
Abstract
Inflammatory changes have been postulated to contribute to secondary brain injury after aneurysmal subarachnoid hemorrhage (SAH). In human specimens after SAH as well as in experimental SAH using mice, we show an intracerebral accumulation of inflammatory cells between days 4 and 28 after the bleeding. Using bone marrow chimeric mice allowing tracing of all peripherally derived immune cells, we confirm a truly CNS-intrinsic, microglial origin of these immune cells, exhibiting an inflammatory state, and rule out invasion of myeloid cells from the periphery into the brain. Furthermore, we detect secondary neuro-axonal injury throughout the time course of SAH. Since neuronal cell death and microglia accumulation follow a similar time course, we addressed whether the occurrence of activated microglia and neuro-axonal injury upon SAH are causally linked by depleting microglia in vivo. Given that the amount of neuronal cell death was significantly reduced after microglia depletion, we conclude that microglia accumulation inflicts secondary brain injury after SAH.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a devastating disease, which occurs through the rupture of an intracranial aneurysm. This bleeding usually takes place in the extraparenchymal compartment—the subarachnoid space. Brain injury after SAH occurs in two phases. Direct brain injury in the early phase starts with the onset of the bleeding and is mediated mainly through physical stress. A rise in intracranial pressure (ICP) leads to a decrease in cerebral perfusion, which can induce hypoxia [40, 61]. Furthermore, blood elicits a deleterious effect on the brain parenchyma, leading to a swelling of the brain within the first days after SAH. Secondary brain injury in the delayed phase after SAH starts around days 4–7. The mechanisms of this secondary brain injury have been a matter of research for many decades. Since the time window between onset of the bleeding and development of delayed brain injury may give rise to preventive or therapeutical options, there is an obvious medical need for a better understanding of the pathophysiology of SAH, including its secondary effects. Delayed cerebral ischemia, following posthemorrhagic cerebral vasospasm is the foremost and best described of all potential mechanisms of delayed brain injury. Therefore, prevention and treatment of cerebral vasospasm has been a major target within decades of scientific effort. After failure of recent pharmacological anti-vasospasm approaches to improve clinical outcome after SAH—despite the fact that significant reduction of cerebral vasospasm had been achieved—other possible mechanisms of delayed brain injury have been brought up [27, 41, 42, 73]. Cortical spreading depolarisations (CSD), toxic effects of hemoglobin metabolites, microthromboembolism and the role of the blood brain barrier (BBB) have been discussed [14, 19, 48, 64, 66]. Inflammatory changes (i.e., an activation of innate immunity) have also been hypothesized as possible contributor to secondary brain injury, predisposal for cerebral vasospasm or aneurysm formation and rupture within the past years [13, 28, 53, 55, 68]. An inflammatory milieu in the subarachnoid space after SAH has already been well characterized by our group and others [17, 23, 60, 70]. In the CSF of patients experiencing SAH, we found pro-inflammatory properties to be increased, which comprised an up-regulation of the leukocyte–endothelial interaction and transmigration [60]. Furthermore, we could show a positive correlation of the amount of inflammatory cytokines in posthemorrhagic CSF correlated directly with the severity of the hemorrhage and with the intracerebral accumulation of Interleukin-6 (as assessed by microdialysate), suggesting that the inflammatory milieu of the subarachnoid space elicited also an intraparenchymal reaction [59]. Despite the latter finding, it has not been defined, if the extraparenchymal inflammatory milieu does also affect the central nervous system (CNS) tissue, and if inflammation within the CNS may also occur upon SAH.
Inflammatory cell invasion into and inflammatory cell accumulation within the CNS has been documented in a variety of other pathologies. While astroglia are of neuroectodermal origin, microglia (MG) rely on lineage-committed precursors in the yolk sac and are considered the brain’s residential immune cells [1, 36, 57]. Microglia have been discussed to play pivotal roles in CNS pathologies like Alzheimer’s disease, autoimmune encephalomyelitis, traumatic brain injury and ischemic stroke [4, 8, 32, 38], where microglia fulfill their pleiotropic roles as cells of the innate immune system, including antigen presentation, phagocytosis and production of a variety of cytokines and chemokines. They have furthermore been described to be attracted to and activated by a variety of toxic or inflammatory stimuli and have been shown to initiate gliotic scar formation via secretion of respective cytokines.
In contrast to these cerebral diseases, the primary pathology of SAH takes place in the extraparenchymal compartment. Apart from a minority of SAH that are accompanied by intraparenchymal hematomas, the subarachnoid bleeding typically occurs in the basal cisterns.
In the present study, we have tested the hypothesis whether SAH causes a delayed inflammatory response within the brain parenchyma. We can show that the brain parenchyma upon SAH is affected by microglia, which get activated and release various pro-inflammatory cytokines, ultimately leading to axonal injury and neuronal cell loss, thus aggravating secondary brain injury after SAH.
Materials and methods
Human specimens
Records of all human patients, who had died in the course of SAH between 1983 and 2005 and were sectioned in the Department of Neuropathology at the University of Göttingen, Germany, were retrospectively reviewed. In this department, routine sectioning is offered and recommended for all in-hospital mortality. Care was taken that these patients did not suffer from CNS infection, intracerebral hematoma, cerebral vasospasm or respective infarctions. Sections of the brain parenchyma in various positions (near site of hemorrhage, far from site of hemorrhage, hippocampus) were obtained for histological analysis. To obtain appropriate sample sizes, the human specimens were pooled for the following time intervals (from onset of SAH until death): 1–4 days (n = 6), 5–8 days (n = 3), 9–15 days (n = 7) and more than 16 days (n = 5). The clinical course of these patients was retrospectively reviewed.
Histological analysis of the human brain sections for microglia/macrophages was done after deparaffinization and rehydration of the slides (10 µm), taken from tissue blocks near the aneurysm location and far from the aneurysm location. The slides were incubated overnight at 4 °C with a primary antibody against CD68 (KiM1P) to detect microglia/macrophage cells (Abcam, 1:50,000 in 10 % FCS/PBS). The slides were incubated with the secondary antibody (biotinylated anti-mouse, Amersham, 1:200 in 10 % FCS/PBS) for 1 h at room temperature, followed by visualization with ExtrAvidin Peroxidase (Sigma, 1:1000 in 10 % FCS/PBS, 1 h at room temperature) and DAB (25 mg/50 ml PBS + 25 µl 30 % H2O2). Counterstaining was done with hemalaun for 30 s, before the slides were rinsed in tab water and mounted with permanent mounting media.
Animal experiments
C57Bl/6-wild type and C57Bl/6-GFP-mice as well as CD11b-HSVTK mice were kept at the Forschungszentrum für Experimentelle Medizin, Charité, Universitätsmedizin Berlin. All animals used for induction of SAH or irradiation experiments were 12–14 weeks old (total body weight: app. 22–25 g) at the start of the experiments.
Study approval
All animal experiments were approved by the local ethics committee on animal research (Landesamt für Gesundheit und Soziales Berlin, Germany, RegNr G240/08) and were committed in conformity with the German law of animal protection and the National Institute of Health guidelines for care and use of laboratory animals.
