
Abstract
Viral neuroinvasion is a critical step in the pathogenesis of viral encephalitis. Multiple mechanisms of neuroinvasion have been identified, but their relative contribution to central nervous system (CNS) infection remains unclear for many viruses. In this study, we examined neuroinvasion of the mosquito-borne bunyavirus La Crosse (LACV), the leading cause of pediatric viral encephalitis in the USA. We found that the olfactory bulb (OB) and tract were the initial areas of CNS virus infection in mice. Removal of the OB reduced the incidence of LACV-induced disease demonstrating the importance of this area to neuroinvasion. However, we determined that infection of the OB was not due to axonal transport of virus from olfactory sensory neurons as ablation of these cells did not affect viral pathogenesis. Instead, we found that OB capillaries were compromised allowing leakage of virus-sized particles into the brain. Analysis of OB capillaries demonstrated specific alterations in cytoskeletal and Rho GTPase protein expression not observed in capillaries from other brain areas such as the cortex where leakage did not occur. Collectively, these findings indicate that LACV neuroinvasion occurs through hematogenous spread in specific brain regions where capillaries are prone to virus-induced activation such as the OB. Capillaries in these areas may be “hot spots” that are more susceptible to neuroinvasion not only for LACV, but other neurovirulent viruses as well.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The central nervous system (CNS) is protected from invading pathogens by the blood–brain barrier (BBB), which is formed by brain capillary endothelial cell (BCEC) tight junctions and reinforced by perivascular astrocytic endfeet [1, 32]. Neurotropic encephalitic viruses have evolved several mechanisms for neuroinvasion that circumvent this barrier. Many viruses, including herpes simplex, polio and rabies, spread to the CNS via anterograde transport along peripheral sensory neurons [38]. Olfactory sensory neurons (OSNs) which reside in the epithelium of the nasal turbinates may also contribute to this form of neuroninvasion [48] and can be infected by certain neurotropic viruses [21, 47]. Additionally, some viruses such as West Nile and HIV can enter the CNS by virus-induced recruitment and transmigration of infected immune cells across a compromised BBB [28]. Furthermore, the activation of BCECs cells by virus-induced innate immune signals modulates BBB permeability and can promote viral invasion directly across endothelial surfaces in the CNS [8, 30]. It has been proposed that some viruses may utilize multiple neuroinvasive mechanisms and enter the CNS both through infected OSNs and through compromised brain capillaries [37, 39]. However, the relative contribution of these mechanisms to peripheral virus neuroinvasion has yet to be determined and a direct comparison of their putative role in this process would further our understanding of encephalitic virus pathogenesis.
To examine mechanisms of neuroinvasion, we utilized a mouse model of La Crosse (LACV) infection. LACV is a neuroinvasive mosquito-borne bunyavirus of the California encephalitis serogroup that infects humans when transmitted during a blood meal. While LACV infection rarely causes neurological disease in adults [40], it is the leading cause of pediatric viral encephalitis in the United States [14, 15]. LACV neuroinvasion in humans and animal models results in the infection, dysfunction and death of neurons [3, 27, 29, 33]. Virus-induced brain pathology can lead to severe neurological disease with symptoms such as seizures, coma and paralysis [27]. In rare cases, LACV-induced neurological disease is lethal particularly in younger patients. The ability of LACV to invade the CNS may be one of the limiting steps of LACV disease pathogenesis and may be predictive of clinical outcome. However, the mechanisms or restrictions that influence the ability of LACV to enter the brain are largely unknown.
The established murine model of LACV infection is ideal for studying neuroinvasion and neuropathogenesis as it recapitulates the age-dependent susceptibility observed in humans [3, 19, 20, 43, 44]. Adult mice are resistant, while weanlings are susceptible to neurological disease following peripheral inoculation. Adult animals effectively clear LACV via innate immune mechanisms during peripheral infection [44]. However, adults develop disease when LACV is administered intracerebrally (i.c.) demonstrating that LACV retains neurovirulence but is not neuroinvasive in adults [19]. Therefore, comparison of these routes in adult and young animals may provide mechanistic insight into neuroinvasion and disease pathogenesis.
