established LC brainstem afferents as well as the major efferent projections of the LC to telencephalic regions.

Schematic diagram of the LC connectome and the retrograde and anterograde transmission of tau pathology from the tau PFF-injected LC. a Schematic representation of LC and anatomically connected brain regions. As shown, LC receives its afferent from prepositus hypoglossi (PrH) and nucleus paragigantocellularis (PGi), and projects to almost the entire CNS, including hypothalamus, cortex, bed nucleus of the stria terminalis (BNST), hippocampus and amygdala. Shown here are AT8-positive tau inclusions in the PrH (b) and PGi (c) at 2 weeks post-injection and the increasing amount of tau pathology that developed with longer post-injection periods. Scale bar is 100 µm
We first examined PrH and PGi which project to the LC, and found AT8-immunoreactive neurons in both PrH (Fig. 3b) and PGi (Fig. 3c) within 2 weeks post-injection. We also found that these AT8-positive inclusions became more numerous with time as shown at 1, 3 and 6 months post-injection.
We also examined the major brain regions to which the LC projects, such as hypothalamus, amygdala, bed nucleus of the stria terminalis (BNST), frontal cortex (Fig. 4a) as well as the hippocampus and entorhinal cortex (see below for details). A small number of AT8-immunoreactive neurons were found in the hypothalamus, amygdala and BNST within 2 weeks post-injection (data not shown). The amount of tau pathology in these structures gradually increased at longer post-injection time periods with prominent hyperphosphorylated tau accumulations in the perikarya of neurons observed at 3 and 6 months post-injection (Fig. 4). Notably, the frontal cortex, which is ~8 mm rostral to the injection site, and spinal cord neurons, which were at an equivalent distance from the injection site, both showed AT8-positive neurons within 3 months (Fig. 4a). Quantitative analysis of MC1-positive cells in amygdala (Fig. 4b) and BNST (Fig. 4c) showed time-dependent increases in the number of MC1-positive cells in both amygdala and BNST until 6 months post-injection. To clearly demonstrate the pattern of tau pathology spread initiated by tau PFF injections in LC, we generated comprehensive heat maps to illustrate both the distribution and burden of tau pathology across different coronal sections at different time points post-injection (Fig. 5). Importantly, tau pathology burden at the level of LC increased from 1 to 3 months but decreased at 6 months due to loss of TH-positive neurons.

Propagation of tau pathology to brain regions innervated by efferent projections from LC neurons. a Shown are microscopic images of sections immunostained with AT8 at 3 and 6 months post-injection from top to bottom in 4 different brain regions (hypothalamus, amygdala, BNST, frontal cortex) and spinal cord. The far left column illustrates sagittal views of the brain with red lines indicating the coronal planes selected for staining the indicated structures, and red dots indicating the location of LC. Scale bar is 100 µm. b and c show quantitative data on MC1-positive neurons in amygdala (b) and, BNST (c). d shows the lack of tau pathology in hippocampus dentate gyrus (DG) region and entorhinal cortex (ENT) at 3 and 6 months post-injection. Scale bar is 100 µm

Heat maps of tau pathology in coronal sections immunostained with AT8. Semi-quantitative analysis of tau pathology was conducted by grading the extent and abundance of AT8 immunoreactivities. Each panel represents heat map pathology distributed on one of the nine different coronal planes (bregma 2.10, −0.22, −1.22, −2.18, −3.52, −4.48, −5.52, −6.36, and −6.84 mm) at 1 month (n = 8), 3 months (n = 8) and 6 months (n = 10) post-injection period. Left column shows sagittal view of the selected coronal planes. Blue dot indicate injection sites
Further, since the tau inclusions that form as a result of transgene-driven mutant P301S human tau in aged PS19 mice do not show the distinct signatures of mature AD-like tangles, including immunoreactivities for ac-K280 and TG3 [25], we asked if the tau pathologies induced by PFF injections into the LC were recognized by these two antibodies since this would indicate that this tau pathology must have originated from the tau PFF injected into the LC. Consistent with templated transmission of PFF-seeded tau pathology from the LC, we found ac-K280 and TG3 immunoreactivities in numerous brain regions connected with LC, including but not limited to BNST, anterior cingulate cortex (ACC), midbrain (MD) and hypothalamus (HT) at varying intervals post-injection (Fig. 6).