Animal model of experimental SAH
A filament perforation model was applied to initiate SAH in female C57Bl/6 mice. To this end, the mice were placed in supine position after induction of an intraperitoneal ketamine/xylazine anesthesia. A midline incision was performed to expose the trachea, the vagus nerve and the carotid artery. A 5–0 non-absorbable monofilament polypropylene suture (Prolene, Ethicon, Norderstedt, Germany) was introduced into the external carotid artery in a retrograde fashion and then flipped to turn anterograde into the internal carotid artery. Under continuous blood flow through the common carotid artery the filament was pushed forward intracranially until puncture at the intracranial carotid bifurcation was achieved. This was accomplished 12 mm distally from the extracranial carotid bifurcation. Animals suffering from hemiparesis after the operation were excluded due to the expected occurrence of intraparenchymal hemorrhage or stroke. At the end of the experiments, the animals were perfused with ice-cold formaldehyde (4 %) under deep anesthesia and the brains were harvested for later immunohistochemical analysis.
Immunohistochemical and immunofluorescence analysis of the murine brains
Immunohistochemical and immunofluorescence analysis of all murine brain specimens was performed in coronary sections (thickness 10 µm). Standard hematoxylin/eosin and iron stainings were performed in all animals [58]. In contrast to KiM1P-stainings in our human experiments, microglia/macrophage cells herein were visualized by staining for ionized calcium-binding adaptor molecule-1 (Iba-1) in the murine experiments. Axonal injury was stained with extracellular amyloid precursor protein (APP).
Immunohistochemistry stainings for light microscopy were done in an automated staining machine (Ventana NexES v9.30, Ventana Medical, Tucson, USA). Primary antibodies included rabbit anti Iba-1 polyclonal antibody (1:500 WAKO Pure Chemical Industries) and mouse anti-APP (1:1000 Millipore). Secondary antibodies were DyLight 488 donkey anti rabbit (1:200 Jackson ImmunoResearch Lab) and FITC donkey anti-mouse (1:100 Jackson ImmunoResearch Lab). Complete automate staining protocols (92 steps) can be provided upon request.
For evaluation a camera-equipped microscope was used (Olympus Bx53 equipped with Olympus DP21, Olympus K.K., Tokyo, Japan).
Quantification of cell accumulation was difficult due to the extremely dense accumulation of the cells—especially on day 14. For counting of cells, five HPFs around the area of highest density were chosen, knowing that this method leads to an underestimation of the actual cell number. To confirm the results obtained in these cell counts, we furthermore measured the area covered by Iba-1-positive cells in relation to the total brain section surface.
Immunofluorescence stainings were performed for Iba-1, NeuN, cell death, IL-6 and TNF-α. Slides were protected and nuclei were labeled with DAPI mounting medium in all protocols (Dianova). The slides were incubated with the primary antibodies at 4° overnight. The secondary antibodies were incubated for 2 h at room temperature. Antibodies used include rabbit anti-Iba-1 (1:250 WAKO Pure Chemical Industries), mouse anti-NeuN (1:200 Millipore), goat anti-Il-6 (1:200 Abcam), goat anti-TNF-α (1:200 R&D), DyLight 488 donkey anti rabbit (1:200 Jackson ImmunoResearch Lab), FITC donkey anti mouse (1:100 Jackson ImmunoResearch Lab) and DyLight 488 donkey anti goat (1:200 Dianova). Neuronal cell death was detected by terminal deoxyuridine triphosphate nickend labeling (TUNEL, ApopTag© Red In Situ, Apoptosis Detection Kit, Millipore), according to the manufacturer’s protocol.
For quantitative analysis, whole sections were mounted and analyzed under a fluorescence microscope (Zeiss, Axio Observer Z1, Carl Zeiss, Microimaging GmbH, Munich, Germany) equipped with a digital camera (AxioCam MRc) and hard drive for later offline freeware image analysis (ImageJ.net). Immunofluorescence sections were divided into 6–10 high power fields, allowing for total cell counts per brain section on three different levels of the brain (bregma, and 1.5 mm before and behind).
Green fluorescent protein (GFP)–bone marrow chimera model
To determine the CNS origin of Iba-1 positive cells in the brain, and thus to distinguish between microglia and secondarily invaded macrophages, a chimeric mouse model was utilized. C57Bl/6 mice were lethally irradiated (11.5 Gy) and reconstituted with preseparated donor bone marrow cells (1 × 107 cells per recipient, i.v. injection, maximum 3 h after irradiation) from GFP-positive mice. During irradiation the animals’ heads were covered with a lead chamber, developed by our group [46], to protect the CNS and the BBB from irradiation effects. The animals were then kept in individually ventilated cages under antibiotic prophylaxis (Baytril 0.01 % via drinking water, Bayer Animal Health) and meticulous clinical control. After 6–8 weeks, the reconstitution was verified by FACS analysis of the peripheral blood. The mice were then further processed in the experimental SAH procedures as described above. Evaluation of the origin of the Iba-1-positive cells was performed on day 14 after SAH. These GFP–bone marrow chimeric mice allowed for differentiation of GFP-positive cells (peripheral blood pool-derived macrophages) and primary brain derived, GFP-negative cells (microglia).
Isolation of microglia from the murine brains
To further analyze the microglia cell fraction for inflammatory properties, a cell extraction assay was established in our laboratory. In a separate set of experiments, murine brains after SAH were brought into suspension, taking advantage of the MACS© Neural Tissue Dissociation Kit (Miteny Biotec), according to the manufacturer’s recommendation.
For the isolation of microglia cells from the suspensions, six hemispheres were pooled for a higher cell yield. The experimental setup was repeated twice. Thus, a total of 12 hemispheres were analyzed. To this suspension a magnetic microbead-labeled CD11b-antibody (CD11b MicroBeads, Miteny Biotec, Germany) was added, normalized to the respective total volume of the suspension. After incubation at 4° for 15 min, the respective cell pools were added to a pre-cooled column placed within a magnet, in which only the magnetically labeled cell fraction was held, while the non-labeled cells passed the column. After elution, a second column was used to improve the purity of the CD11b-positive cell fraction. The purity of this CD11b-positive microglia cell fraction was evaluated by FACS, using antibodies against CD11b and CD45 (BD Bioscience, FITC-coupled, M1/70, APC-coupled 30-F11). DAPI (1:100, Sigma Aldrich, USA) was added to the cell suspension to exclude dead cells before samples were measured on the FACS (BD FACSCanto™ II, BD Bioscience, Germany).
Quantification of inflammatory cytokine mRNA and cDNA in the isolated microglia
RNA extraction was performed from the isolated microglia cell fraction, using RNA Extraction Kit (NucleoSpin© RNA II, Fisher Scientific, UK). The amount of RNA was evaluated at 260 nm filter range. From RNA, cDNA was reversely transcribed using QuantiTec© Reverse Transcription Kit (Quiagen), according to the manufacturer’s recommendation. Quantitative real-time PCR (RTq-PCR) was then performed for Il-1α, Il-1β, TNF-α, Il-6 and the corresponding receptors Il1r1, Il1r2, Il6r, Tnfr1 and Tnfr2 using QuantiTect SYBR Green PCR Kits (Quiagen) and the, respectively, designed primers (Primer3, freeare, simgene.com). Primer sequences are displayed in Table 1. All primers were produced by TIB MOLBIOL (Berlin, Germany).
Iba-1 and intracellular protein staining for FACS analysis
Iba-1 staining was realized after fixation of brain homogenates with 2 % paraformaldehyde (Sigma, USA) using the following antibodies: V450 anti-CD11b (M1/70, BD Pharmingen), allophycocyanin-conjugated anti-CD45 (30-F11, BD Pharmingen) and rabbit anti-mouse Iba-1 (Wako, Japan). As secondary antibody Dylight 549-conjugated anti-rabbit IgG (Dianova) was added. Here, CD11b+CD45+ cells were gated and afterwards the percentage of Iba-1-positive cells was calculated.