The mechanisms of LACV entry into the CNS are not clear. It has been suggested that hematogenous spread and/or anterograde transport from the nasal turbinates may be involved. Peripherally inoculated weanling mice develop viremia prior to disease [3, 20] suggesting a hematogenous route. However, LACV is found in the nasal turbinates of intraperitoneally (i.p.) inoculated weanling mice early during infection suggesting that peripherally virus might be able to infect OSNs and enter the CNS from this site [3]. Furthermore, LACV is pathogenic if given intranasally (i.n.) in weanlings indicating that nasal turbinate infection may lead to neuroinvasion [3].
In this study, LACV neuroinvasion was examined by comparing two routes of virus inoculation, direct (i.n.) and peripheral (i.p.). Our findings indicated that retrograde transport of virus along OSNs was possible, but did not occur following i.p. inoculation. Instead, peripheral inoculation of LACV induced vascular leakage in specific regions of the brain, including the olfactory bulb (OB), anterior olfactory nucleus (AON) and lateral olfactory tract (OT) that coincided with the first areas of virus infection in the brain. Furthermore, LACV-induced vascular leak at this early time point was sufficient to allow virus-sized particles to invade the brain. Proteomic analysis of OB brain capillaries demonstrated specific changes in proteins involved in cytoskeletal rearrangement suggesting a possible mechanism of virus-induced tight junctional disruption of the BBB. Collectively, this study demonstrates that LACV selectively utilizes a hematogenous route of neuroinvasion during peripheral infection that is localized to specific susceptible brain regions such as the OB.
Methods
Infection of mice with LACV
All animal studies were conducted under animal protocol RML2013-062 with strict adherence to the Principles of Laboratory Animal Care and in accordance and approval by the NIH/NIAID/RML Institutional Animal Care and Use Committee. OMP-YFP and C57BL/6 mice obtained from Jackson Laboratories were maintained in a breeding colony at RML. LACV human 1978 stock was a kind gift from Dr. Richard Bennett and has previously been described [3]. Mice at 3 (weanling) or 6–8 (adult) weeks of age were inoculated with 103 or 105 PFU of LACV in phosphate-buffered saline (PBS) in a volume of 200 µl/mouse i.p. or 10 µl/mouse i.n. Mice were observed daily for signs of neurological disease characterized by hunched posture, seizures, reluctance or inability to move normally, or paralysis. Animals with clear clinical signs of neurological disease were scored as clinical and euthanized immediately.
Immunohistochemistry
At set time points, mice were perfused transcardially with heparin saline (100 U/ml) followed by 10 % neutral buffer formalin. Whole brain or decalcified whole skulls were serially sectioned at 5 μm. Sections were blocked (5 % BSA, 0.05 % Triton in PBS) at room temperature (rt) for 1 h. Primary antibodies against NeuN (1:100, Millipore), LACV (1:1000, gifted by Dr. Robert Tesh), GFP (1:1000, Ab cam) and/or CD31 (1:200, Ab cam) were incubated overnight at 4 °C in PBS. Secondary antibodies (donkey anti-goat AF488, goat anti-mouse AF594 or donkey anti-rabbit AF594, Invitrogen) were incubated for 1 h at rt. Slides were cover slipped with Prolong Gold mounting media containing DAPI and imaged using (1) a Zeiss 710 LSM (Carl Zeiss) with a Plan Apochromat 63X oil immersion objective (NA 1.40) with a pinhole of 90 and a 0.5 or 0.25 µm step controlled by Zen software, (2) an Aperio ScanScope FL (fluorescent) slide scanner (Leica Biosystems) with a UPLSAPO 20X objective (NA 0.75) controlled by ScanScope software, (3) an Aperio XT (bright field) slide scanner with the same objective also controlled by ScanScope software, or (4) an epifluorescent Nikon Eclipse 55i clinical microscope (Nikon) with a Plan Fluor 40X objective (NA 0.75) mounted with a Nikon DS-Ri1 digital camera operated by NIS Elementals v3.2 software. Representative images were exported to TIFF format for subsequent processing included Huygens deconvolution (Figs. 4, 6) with Scientific Volume Imaging (SVI) software and orthogonal reconstruction (Figs. 4a, b, 6c, d) in Imaris v7.7.4. All figures were built using Canvas 14 (ACD Systems).