Mature tau tangle-bearing neurons increased in regions to which the LC projects or that project to the LC. Lower and higher power microscopic images of sections immunostained with ac-K280 (a) and TG3 (b) in BNST at 1 month post-injection, anterior cingulate cortex (ACC) at 3 months post-injection, midbrain (MD) at 3 months post-injection, and hypothalamus (HT) at 6 months post-injection from top to bottom. Shown are examples of brain regions with ac-K280 and TG3 immunoreactivities. Scale bars are 100 µm
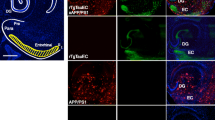
Since the findings reported above suggest that the PFF-templated tau pathology was transmitted anterogradely from the LC to its efferent projection sites mentioned above, we interpret this to signify transneuronal spreading of tau pathology to induce pathological tau in the perikaya of neurons innervated by the LC. We examined this possibility further by double-label immunostaining with the anti-TH antibody and AT8 in regions receiving efferent projections from LC (Fig. 7). Notably, we observed a partial co-localization of AT8-immunoreactive tau pathology with TH-positive axons arising from LC neurons which support our view that the perikaryal tau pathology at these LC projection sites reflects transneuronal spread of tau pathology.

TH- and AT8-positive axons can be detected in the thalamus and hypothalamus. Double immunofluorescence staining with AT8 (green) and TH (red) in thalamus (a) and hypothalamus (b) in 6 months post-injection mouse. White arrows identify neuronal processes that are double labeled for both AT8 and TH. Scale bar is 100 µm
Finally, our previous study showed that tau PFF injection into hippocampus consistently led to tau pathology in the LC within 2 weeks post-injection [25], indicating efficient retrograde transmission of pathological tau from the hippocampus to the LC (see Figure 4A in [25]). Surprisingly, our current study of tau PFF injections into the LC did not induce any AT8- or MC1-positive tau inclusions in the hippocampus even at 6 months post-injection (Fig. 4d), although we did observe transgene-driven endogenous tau pathology 12 months post-injection (data not shown), suggesting that the transmission of pathological tau may exhibit specific directionality and does not undergo transmission bi-directionally or that the hippocampus is otherwise resistant to accumulating tau pathology at the doses of PFFs injected here, the post-injection time intervals studied here or for other unknown reasons. Meanwhile, entorhinal cortex, which also receives efferent projections from LC, also failed to develop any appreciable tau pathology within our experimental time window (Fig. 4d). Since the transentorhinal cortex and the hippocampal formation are thought to be the earliest telencephalic structures that develop tau positive and argyrophilic NFTs in human brains at what is believed to be the earliest stages of the onset of tau pathology that subsequently culminates in widespread AD tau pathology [9, 11], our paradigm of tau PFF injections into the LC of PS19 mice did not completely recapitulate the spatiotemporal progression pattern of NFTs that has been proposed to reflect the stage-wise spread of pathological tau in subjects who develop dementia due to AD.
Discussion
Here, we provided additional compelling evidence of pathological tau transmission via anatomically connected brain regions [1, 16, 25, 37] by injecting synthetic tau PFFs into LC of PS19 mice. Similar to our previous study of synthetic tau PFF injections into hippocampus or striatum/overlying cortex [25], we observed that the injected tau PFFs rapidly seeded the formation of tau pathology near the injection site, i.e., in LC neurons in this study, and this tau pathology was subsequently propagated to widespread brain regions that are anatomically interconnected with LC neurons through either efferent or afferent projections, such as those that comprise the PGi, amygdala and BNST. Also similar to our prior study [25], tau pathology observed in the LC-injected mice more closely resembles mature AD NFTs than that developed in the aged un-injected PS19 mice. We also found significant neuron loss in the ipsilateral LC starting 6 months post-injection, while neurons in the contralateral LC appeared to be capable of clearing tau pathology without showing evidence of neurodegeneration.
LC is a nucleus located in the ventral part of 4th ventricle of the brainstem, and is known as the largest nucleus containing noradrenergic neurons [3, 6, 32]. Anatomical studies using HRP tracing with DAB as a substrate in the late 1970s showed that many structures in CNS project to LC [14, 17, 33]. However, Aston-Jones et al. [4] demonstrated using retrograde tracers Fluoro-Gold and wheat-germ agglutinin conjugated with HRP (WGA-HRP) that LC receives afferents from two limited nuclei, i.e., paragigantocellularis (PGi), and prepositus hypoglossi (PrH). This same group reported later using Cholera-toxin B (CTb) as a tracer that LC also receives some afferents from the preoptic area dorsal to the supraoptic nucleus, the posterior hypothalamus, the Kolliker-Fuse nucleus, and the mesencephalic reticular formation, lesser amount of afferents from hypothalamic paraventricular nucleus, dorsal and median raphe nucleus, dorsal part of the periaqueductal gray, the area of the noradrenergic A5 group, the lateral parabrachial nucleus, and caudoventrolateral reticular nucleus, and virtually none or occasional projections from cortex, central nucleus of the amygdala, lateral part of the bed nucleus of the stria terminalis, vestibular nuclei, nucleus of the solitary tract, and spinal cord [31]. In our study, we observed tau pathology in the PGi and PrH as well as hypothalamus, amygdala and BNST, which receives efferents from LC. Although we cannot clearly distinguish whether those tau pathologies in the hypothalamus, amygdala and BNST arise from efferent or afferent transmission, we consider that the abundance of tau pathology detected in these regions are a consequence of anterograde tau propagation.