The protein analyses of IL-6 and TNF-α were done quantitatively by FACS. Briefly, cells of brain homogenates were cultured in 1 mL DMEM-media (GIBCO; containing 10 % FCS) with 5 µg/ml Brefeldin A (SigmaAldrich, USA) at 37° for 5 h, allowing for accumulation of intracellular proteins. After washing in PBS, the cells were fixed in 2 % formaldehyde at room temperature for 20 min. To allow for intracellular staining, the membrane permeability of cells was induced using Saponin buffer (0.5 %). Cells were stained with FITC anti-CD11b (M1/70, Biolegend), allophycocyanin-conjugated anti-CD45 (30-F11) and with PE anti-IL-6 or PE anti-TNF-α (BD Pharmingen), All samples were measured on a BD FACSCanto II (BD Pharmingen™, Heidelberg, Germany) and evaluated with FlowJo software (Tree Star).
CD11b-HSVTK-mouse model with intraventricular application of ganciclovir
To evaluate the impact of microglia activation on secondary brain injury after experimental SAH, we used Nestin-GFP HSVTK-mice for our experiments. In these animals, Nestin-positive cells (e.g., neuronal precursors) are GFP-positive as well. To rule out a contribution of Nestin-positive cells to our cell counts of Iba-1- and NeuN-positive cells, unstained sections were evaluated for Nestin-GFP signal before further processing. No native Nestin-GFP signal was found in our mice.
CD11b-HSVTK mice have been described earlier [32]. In brief, CD11b-positive cells (e.g., monocytes, macrophages and microglia) harbor the HSVTK suicide gene, which is sensitive to ganciclovir. Application via an osmotic mini pump into the ventricular system targets intracerebral CD11b-positive cells (i.e., microglia) in the CNS predominantly similarly as described earlier [24, 69]. Experimental SAH was induced in CD11b-HSVTK mice as described above. Thereafter, animals were turned into a prone position, the scalp was incised, and a navigated burr hole craniotomy was established 0.8 mm parasagittal on the level of the bregma. Via this craniotomy the ventricular catheter of an osmotic mini pump (Alzet© Brain Infusion Kit, Charles River) filled with ganciclovir (2 mg/ml; 230 µl/pump) was introduced and connected proximally to the pump. Negative littermate CD11b-HSVTK mice served as controls.
After 9 days of infusion, the brains of these mice were harvested, cut in coronary slices (10 µm) and stained for Iba-1 and neuronal cell death, as described above.
Statistical analysis
Statistical analyses were performed using GraphPad PRISM (GraphPad Software, version 6.0). All data were analyzed by Student’s t test or ANOVA (as displayed in the respective figure legends) to detect statistical differences. All cell count values are displayed as means ± standard deviations. p values of p < 0.05 were considered statistically significant. For cytokine expression analysis (Fig. 5c, d), no suitable statistical comparison could be done. Although 12 hemispheres were analyzed, due to the pooling, only two values resulted per time point.
Results
Characterization of cellular inflammation in human brain specimens after SAH
Brain specimens of 21 patients were identified, that had died within the course of SAH, and that had neither suffered from CNS infection nor from cerebral vasospasm. Patient characteristics are displayed in Table 2. Cases with intraparenchymal bleeding and/or territorial infarctions were excluded. An illustrative case with severe SAH (Fisher 3) from an aneurysm of the tip of the basilar artery is depicted in Fig. 1a, b. Extraparenchymal bleeding was exclusively seen in the subarachnoid compartment at the brain base and surface of the investigated brain specimens (Fig. 1c). An intraparenchymal accumulation of microglia/macrophage cells was evident in the respective sections (Fig. 1d). This was even more pronounced in samples that were taken near the site of the ruptured aneurysm. Here, a significantly larger number of microglia/macrophage cells per high power field were documented between days 5 and 15 after the bleeding when compared to earlier time points (Fig. 1e). At later time points, a subsequent decrease of microglia/macrophages was seen, which was comparable to days 1–4. The peak of this cellular inflammatory reaction occurred in a phase, in which secondary brain injury typically takes place. The retrospective review of the patient data revealed an initially high mortality rate of 29 % within the first 4 days (Fig. 1e), representing the devastating course of early brain injury after severe SAH. Throughout the following period, the mortality decreased to 14 %, i.e., almost halving its value before it rose again to 33 % coinciding with the highest microglia/macrophage accumulation in the parenchyma, which led us to further investigate the observed phenomenon. Representative histological images for the microglia/macrophage accumulation are displayed in the addendum to Fig. 1g–j.


Human data. a Illustrative image of an intracranial aneurysm at the basilar tip (arrow tip), displayed through digital subtraction angiography. b The rupture of the aneurysm caused subarachnoid hemorrhage, displayed within the basal cisterns of the central nervous system on computed tomography. c The bleeding typically occurs outside the brain parenchyma, in the subarachnoid compartment (arrows, scalebar 100 µm)—hematoxylin and eosin staining. d Based on immunohistochemical stainings of brain specimen of patients who had died in the course of SAH, an accumulation of KiM1P-positive microglia/macrophage cells inside the brain parenchyma was found. This reaction was primarily seen in regions near the site of hemorrhage (illustrative image of a patient deceased 12 days after the bleeding; scalebar 100 µm). e The time course showed very limited appearance of KiM1P-positive cell accumulation until day 4 after the bleeding. A significant increase over days 5–8, and a climax between days 9 and 15 was documented. After day 16, a subsequent decline was found. ns not significant, **p < 0.01, ***p < 0.001 ANOVA. f The clinical course of the respective patients was retrospectively reviewed. In our patient collective the clinical course paralleled the occurrence of microglia cells. After an initial phase of early brain injury (day 1–4), in which the mortality rate was 29 % (6 patients), a lower mortality rate of only 14 % was documented until day 9 (3 patients). A second peak in mortality of 33 % was documented between days 9 and 15 (7 patients), the respective time interval, when microglia accumulation had been shown to be most pronounced. At later time points (after day 16), the mortality rate declined again to 24 % (5 patients). Figure 1 addendum: Representative histological stainings for KiM1P in sections of patients who had deceased within the respective time intervals, displayed in e and f (days 1–4, days 5–8, days 9–15, days 16 and following). The images show a significant increase in the number of microglia/macrophages in the brain tissue on days within these chosen time intervals. g 1 day, h 8 days, i 14 days, j 25 days after the hemorrhage
Characterization of cellular inflammation in murine brain specimens after experimental SAH
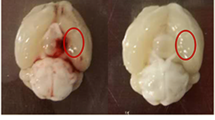
To induce SAH in mice, we used intravascular filament perforation that resulted exclusively in extraparenchymal bleeding. While the procedure of SAH induction in mice was highly standardized, the consequences including the clinical course were less predictable. Therefore, at the end of experiments all animals were thoroughly assessed; mice presenting with intracerebral hemorrhage were thus excluded from further studies. This type of evaluation consisted of a macroscopical analysis of bleeding into the subarachnoid space at the brain base at different time points (Fig. 2a) and a microscopic assessment of hematoxylin/eosin (HE)-stained brain sections at early time points (Fig. 2b). An iron stain proved the occurrence of SAH while excluding intraparenchymal bleeding, also at later stages (Fig. 2b, right).