Chemical depletion of OSNs
Dichlobenil (2,6 Dichlorobenzonitrile) was dissolved in cell culture grade dimethyl sulfoxide (DMSO). Dichlobenil was administered 3 days prior to LACV infection at 50 mg/kg i.p. at 100 µl/mouse [5]. Control mice were given 100 µl DMSO i.p.
Plasma and cell transfers
Blood and spleen tissues were removed from C57BL/6 mice infected with 103 PFU LACV, i.p. at 2 dpi. Blood was spun at 5000 g for 10 min to isolate plasma. Plasma was stored on ice. The remaining pellet was suspended in 70 % percoll/PBS and underlayed on a 0–30 % step percoll gradient which was centrifuged at 1500g for 20 min at 4 °C. Peripheral blood mononuclear cells (PBMCs) were recovered at the 30–70 % interface, rinsed in PBS, and resuspended at 5 × 107 cells/ml in phosphate-buffered balanced salt solution (PBBS) on ice. Spleens were homogenized through a 70 µm filter and fractionated using the same percoll gradient protocol, and then resuspended at 5 × 108 cells/ml in PBBS on ice. 100 µl of cell suspension or neat plasma was given intravenously (i.v.) to individual recipient mice.
Flow cytometry
Immune cells isolated from homogenized whole brain [6], spleen or blood as described above were processed for flow cytometry as previously described [44] with the exception that the cells were also stained with LACV primary antibody conjugated to Pacific Blue™ dye using an APEX® Antibody labeling kit (Life Technologies).
Real-time PCR
Immune cells isolated from homogenized whole brain [6] were processed for quantitative Real-time PCR as previously described [44].
Measuring BBB permeability
Mice were given Evan Blue dye (200 μl of 20 mg/ml i.v.) in PBS at 1–5 dpi or 2000 kd fluorescein–dextran (200 ul of 200 mg/ml i.v.) in PBS at 3 dpi. 30 min later, mice were perfused transcardially with 5 ml of heparinized saline (100 U/ml). Brains were immediately removed and placed on ice in a sealed tube. Dextran-treated mice were used for immunohistochemistry, as described above. Brains for Evans Blue dye studies were homogenized in N, N-dimethylformamide, spun down and 200 μl of the resulting clarified supernatants measured at 620 nm in triplicate on a Biotek Synergy 4 plate reader in a flat bottom 96-well plate. A standard curve of 620 nm absorbance of known Evans Blue dye concentrations in N, N-dimethylformamdie was obtained to determine sample concentration by linear regression.
Bulbectomy surgery
Mice were anesthetized with inhaled isoflurane, placed in stereotactic device and had their heads shaved and sterilized. A retracted midline incision in the skin exposed the skull above the OB. Bilateral holes were drilled through the skull above each lobe and the entire bulb was removed using a sterile pipette tip connected to a vacuum. The skin was closed by sutures and mice were placed in heated cages and monitored until awake and active. Ketoprofen (5 mg/kg) was administered i.p. pre-operatively. Buprenorphine (0.1 mg/kg) was administered i.p. pre-operatively and post-operatively every 8–12 h for 2 days. Ceftriaxone (30–50 mg/kg) was administered subcutaneously pre-operatively and again post-operatively every 24 h for 2 days to prevent infection. Sham surgery mice received the same treatment with the exception that a sterile needle was used to scratch the skull over the OB (no holes were drilled) prior to suturing the skin.