Intriguingly, recent studies suggest that LC may be the first brain structure to develop tau lesions, long before the appearance of NFTs in the transentorhinal cortex and hippocampus, which are brain regions thought to be affected in the earliest stages of AD [10, 11]. Therefore, LC neurons may be the most vulnerable to abnormal tau accumulations in the entire brain. This selective vulnerability of LC neurons to pathological tau also is supported by our previous study of synthetic tau PFF injections into either hippocampus or striatum plus the overlying neocortex of young PS19 mice, whereby tau inclusions were rapidly induced in LC neurons following injections of tau PFF into these sites [25]. Furthermore, the widespread efferent projections from LC to various brain structures led us to speculate that brain regions involved in the later stages of AD may have received the initial pathological tau seeds from LC through synaptic connections, and among them, transentorhinal cortex and hippocampus are the most vulnerable to tangle formation; these regions may in turn transmit misfolded tau to other interconnected brain structures, thereby implicating more brain areas. Thus, the primary goal of our current study was to test the hypothesis that the LC is the critical structure that initiates the stereotypical propagation of NFTs in AD brains.
Contrary to our expectation, although tau PFF-induced pathology in the LC did lead to pathological tau transmission to numerous brain regions interconnected with LC, the pattern of spreading did not match NFT staging in human AD brains [9, 11]. Specifically, entorhinal cortex and hippocampus failed to show any appreciable tau lesions even after 6 months post-injection survival intervals, whereas brain regions that are only affected in the more advanced stages of AD, such as frontal cortex (stage V), developed tau pathology more rapidly after tau PFF injections into the LC. While LC neurons projecting to hippocampus and entorhinal cortex may be somehow more vulnerable and degenerated before transmission to these regions occurred, our results more likely argue against a simple model of LC being the sole origin of AD NFTs. Other brainstem structures that display early tau lesions, such as nuclei of the upper raphe system and magnocellular nuclei of the basal forebrain [11], might play more important roles in transmitting tau pathology to stage I and II regions. Alternatively, NFT formation in transentorhinal cortex and hippocampus may be an independent event not causally linked to pretangle formation in the brainstem; while the former signifies the start of true pathogenic process eventually leading to full-blown AD, the latter may be a relatively innocuous process taking place in almost all individuals. This possibility is supported by a recent study showing AD-like spreading of tau pathology initiated by tau PFF injection into the entorhinal cortex of the same tau transgenic mice we are using [39]. On the other hand, the overexpression of human mutant P301S tau in PS19 mice may have accelerated propagation of tau pathology to neocortex which is typically affected only in later stages of AD. Lastly, we cannot rule out the possibility that synthetic tau fibrils, which were shown to be conformationally distinct from the tau fibrils that develop in AD brains [34], may display a different transmission pattern that is further complicated by the overexpression of mutant tau.
Injection of tau PFFs into the LC led to progressive loss of tangle-bearing LC neurons at 6–12 months post-injection on the ipsilateral side, but there was no significant loss of tangle-bearing neurons in other interconnected brain regions including the contralateral LC. While the toxicity of NFTs has been a source of controversy almost since their discovery, human studies showed that NFTs are always found in brain regions undergoing neurodegeneration, and the burden of NFTs correlates well with severity of dementia in AD patients [2, 41]. These observations led to the traditional view that filamentous tau aggregates are toxic to cells by creating physical obstacles for important cellular processes and sequestering critical cytoplasmic proteins, including their soluble counterparts, to prevent them from performing crucial cellular functions [5]. However, numerous tau transgenic mouse studies have demonstrated that neuronal dysfunction or death can occur in the absence of or before NFT formation, leading to the hypothesis that early species in the tau aggregation pathway, such as soluble oligomers, may be the real neurotoxic species [28, 38, 40, 42, 43]. In addition, our previous study of telencephalic tau PFF injections in PS19 mice [25], and an earlier study [16] using brain lysates containing tau aggregates to induce pathology in Tg mice overexpressing WT tau showed lack of neuron loss after seeded NFT induction, suggesting a dissociation between transmissible species and toxic species of tau. However, since a more recent study using much higher doses of tau PFFs [37] or human AD lysate [8] demonstrated neuron loss following injections into the hippocampus, it is clear that many variables must be considered in the interpretation of tau transmission studies.