Murine experiments. a Illustrative macroscopic findings in perfused brains. After sham operation, no subarachnoid blood could be detected around the brain (left). Early after induction of experimental SAH (day 2), a diffuse bleeding was detected in the subarachnoid compartment (middle), where it remained after washout by intra-arterial perfusion. CSF-circulation subsequently washed out blood in the subarachnoid space. No residual blood could be detected macroscopically at day 28 (right). b To prove the occurrence of subarachnoid hemorrhage, hematoxylin/eosin stainings were performed at day 2 after the bleeding (left, arrow pointing at SAH). To rule out parenchymal damage by ischemia or intraparenchymal bleeding, hematoxylin/eosin staining was performed at day 28 (middle, arrow pointing at site of former hemorrhage). To prove the occurrence of the hemorrhage at the end of the experiments, when no residual blood could be seen macroscopically, iron staining for detection of hemoglobin was done (right), showing hem residuals in the ambient cistern (arrows). Scalebars 500 µm. c Staining for Iba-1-positive cells in the brain parenchyma after experimental SAH showed the initiation of an early accumulation near the site of the induction of the hemorrhage (left). The highest accumulation of Iba-1-positive cells was seen on day 14 after the hemorrhage, surpassing basal ganglia, reaching the corpus callosum and even cortical brain areas (middle). At the end of the observation period, a less dense but global accumulation of Iba-1-positive cells was seen throughout both hemispheres (right). d Stainings for extracellular APP revealed an accumulation around the site of cellular accumulation near the brain base on day 4 (left). This accumulation culminated around day 14, showing massive extracellular APP accumulation, that paralleled the cellular reaction (middle). Towards day 28, less extracellular APP could be stained (right). Scalebars (lower magnification) = 500 µm, scalebars (higher magnification) = 100 µm, n = 6 per time point. e Quantification of Iba-1-positive cells per HPF (left), and evaluation of the total covered area of the respective brain sections by Iba-1-positive cells (middle) and extracellular APP (right). ANOVA: **p < 0.01, ***p < 0.001, ****p < 0.0001
A marked accumulation of Iba-1-positive cells was detected within the brain parenchyma, starting at the brain base around day 4 (Fig. 2c, left), suggesting an accumulation of immune cells of myeloid origin—i.e., resident microglia or peripherally derived macrophages. This intraparenchymal accumulation culminated into a massive reaction around day 14. It surpassed the basal ganglia, reached the corpus callosum and in some slices even subcortical and cortical areas (Fig. 2c, middle). This very dense and focal accumulation of Iba-1-positive cells seemed to fade into a significantly less dense, but more global spread of inflammatory cells until day 28 after the bleeding (Fig. 2c, right). Evaluation of the number of Iba-1-positive cells per HPF and the covered area showed a significant increase over time (Fig. 2e). Thus, SAH resulted in a centrifugal spreading of microglia/macrophage accumulation from the base to the cortex of both hemispheres, resembling a wave of intracerebral immune cell activation.
Neuro-axonal injury after experimental SAH
Coinciding with this cellular inflammatory response, we found signs for axonal injury, displayed by an intraparenchymal accumulation of extracellular amyloid precursor protein (APP). Interestingly, the APP accumulation followed the time course and spatial distribution pattern of the intracerebral immune cell activation (from the base to the cortex of both hemispheres with a peak at 14 days after SAH). The APP-covered area increased significantly over time (Fig. 2d, e).
We furthermore assessed whether neuronal cell death occurs and co-localized NeuN-immunoreactivity and TUNEL-positivity. Here, we identified a significantly higher activity of DNA fragmentation in neurons in the time course of the experiment (Fig. 3a). In cell counts, an increase of dying neurons could be detected from day 4 on. This became highly significant on day 14, before the number declined again towards day 28 after SAH (Fig. 3b). Correspondingly, the absolute number of vital neurons declined significantly until day 28 (Fig. 3c).

Neuronal apoptosis in mice after experimental subarachnoid hemorrhage. a Representative immunofluorescence staining of neurons (NeuN, green) and apoptosis (TUNEL, red). Nuclei were stained with DAPI (blue). An increased number of apoptotic neurons could be detected throughout the time-course. At day 4 (d4, upper), a small number of neurons were stained positive for TUNEL as well. At day 14 (d14, lower), a higher number of neurons also expressed TUNEL-positivity (scalebars = 100 µm). b In control animals, almost no apoptosis in neurons was detected. On subsequent days 4, 14 and 28, a significant increase in the number of apoptotic neurons could be found. c Accordingly, the total number of neurons decreased significantly over time, when compared to controls *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA, n = 6 per time point
Summarized, we confirmed neuro-axonal injury in the time-course of experimental SAH that closely followed the time course of microglia/macrophage accumulation in the brain tissue.
Accumulating Iba-1-positive cells originate from resident microglia pool
Next, we addressed the origin of the observed Iba-1-positive cells that accumulated in the brain parenchyma. To determine, whether our Iba-1-positive cells were resident microglia or were derived from the periphery, invading the brain parenchyma, possibly through a compromised blood brain barrier, we generated chimeric mice harboring green-fluorescent-protein (GFP) bone marrow. In these mice, peripheral cells including those of myeloid origin are GFP-positive, while radio-resistant resident microglia are GFP negative. Since there are some reports suggesting that whole body irradiation in the absence of head protection may influence the amount of peripherally derived myeloid cells [1, 34]—an issue which is still a matter of an ongoing debate in the field [22, 52]—we shielded the head (Fig. 4a). The reconstitution rates of the myeloid cell lineage in the blood after GFP–bone marrow transfer were up to 85 % (measured by the amount of GFP-positive cells in the blood of recipient mice) and thus comparable to the amount of GPF-positive cells upon total body irradiation without shielding the head [46]. Thus, a precise discrimination of infiltrating peripherally derived presumably GFP-positive macrophages from resident GFP-negative microglia in the brain was feasible. When analyzing the amount of Iba-1-positive and GFP-positive myeloid cells, we detected only few Iba-1-/GFP double-positive cells, while most of the iba-1-positive cells lacked a GFP signal, demonstrating that resident microglia but not peripherally derived myeloid cells are the source of the accumulating inflammatory cells (Fig. 4b, c).

Determination of the origin of Iba-1-positive cells on day 14 after experimental subarachnoid hemorrhage in mice. a Illustration of our chimeric mouse model for green fluorescent protein-labeled (GFP) peripheral leukocytes. Mice were lethally irradiated using a head-shielding device. Reconstitution was achieved by transplantation of GFP-positive bone marrow. Using this model, we were able to distinguish Iba-1-positive microglia within the brain parenchyma from monocytes, derived from the peripheral blood pool, and primary brain residential microglia. b Representative immunofluorescence staining at the brain base. The white interrupted line outlines the border between brain parenchyma and subarachnoid cistern. Inside the brain, an accumulation of Iba-1-positive cells (red) is seen. Even at higher magnification, only a few GFP-positive cells from the peripheral blood pool could be detected (green). Marginal amounts of double-positive cells (yellow) could be seen. Nucleus staining was done with DAPI (blue). Scalebars = 100 µm. c Cell count of Iba-1-positive cells and of Iba-1 GFP-double-positive cells per high power field. Iba-1 GFP-double-positive cells could hardly be found ***p < 0.001. t test, n = 6
Pro-inflammatory cytokine and receptor profiles of the microglia cells during SAH
Our experiments showed that resident microglia and not peripheral macrophages accumulate in the brain tissue after induction of SAH. Beside Iba-1, microglia also express the surface molecule CD11b. Therefore, we analyzed the proportion of microglia within brain homogenates of mice after SAH by CD11b+CD45+ expression via FACS. Here, we observed a slightly increased percentage of CD11b+CD45+ cells within the whole brain cell suspensions of animals on day 4 and 14 following SAH, while towards day 28 a decline was seen (Fig. 5a). Additionally, a co-staining with Iba-1 was performed to verify their factual CNS-intrinsic microglial origin (Fig. 5b). At all time points, the amount of Iba-1-positive cells retained the high levels of naïve mice.