BCEC isolation, 2D-DIGE protein expression profiling and mass spectrometry
Primary BCEC microvessels were obtained from LACV and mock-infected mice at 3 dpi according to a previously published protocol [49] with the exception that the OB and tract were processed separately from cortices. Isolation of microvessels was confirmed by microscopic analysis. Isolated microvessels were solubilized in 2-D cell lysis buffer (30 mM Tris–HCl, pH 8.8, 7 M urea, 2 M thiourea, 4 % CHAPS), frozen in liquid nitrogen and sent to Applied Biomics (Hayward, CA, USA) for CyDye labeling and two-dimensional difference gel electrophoresis (2D-DIGE) analysis. Briefly, 30 µg of protein lysate sample was labeled with 1 µl of CyDye (containing Cy2, Cy3 or Cy5 minimal dyes, diluted 1:5 with DMF) for 30 min on ice. 250 µl of sample was placed in a 13 cm immobilized pH gradient strip (Amersham BioSciences) and isoelectric focusing was run in the dark at 20 °C. Samples were transferred into a 12 % SDS-Gel and run at 15 °C until the dye front ran off the gel. Gel image scans were done using a Typhoon TRIO (GE Healthcare) following the manufacture’s protocol. The scanned images were analyzed by Image QuantTL software (GE Healthcare), and subjected to in-gel analysis and cross-gel analysis using DeCyder software version 6.5 (GE Healthcare). Protein expression fold change was determined by comparison of volume ratios of spots from LACV-OB vs mock-OB and LACV-OB vs LACV-cortex using in-gel DeCyder analysis software (Supplemental Fig. 1). Similar expression changes were observed in two sample protein preparations obtained independently. 29 spots with greater than 1.5-fold change were captured by an Ettan Spot Picker (GE Healthcare), digested in-gel (Trypsin Gold, Promega) and desalted by Zip-tip C18 (Millipore). MALDI-TOF (MS) and TOF/TOF (tandem MS/MS) were performed on a 5800 mass spectrometer (AB Sciex). Resulting peptide mass and associated fragmentation spectra were searched by MASCOT in NCBInr or Swiss Protein databases. Candidates with either protein score C.I. % or Ion C.I. % greater than 95 were considered significant. Proteomic and signaling pathway analysis of mass spectrometry identified proteins was performed using Ingenuity Pathway analysis (IPA, Qiagen).
Results
LACV infectivity is contained in plasma
LACV is naturally transmitted by a mosquito during a blood meal indicating infection originates in the periphery. Weanling mice inoculated i.p. have infectious virus in their blood suggesting that blood or blood cells may be possible sources of viral transmission to the CNS [3]. To determine if blood-borne virus alone or circulating blood cells were capable of inducing neurological disease, we transferred plasma, PBMCs or splenocytes from i.p. infected weanling mice at 2 dpi to naïve age-matched hosts. All mice receiving plasma developed neurological disease within 5–7 days (Fig. 1), while mice receiving PBMCs or splenocytes did not. Immunohistochemical analysis of clinical animals confirmed virus-infected neurons in these mice [29] (data not shown). Flow cytometric, qRT-PCR and immunohistochemical analysis of PBMCs and splenocytes revealed no viral antigen or RNA in these cells [44] (data not shown) confirming they are non-infectious. Therefore, blood-borne leukocytes do not appear to be responsible for neuroinvasion. Instead, free virus in the blood is necessary for viral entry into the CNS.

Mice infected i.p. with LACV develop transferable levels of infectious viremia. Neat plasma, peripheral blood mononuclear cells (PBMCs) and splenocytes were taken at 2 dpi from donor weanling mice inoculated i.p. with 103 particle forming units (PFU) LACV. Sample fractions [5 × 106 PBMCs (filled square), 50 × 106 splenocytes (filled circle) or neat plasma (filled triangle)] were independently transferred to a single naïve recipient mouse i.v. in a 200 µl volume. Mice were then monitored for the development of terminal neurological disease. Groups were compared using a Log-rank test (n = 6 mice per group, ***p < 0.005)
LACV infection causes significant CNS vascular leak
Hematogenous neuroinvasion would likely require BBB breakdown and increased vascular leak during a peripheral viral infection. To determine if i.p. infection of LACV induced BBB breakdown, Evans Blue dye leakage into the CNS was measured at 1–5 dpi (Fig. 2a). Leakage was significantly higher in brain tissue of LACV-infected animals compared to mock-infected controls at 3 and 5 dpi (Fig. 2a). Gross histological examination of CNS tissue from mock-infected animals demonstrated slight Evans Blue staining in the OB relative to the rest of the brain. In comparison, LACV induced visually stronger Evans Blue staining in the OB at 3 dpi (Fig. 2b upper, white arrow) which spread to multiple areas by 5 dpi (Fig. 2b lower, white arrows). These data indicate that LACV infection induces BBB breakdown and vascular leakage. Furthermore, leakage in the OB occurs as early as 3 dpi, suggesting that this may be a region susceptible to viral neuroinvasion.