What accounts for the progressive loss of TH-positive neurons in the ipsilateral LC, which rapidly developed mature tangles following tau PFF injection is unclear. One possibility is the higher vulnerability of LC neurons to NFT-induced cell death than hippocampal neurons that were the initial target of our previous study. Another possibility is that NFT-mediated neurodegeneration may be a dose-dependent process, whereby injection of 4 µg tau PFFs into the small LC structure resulted in a higher effective concentration of seeds for more efficient templated misfolding of tau near the injection site than injection of 5 µg tau PFFs into the hippocampus as performed in our previous study [25]. The second possibility, as noted above, is also supported by a recent study reporting loss of hippocampal CA1 neurons following 25 µg tau PFF injection into the hippocampus of Tg overexpressing tau with P301L mutation [37]. Together with studies estimating that NFT-bearing neurons can survive for years or even decades in human brains [7, 27, 35], our current data suggest that NFTs are capable of causing neurodegeneration even though they normally do not kill cells in an acute manner unless a sudden burst of high NFT burden is induced under certain experimental paradigms.
Interestingly, the contralateral LC, which also developed tau inclusions, did not show detectable loss of TH-positive neurons up to 12 months post-injection, but instead appeared to have partially cleared tau pathology over time. Since the burden of tau pathology induced in the contralateral LC was much lower than that in the ipsilateral LC (Fig. 2a), presumably due to lower abundance of the available misfolded tau seeds, this result suggests NFTs may be degradable structures as long as the burden of tau pathology does not reach a critical threshold and the clearance of NFTs may help to protect neurons from undergoing degeneration.
Taken together with our previous results and those of other investigators cited above, we provide strong evidence for the transmission of misfolded tau through neuroanatomical connections from the injected brain regions, offering further experimental support for the progressive spread of tau pathology in the human CNS. More importantly, the novel model of tau pathology propagation described here opens up new avenues for investigating the role of LC neurons in the progression of NFTs in AD brains and will facilitate efforts to interrogate critical aspects of tauopathies, such as selective predilection of specific neuronal populations to acquire tau pathology and become vulnerable to NFT-mediated neurodegeneration as well as the ability of cells to acquire resilience to the potential toxicity of pathological tau in part by being able to eliminate tau pathology before it triggers a pathway that culminates in neurodegeneration.
References
Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S et al (2014) A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 127:667–683. doi:10.1007/s00401-014-1254-6
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42:631–639
Aston-Jones G, Cohen JD (2005) An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci 28:403–450. doi:10.1146/annurev.neuro.28.061604.135709
Aston-Jones G, Ennis M, Pieribone VA, Nickell WT, Shipley MT (1986) The brain nucleus locus coeruleus: restricted afferent control of a broad efferent network. Science 234:734–737
Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 8:663–672. doi:10.1038/nrn2194
Berridge CW, Waterhouse BD (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev 42:33–84. doi:10.1016/S0165-0173(03)00143-7
Bobinski M, Wegiel J, Tarnawski M, de Leon MJ, Reisberg B, Miller DC, Wisniewski HM (1998) Duration of neurofibrillary changes in the hippocampal pyramidal neurons. Brain Res 799:156–158 (pii:S0006-8993(98)00441-7)
Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ (2015) Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol 129:221–237. doi:10.1007/s00401-014-1373-0
Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Del Tredici K (2011) The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 121:171–181. doi:10.1007/s00401-010-0789-4
Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70:960–969. doi:10.1097/NEN.0b013e318232a379
Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16:109–120. doi:10.1038/nrn3887
Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K, de Lecea L (2010) Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci 13:1526–1533. doi:10.1038/nn.2682
Cedarbaum JM, Aghajanian GK (1978) Afferent projections to the rat locus coeruleus as determined by a retrograde tracing technique. J Comp Neurol 178:1–16. doi:10.1002/cne.901780102
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M et al (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 110:9535–9540. doi:10.1073/pnas.1301175110
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M et al (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11:909–913. doi:10.1038/ncb1901
Clavier RM (1979) Afferent projections to the self-stimulation regions of the dorsal pons, including the locus coeruleus, in the rat as demonstrated by the horseradish peroxidase technique. Brain Res Bull 4:497–504