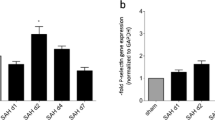
Analysis of the isolated microglia from the murine brains after experimental SAH. a The percentage of CD11b+CD45+ cells of brain homogenates increased slightly throughout the time course of the bleeding until day 14 while on day 28 the amount was diminished (FACS). b Representative Dot Plot of a brain cell suspension 14 days after SAH. Low numbers of Iba-1 negative cells were detected indicating almost no granulocytes within the analyzed CD11b+CD45+ cell fraction. Percentages of CD11b+CD45+Iba-1− cells at all time points were comparable to controls without SAH. c Relative expression values of the inflammatory cytokines in the isolated microglia followed a time course comparable to the cellular inflammation. In most of the cytokines, a marked increase (up to tenfold of baseline) could be documented, confirming a pro-inflammatory state of the accumulated microglia. d The respective cytokine receptors (except for Il-1r1) were up-regulated throughout the observational period. Statistical analysis not feasible (for details see “Materials and methods” section). e Although the percentage of Il-6+CD11b+ cells was low, a significant increase could be found towards day 14 after SAH **p < 0.01, ***p < 0.001. f Suggesting the microglia to be a source of Il-6-production, this finding could be supported by immunofluorescence stainings, showing Iba-1+Il-6+ cells (yellow) in murine brain sections, scalebars = 100 µm. g The percentage of CD11b+ CD45+ TNF-α+ cells was found to be significantly increased with a time course comparable to the cellular inflammation, suggesting the microglia to be a source of TNF-α-production. In addition, the amount of TNF-α per cell was significantly up-regulated, defined by the mean fluorescence intensity (mfi) *p < 0.05, **p < 0.01. ANOVA
In the next step, primary microglia were isolated by CD11b via MACS from brains of mice at various time points after SAH to investigate their genetic pro-inflammatory cytokine profile and the corresponding receptors. The analyzed cytokine genes Il6, Tnfα and Il1α/β can be up-regulated by activated macrophages. All investigated molecules showed an up-regulation within the microglia cell population after SAH induction. While the increase of Il6 and Il1α was only gradual during the course of disease, Tnfα exhibited high-level expression on day 14 only. Il1β was strongly up-regulated at day 4 after SAH and maintained a stable expression level over time (Fig. 5c). Therefore, we conclude that microglia were able to up-regulate pro-inflammatory cytokines in response to SAH.
In addition, the expression of the cytokine receptors was elevated except for Il1r1 that displayed no regulation. For Il1r2, the highest relative expression levels were seen on day 4. For Il6r and Tnfr the relative expression levels were constantly rising until day 28 (Fig. 5d). These results show a sustained expression of both, pro-inflammatory cytokines and their corresponding receptors, in resident microglia isolated upon SAH at various time points, thus verifying an inflammatory state of the cells.
Since we found Il6 and Tnfα-genes strongly regulated, we aimed at assessing the respective protein levels by means of a quantitative FACS-based analysis, which revealed an up-regulation also on protein level. A significant increase of IL6-producing microglia was found throughout the time course of the experiments until day 14 after SAH. At day 28, a decline was noted, but the percentage of IL-6-producing cells was still significantly higher than in controls (Fig. 5e). By immunohistochemistry we confirmed that Iba-1-positive cells were the source of IL-6 on day 14 (Fig. 5f). The percentage of cells producing IL-6 was low. Yet, no other source of IL-6-production could be identified. On the other hand, the percentage of TNF-α-producing microglia was much higher (around 30 %). The rise over time also showed significant values on day 14, similar to the course of RNA expression. More interestingly, also the expression per cell was significantly higher for TNF-α (Fig. 5g).
Attenuation of neuronal cell death after SAH by microglia depletion
Taken together we show that SAH in mice and humans leads to a secondary wave of intracerebral inflammation, mediated mainly via an accumulation of resident microglia cells, which coincided with a presumably secondary neuro-axonal injury. In order to further test the hypothesis, that the brain becomes a target of its own resident (microglia-based) immune system after SAH, i.e., that resident microglia mediate this neuro-axonal injury, we depleted microglia in vivo using CD11b HSVTK+/− mice [32]. Here, intraventricular infusion of ganciclovir, as also reported elsewhere [24, 69], leads to a substantial reduction of CD11b-positive cells (Fig. 6a), resulting in a decrease of microglia by 76 % compared to the CD11b HSVTK−/− littermate control animals (Fig. 6b).

Microglia induce neuronal apoptosis in experimental subarachnoid hemorrhage. a Illustration of the experimental protocol. Ganciclovir (GCV)-loaded minipumps were intraventricularly implanted to deplete microglia for 9 days in the animal model of Herpes simplex virus-thymidine kinase (TK+)—following SAH induction, as well as in their respective littermates (TK−). In the TK+ animals, all CD11b-positive cells harbor the thymidine kinase, which could be targeted by ganciclovir and leads to a depletion of the respective cells. Illustrative IF images show a significant reduction of Iba-1-positive cells in the depleted animals. b Through quantification of Iba-1-positive cells, a decline of about two-thirds could be documented at the time point of analysis. c Illustrative immunofluorescence staining of TK− (upper image) and TK+ animals (lower image). Scalebars = 100 µm. d In the non-depleted animals (TK−), a significant number of apoptotic neurons was detected on day 9 after induction of the hemorrhage, while in comparison, in TK+ animals the number of apoptotic neurons was significantly reduced. e Also the number of the survived neurons was significantly higher in TK+ (microglia-depleted) animals than in TK− (non-depleted) *p < 0.05, **p < 0.01, ***p < 0.001. t test, n = 4 per group
In order to address, whether microglia accumulation is cause or consequence of delayed neuronal injury after SAH, we found the number of dying neurons upon SAH in the microglia-depleted animals significantly reduced by two-thirds after SAH in comparison to their CD11b HSVTK−/− littermates with unaffected microglia (Fig. 6c, d). Accordingly, the number of residual, vital neurons was significantly higher in microglia-depleted animals (Fig. 6e).
Discussion
Recently, inflammatory processes have been discussed as a possible mechanism for brain injury after SAH [15, 25, 43, 59, 60, 66, 71]. Up to date, the mechanisms leading to secondary brain injury remained unclear. For the first time, we report an intracerebral immune cell accumulation following aneurysmal SAH in human brain specimens. To gain further insight into the underlying pathogenetic mechanisms, we established an experimental SAH model in mice, in which we confirmed our principle novel finding, that an arterial bleeding into the subarachnoid space at the base of the brain induces a wave of immune cells within the cerebral parenchyma. The major contributing immune cell was identified as resident microglia, the brain’s intrinsic innate immune cell. Following SAH, microglia produced a significantly increased amount of pro-inflammatory cytokines such as IL-6 and TNFα. Neuro-axonal injury coincided with the accumulation of activated microglia, reaching its maximum extent between day 7 and 14 after the bleeding, both in murine and in human samples.
Given that depletion of microglia in vivo led to an attenuation of neuronal cell death, microglia appear not only to be secondarily attracted to the site of injury, but do indeed support and exacerbate the SAH-related brain damage. Thus, our results show that a spreading microglia-mediated inflammation within the brain tissue is a significant contributor to secondary brain injury after SAH.
Many concepts of brain injury after SAH have been described before. According to the timing and occurrence of damage, they have been assigned to an early and delayed brain injury. In the early phase of SAH, brain injury has been discussed to take place via apoptotic pathways, induced by the sudden rise of intracranial pressure and the subsequent impairment of the cerebral perfusion. Several downstream pathways of this concept have been described within the past years [16, 27, 29, 47, 56, 60, 61, 71, 73].