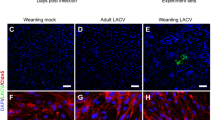
Peripheral LACV infection induces progressive BBB breakdown and neuronal infection starting in the OB. Weanling mice inoculated i.p. with 103 PFU LACV were injected i.v. with Evans Blue dye and their brains assayed for dye leakage at 620 nm absorbance (a). Mean ± standard error of the means is plotted and a one-way ANOVA with Bonferonni’s multiple comparisons was run to compare significance between days (n = 6 mice per time point, **p < 0.01) (b). Gross morphological images of brains from 3 dpi mock (b, top left) and LACV (b, top right) mice injected with Evans Blue demonstrate the initial site of BBB leakage is the OB (b, top white arrow). Similar images from 5 dpi brains demonstrate that BBB leakage has spread to other regions of the brain including the cortex (b, bottom white arrows). Representative immunohistochemical staining of LACV (Alexa Fluor 594, pseudo-colored white) and DAPI (blue) in the brains of 3 dpi (c) and 6 dpi (d) C57/B6 mice inoculated i.p with 103 PFU of LACV are shown. Virus was found earliest in the olfactory bulb (OB), lateral olfactory tract (OT), anterior olfactory nucleus (AON) (c, red arrows) and thalamus (not shown). Inset in (c) is a high magnification image of an adjacent section labeled for a neuronal marker (NeuN, red), LACV (green) and DAPI (blue) that demonstrates infection solely of neurons. By 6 dpi, virus had spread to the majority of the brain; however, staining remains in areas of early infection (n = 8–12 animals per time point)
The OB is an early site of LACV CNS infection
The leakage in the OB suggests that this region may be a primary site of neuroinvasion. Therefore, we analyzed brain tissues from i.p. infected mice at 1–6 dpi for virus by immunohistochemistry. The earliest detection of virus in the CNS was in neurons at 3 dpi in the OB, AON and lateral OT (Fig. 2c, red arrows and inset). Infection was also observed in the thalamus at 3 dpi in some samples (data not shown). By 6 dpi, when mice showed signs of neurological disease, LACV was distributed throughout the CNS (Fig. 2d). Areas of neuronal infection included the cerebellum, pons, midbrain, thalamus, hypothalamus, striatum, hippocampus and cortex as well as increased infection in the OB, AON and lateral OT (Fig. 2d). Importantly, LACV did not appear to infect brain capillary endothelial cells (BCECs) (Supplemental Fig. 1), suggesting that the virus induces vascular leak through an indirect mechanism. Thus, LACV-induced vascular breakdown and virus infection in the CNS occur in very specific regions during the early stages of virus infection. These primary areas are the OB, AON and lateral OT.
Altered expression of cytoskeletal proteins in OB BCECs following LACV infection
Recent studies associated changes in GTPase signaling within BCECs with virus trafficking [8]. To determine if BCECs from the OB had specific responses to LACV infection, a proteomic analysis of BCECs following LACV infection was conducted. Specifically, OB BCECs from LACV-infected mice at 3 dpi were compared to (1) cortical BCECs removed from the same infected animal and (2) OB BCECs from mock-infected mice. Cortical BCECs were used for comparison as early virus infection was not observed in this region (Fig. 2c). Proteins from purified BCECs were analyzed by 2D DIGE and identified by mass spectrometry. 29 proteins that differed by greater than 1.5-fold between either comparison group were identified (Table 2 and Supplemental Table 1). IPA analysis of these proteins identified significant overlap with three canonical signaling pathways associated with cytoskeletal rearrangement and plasma membrane plasticity (Table 1). Seven of these proteins were involved in actin dynamics and microtubule function (Table 2 and Supplemental Fig. 2) [41]. The direction of the fold change was consistent for both comparison groups (Table 2). Thus, LACV infection induces distinct molecular changes associated with cytoskeletal rearrangement and Rho Family GTPase signaling molecules in OB but not cortical BCECs (Table 2 and Supplemental Fig. 2). These findings suggest that OB BCECs may be particularly susceptible to BBB leakage due to virus-induced changes in cytoskeletal signaling protein expression.
LACV-induced capillary leakage in the OB is sufficient to allow virus-sized particles into the brain
To determine if LACV-induced BBB leakage was sufficient to allow virus-sized particles into the CNS, 2000 kd FITC–dextran was injected i.v. into LACV-infected mice at 3 dpi. This FITC–dextran is similar in hydrodynamic radius to an LACV virion [24, 42]. In mock-infected animals, FITC–dextran was found only within blood vessel (Fig. 3a). In contrast, FITC–dextran was found within the perivascular and parenchymal space adjacent to blood vessels in LACV-infected animals (Fig. 3b). FITC–dextran leakage was only found in the OB, OT and thalamus of infected animals at 3 dpi (Fig. 3c, data not shown). These data correlate with cytoskeletal changes in the OB BCECs at this time point and suggest that BCECs in these regions are responsible for allowing LACV to invade the CNS.