de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA et al (2012) Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73:685–697. doi:10.1016/j.neuron.2011.11.033
Foote SL, Bloom FE, Aston-Jones G (1983) Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol Rev 63:844–914
Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852. doi:10.1074/jbc.M808759200
Guo JL, Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20:130–138. doi:10.1038/nm.3457
Guo JL, Lee VM (2013) Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett 587:717–723. doi:10.1016/j.febslet.2013.01.051
Guo JL, Lee VM (2011) Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 286:15317–15331. doi:10.1074/jbc.M110.209296
Hurtado DE, Molina-Porcel L, Iba M, Aboagye AK, Paul SM, Trojanowski JQ, Lee VM (2010) Aβ accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol 177:1977–1988. doi:10.2353/ajpath.2010.100346
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33:1024–1037. doi:10.1523/JNEUROSCI.2642-12.2013
Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, Lee VM, Trojanowski JQ (2015) Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol 129:469–491. doi:10.1007/s00401-014-1380-1
Kenessey A, Yen SH, Liu WK, Yang XR, Dunlop DS (1995) Detection of d-aspartate in tau proteins associated with Alzheimer paired helical filaments. Brain Res 675:183–189 (pii:0006-8993(95)00061-T)
Kuchibhotla KV, Wegmann S, Kopeikina KJ, Hawkes J, Rudinskiy N, Andermann ML, Spires-Jones TL, Bacskai BJ, Hyman BT (2014) Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc Natl Acad Sci USA 111:510–514. doi:10.1073/pnas.1318807111
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. doi:10.1146/annurev.neuro.24.1.1121
Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K (2012) Trans-synaptic spread of tau pathology in vivo. PLoS One 7:e31302. doi:10.1371/journal.pone.0031302
Luppi PH, Aston-Jones G, Akaoka H, Chouvet G, Jouvet M (1995) Afferent projections to the rat locus coeruleus demonstrated by retrograde and anterograde tracing with cholera-toxin B subunit and Phaseolus vulgaris leucoagglutinin. Neuroscience 65:119–160
Moore RY, Bloom FE (1979) Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci 2:113–168. doi:10.1146/annurev.ne.02.030179.000553
Morgane PJ, Jacobs MS (1979) Raphe projections to the locus coeruleus in the rat. Brain Res Bull 4:519–534
Morozova OA, March ZM, Robinson AS, Colby DW (2013) Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein. Biochemistry 52:6960–6967. doi:10.1021/bi400866w
Morsch R, Simon W, Coleman PD (1999) Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol 58:188–197
Paxinos G, Franklin K (2003) The mouse brain in stereotaxic coordinates, 2nd edn. Academic, Waltham
Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, Duytschaever H, Ver Donck L, Torremans A, Sluydts E et al (2015) Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis 73:83–95. doi:10.1016/j.nbd.2014.08.032
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E et al (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. doi:10.1126/science.1113694
Stancu IC, Vasconcelos B, Ris L, Wang P, Villers A, Peeraer E, Buist A, Terwel D, Baatsen P, Oyelami T et al (2015) Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol 129:875–894. doi:10.1007/s00401-015-1413-4
Van der Jeugd A, Hochgrafe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D’Hooge R et al (2012) Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol 123:787–805. doi:10.1007/s00401-012-0987-3
Wilcock GK, Esiri MM (1982) Plaques, tangles and dementia. A quantitative study. J Neurol Sci 56:343–356
Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714. doi:10.1126/science.1062382
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351. doi:10.1016/j.neuron.2007.01.010
Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, Hogan AM, Xie SX, Ballatore C, Smith AB 3rd et al (2012) The microtubule-stabilizing agent, epothilone d, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci 32:3601–3611. doi:10.1523/JNEUROSCI.4922-11.2012
Acknowledgments
This work was supported by National Institutes of Health Grant AG17586 and the Marian S. Ware Alzheimer Program Cure Alzheimer’s Fund, the Karen Cohen Segal and Christopher S. Segal Alzheimer Drug Discovery Initiative Fund, the Paula C. Schmerler Fund for Alzheimer’s Research, the Barrist Neurodegenerative Disease Research Fund, the Eleanor Margaret Kurtz Endowed Fund, the Mary Rasmus Endowed Fund for Alzheimer’s Research, Gloria J. Miller, and Dr. Arthur Peck. We thank Joshua Daniels, Bryan Zoll and Susan Leight for their assistance in mouse husbandry, Martine White and Chris Chung for counting cell numbers, Theresa Schuck, John Robinson, and Kevin Raible for their technical assistance in immunohistochemistry, and Rui Tong for his computer expertise in constructing the heat maps.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animal were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Iba, M., McBride, J.D., Guo, J.L. et al. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol 130, 349–362 (2015). https://doi.org/10.1007/s00401-015-1458-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1458-4