Concerning the proposed mechanisms of delayed brain injury, the most established one is a hemodynamic compromise due to cerebral vasospasm [51]. For decades, a large number of scientists have put tremendous effort in the prevention of cerebral vasospasm. When, finally, a solution seemed to be found in the endothelin system, the clinical CONSCIOUS trials failed to improve patient outcome after SAH, despite the successful prevention of cerebral vasospasm [42], indicating that cerebral vasospasm is not the major contributor for delayed injury. Thus, other concepts have become more and more popular. For several years, the idea of cortical spreading depolarization has been revived [11, 19]. Spreading depolarization that migrates along the cerebral cortex is accompanied by decreased oxygen availability and linked to ischemic events, and is thus thought to subsidize CNS damage in the course of SAH.
Although systemic inflammatory reactions after SAH have been described earlier [18, 35], the concept of inflammatory changes in the CNS after SAH is rather novel and has been mostly applied to the perivascular and subarachnoid space [20, 53, 55], as well as to aneurysm wall weakening [13, 28]. Only very recently, the modulation of inflammatory processes was correlated with an impaired outcome after SAH [54, 62, 63]. By establishing a reliable mouse model of SAH, we could show an intracerebral microglia accumulation, starting at the site of vessel puncture, as it is similarly detectable in human samples of patients dying in the course of SAH. The maximum extent of the spreading microglia was accompanied by a marked increase in mortality rate of our patient cohort lacking vasospasm or cerebral infarctions 9–12 days after the bleeding. Thus, the phenomenon, which we would like to describe as spreading cerebral inflammation is not only an experimental phenomenon, but seems to be a clinically relevant contributor to secondary brain injury after SAH. Patients after SAH often show a delayed re-orientation phase, which has been widely explained by an initial rise in intracranial pressure and subsequent drop of the cerebral perfusion. A spreading cerebral inflammation might be another explanation for the compromised clinical state of patients after SAH, which often is not detectable by standard neuro-imaging methods.
Different subsets of macrophages have been described to contribute to inflammation in various CNS diseases. Beyond peripherally derived macrophages, perivascular, choroidal and meningeal macrophages have been described as independent entities, as reviewed by Bogie last year [10]. In our model of experimental SAH we do see perivascular and choroidal, as well as meningeal Iba1-positive cells. In general, these cells seem to be highly heterogeneous and only 50 % display microglia markers. Also their origin has only been clarified in EAE [10]. While in EAE, meningeal macrophages seem to be derived from the peripheral blood pool [44], after SAH, they did not display GFP-positivity, rather suggesting a non-peripheral origin. Nevertheless, stimulation or propagation of the intraparenchymal microglia accumulation might occur through these extraparenchymal macrophage subsets in SAH, as it has been proposed in EAE [5, 21].
The question why microglia should inflict additional damage to an already stressed brain remains to be answered. Not only being the major representative of the cerebral immune system, but also being sort of a caretaker of the CNS, microglia have been shown to play an important role in homeostasis between synaptogenesis and developmental apoptosis. Yet, the mechanisms underlying their beneficial versus detrimental dialectic have not been fully discovered. Several proteins (e.g., IL-6, TNF-α, BDNF, KARAP/DAP12) have been hypothesized to unbalance microglial equilibrium to one or the other side [9]. Thus, activation-dependent change of microglial functions may also include insufficient production of trophic factors typically produced by microglia, possibly mimicking conditions of a microglia-specific lack of brain-derived neurotrophic factor (BDNF), which recently was shown to be key for important physiological brain functions in learning and memory by promoting learning-related synapse formation [49]. At the same time, as described for other CNS diseases, prolonged and sustained activation of microglia may result in a negative-feedback loop ultimately resulting in an overshoot-type of microglial reaction similarly as described in animal models of Alzheimer’s disease, where prolonged innate immune activation including the sustained expression of pro-inflammatory cytokines such as interleukin (IL)-12 and IL-23 exacerbates pathology [8, 31, 45, 72]. Only recently, depletion of microglia in experimental SAH led to a beneficial effect on cerebral vasospasm and hippocampal neuronal survival, supposing a rather detrimental effect of microglia in the setting of SAH [26].
Along this line it is of note that the gene expression profile in our isolated microglia upon SAH suggests a pro-inflammatory state. Here, we focused on cytokines that are known for effects on immunomodulation and apoptotic pathways. When characterizing the inflammatory potential of the microglia, we found not only an increase in inflammatory cytokines, but also an up-regulation of corresponding cytokine receptors indicative for a disposition to react to respective stimuli—possibly even in a paracrine–autocrine fashion, as we did not find other sources of Il-6- or Tnf-α-production apart from microglia. The percentage of microglia expressing Tnf-α was high (app. 30 %), while the percentage of microglia that expressed Il-6 was much lower. The roles of these two cytokines in inflammation and apoptosis seem to be variable. Both proteins can induce inflammatory and detrimental effects in brain cells [2, 3, 50, 74]. Nevertheless, both have also been shown to improve neuronal survival under certain circumstances [33, 39, 65]. In previous studies, we had found IL-6 to be up-regulated in the CSF as well as in brain tissue in humans after SAH with a significant correlation to the severity of the disease course [59]. Herein, we identify resident microglia as cellular source for this previously documented increase of IL-6. Since depletion of the microglia as the source of both cytokines led to an attenuation of neuronal cell death, it appears that in the course of SAH IL-6 and TNF-α, at least when expressed over time, may rather play a harmful role. While we cannot exclude that other cytokines and/or immune-relevant factors might also be crucially involved, it is also obvious that the when and where of IL-6 and TNF-α actions are crucial determinants how they impact the disease course.
Many models of experimental SAH for different species have been described. The two most commonly used ones are the blood injection model and the endovascular perforation model [67]. In the injection model, a predefined amount of autologous blood is injected under constant pressure settings into the cerebello-pontine cistern via a burr hole craniotomy. The advantage in that model is that by variability of the blood amount and pressure, the severity of the hemorrhage can be controlled and varied. In the model of endovascular perforation used by us, the extent of the hemorrhage is not as high, leading to a low number of lethal events in the short-term post-operative phase. Yet, a craniotomy is known to induce inflammatory changes in the brain by the procedure itself [37]. Furthermore, the endovascular perforation technique provokes the hemorrhage at the site, where the SAH occurs in humans as well. Furthermore, the actual rupture of a vessel might also play a role in the patho-mechanisms [30]. For these reasons, the model of endovascular perforation—for our questions—offered a closer correlation to SAH in humans.
In our experiments, the microglia activation started unilaterally, near the site of vessel puncture, before it spread throughout both hemispheres. An appropriate explanation for this phenomenon might also be a direct mechanism—e.g., mediated chemically through hemoglobin metabolites or physically through pressure changes—which inflicts the intraparenchymal microglia accumulation. Another possible explanation for asymmetric microglia accumulation after SAH has been discussed by Czosnyka and co-workers, who argue for an asymmetry of blood flow auto-regulation after SAH [12].
Interestingly, in the long-term clinical course after SAH, patients often show some degree of neuro-psychological deficits, subtle mental impairment [7], or even global brain atrophy, as well as distinct hippocampal volume loss [6, 7]. While these findings might be explained by our results demonstrating neuronal loss in the course of SAH, further studies are required to dissect the molecular underpinnings of this phenomenon including the analysis if and to what extent compensatory mechanisms counteracting this neuro-degenerative phenomenon may take place.