Specific brain regions are susceptible to virus-sized particle leakage at 3 dpi. 2000 kd Dextran conjugated to FITC was injected i.v. into 3 dpi weanling mice inoculated i.p. with mock (a) or 103 PFUs of LACV (b, c). Dextran leakage across brain capillaries was assayed using immunohistochemistry where vessels are labeled with CD31 (Alexa Fluor 594, red), cell nuclei by DAPI (blue) and the Dextan-FITC infiltrate is green. Representative 15 µm z-stacks of OB capillaries from mock (a) and LACV (c) infected mice were obtained using a Zeiss LSM 710 confocal microscope. Sites of dextran-FITC (green) leakage was found exclusively in LACV infected mice, primarily in their OB (c, red box enlarged in b and white arrow). c Representative wide-field image from a 3 dpi LACV-infected mouse OB (n = 6 mice). Scale bars represent 20 µm (a, b) and 1 mm (c)
Removing the OB improves survival following LACV infection
To examine the contribution of BCECs within the OB to viral neuroinvasion, the OB was surgically removed (BulbX) from mice prior to infection. BulbX resulted in the complete removal of the OB, and the BCECs contained therein, as evidenced by the absence of glomerular organization (red arrow) in the OB region (Fig. 4b vs a). Vascular leak was examined by Evans Blue dye leakage at 3 days post-surgery and was equivalent between BulbX and sham-treated mice, demonstrating that surgery did not compromise the BBB (data not shown). BulbX mice had increased survival following LACV infection compared to sham controls suggesting removal of the OB impaired neuroinvasion (Fig. 4c). However, most animals developed neurological disease suggesting BCECs in other areas of the brain, possibly the lateral OT, AON and thalamus, also allowed virus invasion. Indeed, increasing the virus dose 100-fold resulted in all animals developing disease (Fig. 4d). Thus, while not the only site of virus entry, the OB has a significant impact on LACV neuroinvasion and resulting neurological disease.

Removing the OB and the capillaries contained within increased survival of i.p. inoculated mice. Representative bright field images of the OB region of weanling mice that underwent sham (a) or bulbectomy (b) surgery are shown. The OB, whose glomerular structure is indicated by the red arrow (a), was completely removed in bulbectomy, but not sham mice. Three days post-surgery, sham (filled circle) and BulbX (filled square) mice were infected i.p. with 103 (c) or 105 (d) PFUs of LACV. Animals were monitored for neurological symptoms until 20 dpi and resulting survival curves compared using a Log-rank test (n = 10–15 mice per group, *p < 0.05)
Peripheral inoculation of LACV does not result in OSN infection
Peripheral i.p. inoculation of LACV induces infectious viremia (Fig. 1) and alters the expression of proteins involved in cytoskeletal signaling in BCECs (Table 2, Supplemental Fig. 2) in brain regions where early virus infection (Fig. 2c) and virus-sized particle leakage are observed (Fig. 3b, c). These findings demonstrate the virus utilizes a hematogenous route of neuroinvasion. However, LACV is also present in the nasal turbinates of i.p. inoculated weanling mice at 2–3 dpi [3], suggesting that LACV could also invade the CNS along OSN pathways following peripheral infection. Therefore, we analyzed whether i.p. infection resulted in infection of OSNs. As a positive control, we utilized a direct i.n. inoculation route that results in neurological disease [3]. Weanling mice infected i.n. developed neurological disease with similar speed and frequency as i.p. inoculated mice (Supplemental Fig. 3a). Interestingly, adult mice, which are more resistant to LACV-induced neurological disease following i.p. infection than weanling mice, were highly susceptible to i.n. infection (Supplemental Fig. 3b). Therefore, we analyzed OSN infection in both weanling and adult mice for both the i.p. and i.n. routes of inoculation. In weanling mice inoculated i.n., virus-infected cells were grouped together in clusters within the neuronal layer of the nasal epithelium and had the morphological appearance of OSNs including projections into the nasal cavity (Fig. 5a, white arrow). In contrast, OSNs were not infected in i.p. inoculated weanling mice (Fig. 5b), but rather other nasal epithelial cells that could include immature epithelial support cells or basal cells (Fig. 5b, white arrows).

OSNs are infected during i.n. but not i.p. inoculation. Weanling C57/BL6 mice were infected either i.n. (a) or i.p. (b) with 103 PFU of LACV and immunohistochemistry staining for virus (Alexa Fluor 594, pseudo-colored magenta) and DAPI (blue) of their olfactory epithelium was performed. Representative 15 µm confocal z-stacks taken as described in “Methods” are shown demonstrating the distinct cell morphologies infected by LACV with either i.n. or i.p. inoculation. The white arrow in a indicates a representative OSN sensory projection into the nasal mucosa. White arrows in b indicate infected non-OSN epithelial cells. Adult OMP-YFP mice were also infected i.n. (c) or i.p. (d) with 103 PFU LACV. Representative 1.75 µm confocal z-stacks of olfactory epithelium immunohistochemically labeled for virus (Alexa Fluor 594, pseudo-colored magenta), YFP-positive OSNs (Alexa Fluor 488, green) and DAPI (blue) are shown. Thick white arrows in c indicate infected OSNs that are confirmed in the orthogonal view. The thin white arrow in c shows LACV in OSN projections to the CNS. Chevrons in c and d indicate infected non-OSN epithelial cells. Scale bars represent 10 µm
To verify that the infected cells following i.p. inoculation were not OSNs, we repeated these experiments in adult transgenic mice with yellow-fluorescent protein (YFP)-tagged olfactory marker protein (OMP), expressed exclusively by OSNs. Virus was found in the cell bodies and projections of YFP+ OSNs as well as in other non-OSN epithelial cells of i.n. inoculated adult mice (Fig. 5c). By comparison, i.p. inoculated adults showed infection of non-OSNs only (Fig. 5d). These findings are consistent with those of the weanling mice. Therefore, LACV infects OSNs in both adult and weanling mice only following direct i.n. and not peripheral i.p. infection.
OSNs are not necessary for LACV-induced neurological disease following i.p. infection
To confirm that OSNs did not contribute to LACV neuroinvasion, we chemically ablated these cells using dichlobenil (DCB) [5]. Three days following a single injection of DCB in weanling and adult mice, the majority of the olfactory neuroepithelium was destroyed along with most OSNs (Fig. 6a vs b). DCB treated adult and weanling mice inoculated i.n. with LACV were more resistance to neuroinvasion compared to DMSO-treated controls (Fig. 6c, d). Similarly, BulbX treatment of adult and weanling mice infected i.n. resulted in inhibition of disease (Supplemental Fig. 4a, b). Thus, direct i.n. inoculation of LACV required OSN connections to the OB for neuroinvasion to occur.

Depleting OSNs impairs neuroinvasion in adult and weanling i.n. but not i.p. inoculated mice. a, b Naïve OMP-YFP mice were treated i.p. with (a) DMSO or (b) 50 mg/kg dichlobenil (DCB) and their olfactory epitheliums were labeled for YFP (Alexa Fluor 488, green) and DAPI (blue). Wide-field representative images were obtained using an Aperio Scanscope FL slide scanner (“Methods”). 40X epifluorescent insets were taken on Nikon Eclipse 55i clinical microscope (“Methods”). DCB-treated mice contained few OSNs as demonstrated by lack of YFP (green) positive cells and the reduced thickness of the nasal epithelium (inset b, epithelial boundary outlined by white arrowheads) while DMSO-treated controls had intact nasal epithelium (inset a). Adult (c) and weanling (d, e) mice were treated with DCB (filled square) to ablate OSNs or DMSO (vehicle, filled circle) and all groups were subsequently infected with 103 PFU LACV either i.n. or i.p as indicated. Resulting survival curves were compared using a Log-rank test (n = 7–13 mice per group, **p < 0.01, ***p < 0.005)
In contrast to the requirement of OSNs for virus neuroinvasion during i.n. inoculation, i.p. infected DBC-treated weanling mice developed neurological disease similarly to DMSO-treated controls (Fig. 6e). Thus, neuroinvasion along OSNs was not necessary to develop neurological disease following i.p. infection. This indicates that the decreased disease observed by removal of the OB (Fig. 4a) was due to removal of the BCECs within the OB and not because of the severed connection between the OB and OSNs in the nasal epithelium. These data, along with the data showing vascular leak of the olfactory BCECs (Fig. 3b, c), the early infection in the OB by LACV (Fig. 2c) and the molecular changes in the OB BCECs (Table 2), indicate that BCECs in the OB become permeable to virus neuroinvasion following peripheral LACV infection and are a primary area of LACV neuroinvasion into the CNS.