In conclusion, our data demonstrate that intraparenchymal activation of accumulated resident microglia follows SAH in humans and in mice, which is characterized by a pro-inflammatory cytokine profile. Accumulation of resident microglia was found to be accountable for neuronal cell death, delineating a new mechanism of delayed brain injury after SAH and offering novel approaches and interventional strategies to treat long-term effects of SAH. In summary our results consolidate the theory of neuro-inflammation after SAH.
References
Ajami B, Bennett JL, Krieger C et al (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14:1142–1149. doi:10.1038/nn.2887
Aktas O, Smorodchenko A, Brocke S et al (2005) Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 46:421–432. doi:10.1016/j.neuron.2005.03.018
Aktas O, Ullrich O, Infante-Duarte C et al (2007) Neuronal damage in brain inflammation. Arch Neurol 64:185–189. doi:10.1001/archneur.64.2.185
Aungst SL, Kabadi SV, Thompson SM et al (2014) Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab 34:1223–1232. doi:10.1038/jcbfm.2014.75
Bauer J, Huitinga I, Zhao W et al (1995) The role of macrophages, perivascular cells, and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia 15:437–446. doi:10.1002/glia.440150407
Bendel P, Koivisto T, Hänninen T et al (2006) Subarachnoid hemorrhage is followed by temporomesial volume loss: MRI volumetric study. Neurology 67:575–582. doi:10.1212/01.wnl.0000230221.95670.bf
Bendel P, Koivisto T, Niskanen E et al (2009) Brain atrophy and neuropsychological outcome after treatment of ruptured anterior cerebral artery aneurysms: a voxel-based morphometric study. Neuroradiology 51:711–722. doi:10.1007/s00234-009-0552-5
Berg Vom J, Prokop S, Miller KR et al (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat Med 18:1812–1819. doi:10.1038/nm.2965
Bessis A, Béchade C, Bernard D, Roumier A (2007) Microglial control of neuronal death and synaptic properties. Glia 55:233–238. doi:10.1002/glia.20459
Bogie JFJ, Stinissen P, Hendriks JJA (2014) Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol 128:191–213. doi:10.1007/s00401-014-1310-2
Bosche B, Graf R, Ernestus R-I et al (2010) Recurrent spreading depolarizations after subarachnoid hemorrhage decreases oxygen availability in human cerebral cortex. Ann Neurol 67:607–617. doi:10.1002/ana.21943
Budohoski KP, Czosnyka M, Smielewski P et al (2013) Cerebral autoregulation after subarachnoid hemorrhage: comparison of three methods. J Cereb Blood Flow Metab 33:449–456. doi:10.1038/jcbfm.2012.189
Chalouhi N, Ali MS, Jabbour PM et al (2012) Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab 32:1659–1676. doi:10.1038/jcbfm.2012.84
Clark JF, Loftspring M, Wurster WL, Pyne-Geithman GJ (2008) Chemical and biochemical oxidations in spinal fluid after subarachnoid hemorrhage. Front Biosci 13:1806–1812
Clark SR, McMahon CJ, Gueorguieva I et al (2008) Interleukin-1 receptor antagonist penetrates human brain at experimentally therapeutic concentrations. J Cereb Blood Flow Metab 28:387–394. doi:10.1038/sj.jcbfm.9600537
Chou SH-Y, Feske SK, Atherton J et al (2012) Early elevation of serum tumor necrosis factor-α is associated with poor outcome in subarachnoid hemorrhage. J Investig Med 60:1054–1058
Dengler J, Schefold JC, Graetz D et al (2008) Point-of-care testing for interleukin-6 in cerebro spinal fluid (CSF) after subarachnoid haemorrhage. Med Sci Monit 14:BR265–8
Dhar R, Diringer MN (2008) The burden of the systemic inflammatory response predicts vasospasm and outcome after subarachnoid hemorrhage. Neurocrit Care 8:404–412. doi:10.1007/s12028-008-9054-2
Dreier JP, Major S, Manning A et al (2009) Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 132:1866–1881. doi:10.1093/brain/awp102
Dumont AS, Dumont RJ, Chow MM et al (2003) Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery 53:123–133 (discussion 133–135)
Fabriek BO, Van Haastert ES, Galea I et al (2005) CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia 51:297–305. doi:10.1002/glia.20208
Goldmann T, Wieghofer P, Müller PF et al (2013) A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16:1618–1626. doi:10.1038/nn.3531
Graetz D, Nagel A, Schlenk F et al (2010) High ICP as trigger of proinflammatory IL-6 cytokine activation in aneurysmal subarachnoid hemorrhage. Neurol Res 32:728–735. doi:10.1179/016164109X12464612122650
Grathwohl SA, Kälin RE, Bolmont T et al (2009) Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci 12:1361–1363. doi:10.1038/nn.2432
Greenhalgh AD, Brough D, Robinson EM et al (2012) Interleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathology. Dis Model Mech 5:823–833. doi:10.1242/dmm.008557
Hanafy KA (2013) The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J Neuroinflammation 10:83. doi:10.1186/1742-2094-10-83
Hansen-Schwartz J, Vajkoczy P, Macdonald RL et al (2007) Cerebral vasospasm: looking beyond vasoconstriction. Trends Pharmacol Sci 28:252–256. doi:10.1016/j.tips.2007.04.002
Hasan DM, Chalouhi N, Jabbour P et al (2013) Evidence that acetylsalicylic acid attenuates inflammation in the walls of human cerebral aneurysms: preliminary results. J Am Heart Assoc 2:e000019. doi:10.1161/JAHA.112.000019
Hasegawa Y, Suzuki H, Sozen T et al (2011) Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl 110:43–48. doi:10.1007/978-3-7091-0353-1_8
Hashimoto T, Meng H, Young WL (2006) Intracranial aneurysms: links among inflammation, hemodynamics and vascular remodeling. Neurol Res 28:372–380. doi:10.1179/016164106X14973
Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477. doi:10.1038/nri3705
Heppner FL, Greter M, Marino D et al (2005) Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 11:146–152. doi:10.1038/nm1177
Hoffmann O, Priller J, Prozorovski T et al (2007) TRAIL limits excessive host immune responses in bacterial meningitis. J Clin Invest 117:2004–2013. doi:10.1172/JCI30356
Johansson CB, Youssef S, Koleckar K et al (2008) Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nat Cell Biol 10:575–583. doi:10.1038/ncb1720
Kasius KM, Frijns CJM, Algra A, Rinkel GJE (2010) Association of platelet and leukocyte counts with delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage. Cerebrovasc Dis 29:576–583. doi:10.1159/000306645
Kierdorf K, Erny D, Goldmann T et al (2013) Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 16:273–280. doi:10.1038/nn.3318
Kozai TDY, Vazquez AL, Weaver CL et al (2012) In vivo two-photon microscopy reveals immediate microglial reaction to implantation of microelectrode through extension of processes. J Neural Eng 9:066001. doi:10.1088/1741-2560/9/6/066001
Krabbe G, Halle A, Matyash V et al (2013) Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 8:e60921. doi:10.1371/journal.pone.0060921
Leibinger M, Müller A, Gobrecht P et al (2013) Interleukin-6 contributes to CNS axon regeneration upon inflammatory stimulation. Cell Death Dis 4:e609. doi:10.1038/cddis.2013.126
Macdonald RL (2014) Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol 10:44–58. doi:10.1038/nrneurol.2013.246
Macdonald RL, Higashida RT, Keller E et al (2010) Preventing vasospasm improves outcome after aneurysmal subarachnoid hemorrhage: rationale and design of CONSCIOUS-2 and CONSCIOUS-3 trials. Neurocrit Care 13:416–424. doi:10.1007/s12028-010-9433-3
Macdonald RL, Kassell NF, Mayer S et al (2008) Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke 39:3015–3021. doi:10.1161/STROKEAHA.108.519942
Mashaly HA, Provencio JJ (2008) Inflammation as a link between brain injury and heart damage: the model of subarachnoid hemorrhage. Cleve Clin J Med 75(suppl 2):S26–S30
McMenamin PG, Wealthall RJ, Deverall M et al (2003) Macrophages and dendritic cells in the rat meninges and choroid plexus: three-dimensional localisation by environmental scanning electron microscopy and confocal microscopy. Cell Tissue Res 313:259–269. doi:10.1007/s00441-003-0779-0
Michaud J-P, Rivest S (2015) Anti-inflammatory signaling in microglia exacerbates Alzheimer’s disease-related pathology. Neuron 85:450–452. doi:10.1016/j.neuron.2015.01.021
Müller A, Brandenburg S, Turkowski K et al (2014) Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int J Cancer. doi:10.1002/ijc.29379
Ostrowski RP, Colohan AR, Zhang JH (2006) Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res 28:399–414. doi:10.1179/016164106X115008
Park S, Yamaguchi M, Zhou C et al (2004) Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke 35:2412–2417. doi:10.1161/01.STR.0000141162.29864.e9
Parkhurst CN, Yang G, Ninan I et al (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–1609. doi:10.1016/j.cell.2013.11.030
Penkowa M, Molinero A, Carrasco J, Hidalgo J (2001) Interleukin-6 deficiency reduces the brain inflammatory response and increases oxidative stress and neurodegeneration after kainic acid-induced seizures. Neuroscience 102:805–818
Pluta RM, Hansen-Schwartz J, Dreier J et al (2009) Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res 31:151–158. doi:10.1179/174313209X393564
Prinz M, Priller J, Sisodia SS, Ransohoff RM (2011) Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14:1227–1235. doi:10.1038/nn.2923
Provencio JJ (2013) Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: a review. Acta Neurochir Suppl 115:233–238. doi:10.1007/978-3-7091-1192-5_42
Provencio JJ, Altay T, Smithason S et al (2011) Depletion of Ly6G/C(+) cells ameliorates delayed cerebral vasospasm in subarachnoid hemorrhage. J Neuroimmunol 232:94–100. doi:10.1016/j.jneuroim.2010.10.016
Provencio JJ, Fu X, Siu A et al (2010) CSF neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit Care 12:244–251. doi:10.1007/s12028-009-9308-7
Prunell GF, Mathiesen T, Diemer NH, Svendgaard N-A (2003) Experimental subarachnoid hemorrhage: subarachnoid blood volume, mortality rate, neuronal death, cerebral blood flow, and perfusion pressure in three different rat models. Neurosurgery 52:165–175 (discussion 175–176)
Ransohoff RM, Cardona AE (2010) The myeloid cells of the central nervous system parenchyma. Nature 468:253–262. doi:10.1038/nature09615
Sakai H, Horinouchi H, Tomiyama K et al (2001) Hemoglobin-vesicles as oxygen carriers: influence on phagocytic activity and histopathological changes in reticuloendothelial system. Am J Pathol 159:1079–1088. doi:10.1016/S0002-9440(10)61783-X
Sarrafzadeh A, Schlenk F, Gericke C, Vajkoczy P (2010) Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocrit Care 13:339–346. doi:10.1007/s12028-010-9432-4
Schneider UC, Schiffler J, Hakiy N et al (2012) Functional analysis of pro-inflammatory properties within the cerebrospinal fluid after subarachnoid hemorrhage in vivo and in vitro. J Neuroinflammation 9:28. doi:10.1186/1742-2094-9-28
Schubert GA, Thomé C (2008) Cerebral blood flow changes in acute subarachnoid hemorrhage. Front Biosci 13:1594–1603
Smithason S, Moore SK, Provencio JJ (2012) Systemic administration of LPS worsens delayed deterioration associated with vasospasm after subarachnoid hemorrhage through a myeloid cell-dependent mechanism. Neurocrit Care 16:327–334. doi:10.1007/s12028-011-9651-3
Smithason S, Moore SK, Provencio JJ (2013) Low-dose lipopolysaccharide injection prior to subarachnoid hemorrhage modulates Delayed Deterioration associated with vasospasm in subarachnoid hemorrhage. Acta Neurochir Suppl 115:253–258. doi:10.1007/978-3-7091-1192-5_45
Stein SC, Browne KD, Chen X-H et al (2006) Thromboembolism and delayed cerebral ischemia after subarachnoid hemorrhage: an autopsy study. Neurosurgery 59:781–787. doi:10.1227/01.NEU.0000227519.27569.45 (discussion 787–788)
Tan Y, Uchida K, Nakajima H et al (2013) Blockade of interleukin 6 signaling improves the survival rate of transplanted bone marrow stromal cells and increases locomotor function in mice with spinal cord injury. J Neuropathol Exp Neurol 72:980–993. doi:10.1097/NEN.0b013e3182a79de9
Tiebosch IACW, Dijkhuizen RM, Cobelens PM et al (2013) Effect of interferon-β on neuroinflammation, brain injury and neurological outcome after experimental subarachnoid hemorrhage. Neurocrit Care 18:96–105. doi:10.1007/s12028-012-9692-2
Titova E, Ostrowski RP, Zhang JH, Tang J (2009) Experimental models of subarachnoid hemorrhage for studies of cerebral vasospasm. Neurol Res 31:568–581. doi:10.1179/174313209X382412
Tulamo R, Frösen J, Junnikkala S et al (2006) Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery 59:1069–1076. doi:10.1227/01.NEU.0000245598.84698.26 (discussion 1076–1077)
Varvel NH, Grathwohl SA, Baumann F et al (2012) Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci USA 109:18150–18155. doi:10.1073/pnas.1210150109
Xie X, Wu X, Cui J et al (2013) Increase ICAM-1 and LFA-1 expression by cerebrospinal fluid of subarachnoid hemorrhage patients: involvement of TNF-α. Brain Res 1512:89–96. doi:10.1016/j.brainres.2013.03.041
You W-C, Wang C-X, Pan Y-X et al (2013) Activation of nuclear factor-κB in the brain after experimental subarachnoid hemorrhage and its potential role in delayed brain injury. PLoS ONE 8:e60290. doi:10.1371/journal.pone.0060290
Zhang B, Gaiteri C, Bodea L-G et al (2013) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153:707–720. doi:10.1016/j.cell.2013.03.030
Zhou Y, Martin RD, Zhang JH (2011) Advances in experimental subarachnoid hemorrhage. Acta Neurochir Suppl 110:15–21. doi:10.1007/978-3-7091-0353-1_3
Zipp F, Aktas O (2006) The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci 29:518–527. doi:10.1016/j.tins.2006.07.006
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (SFB TRR 43 and NeuroCure Exc 257 to FLH and PV, as well as HE 3130/6-1 to FLH) and the Federal Ministry of Education and Research (DLR/BMBF; Kompetenznetz Degenerative Demenzen to FLH). We thank Dr. Jana Glumm and Dr. Kelly Miller for kindly providing GFP-mice and CD11b-HSVTK mice, respectively. We thank Melina Nieminen-Kelhä and Irina Kremenetskaia for their constant technical support in the lab. We thank Adnan Ghori for advice in PCR issues.
Conflict of interest
The authors have declared that no conflict of interest exists.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Schneider, U.C., Davids, AM., Brandenburg, S. et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol 130, 215–231 (2015). https://doi.org/10.1007/s00401-015-1440-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1440-1