Discussion
Neuroinvasion is a necessary step in the induction of neurological disease by neurotropic virus. Understanding the mechanisms of this process is critical to studying pathogenesis. Here, we show that during a peripheral LACV inoculation, the initial sites of CNS infection are areas of the brain associated with olfaction, such as the OB, lateral OT and AON. Although LACV neuroinvasion through OSNs pathways has been suggested, LACV only infected OSNs following direct i.n. inoculation, and not i.p. infection. Instead, free virus in the blood plasma, but not blood cells, was sufficient for neuroinvasion. Specifically, we found that LACV induced BBB breakdown allowing neuroinvasion of virus-sized particles into brain regions correlating with sites of early brain infection, particularly the OB. Surgical removal of the OB significantly reduced the incidence of neurological disease. Thus, brain capillaries in this region were more susceptible to virus-induced leakage and may be areas to focus studies of virus-induced BBB breakdown.
Ex vivo analysis of OB BCECs demonstrated altered expression of proteins involved in cytoskeletal rearrangement, plasma membrane morphology and tight junctional organization. Of the identified proteins, Stathmin 1 (STMN1) inhibits microtubule formation, Rho GDP-dissociation inhibitor (ARHGDIB) blocks GDP removal from Rho proteins and CAPZ (CAPZA1) regulates the growth of both actin filaments and microtubules [7, 31, 46]. Additionally, coronin actin-binding protein 1A (CORO1A) is involved in both invagination and protrusions of plasma membranes [12]. Considering these changes together, alterations of normal BCECs (Fig. 7a) could affect the stability of BBB tight junctions by active cytoskeletal rearrangement (Fig. 7b), resulting in barrier disruption and infectious virus entering the CNS.

Diagram of potential changes in cytoskeletal and cell morphological structure in BCECs that may allow for viral neuroinvasion. a Normal BCECs contain multiple components that are involved in maintaining cellular morphology and possibly tightly interactions. b Proteins that were upregulated or downregulated during infection are associated with microtubule dynamics and actin growth and stability as well as membrane protrusions and invaginations. Collectively, these changes suggest a mechanism for alteration of BBB integrity allowing for viral neuroinvasion. Proteins identified by 2D-DIGE gel analysis and mass spectrometry are labeled and fold expression changes are indicated by arrows (red decreased, green increased)
Cytoskeletal rearrangement and blood vessel permeability in the early stages of LACV infection may be initiated by proinflammatory cytokines. The primary protein changes in OB BCECs were associated with Rho GTPases and actin cytoskeletal signaling (Table 1). Similarly, recent studies of WNV stimulation of BCEC cultures altered RhoA/Rac-GTPase activation [8]. These GTPases are implicated in cytoskeletal rearrangement and cell migration [23, 50]. WNV-induced cytokine responses altered the cellular membrane distribution of B CEC-specific tight junctional proteins Claudin-5 and Occludin-1 [8] suggesting that virus induced inflammatory responses mediated BBB tight junctional integrity. Similarly, cytokines induced by LACV infection may affect the Rho GTPase and actin cytoskeleton signaling pathways found in this study. Since these changes were observed in OB BCECS, but not in cortical BCECs from LACV-infected mice (Table 2), the former may be more sensitive to cytokine-induced cytoskeletal rearrangement resulting in their increased permeability.
Differences in peripheral cytokine responses may also influence susceptibility to neuroinvasion. Peripheral infection by WNV induces expression of type 1 interferons (IFN) as well as the proinflammatory cytokines TNF and IL-1β by CNS BCECs [8]. Type I IFN can prevent T cells infiltration across the BBB [22] and promote endothelial stability following virus infection [8, 26]. In contrast, TNF and IL-1β destabilize the BBB in multiple disease models involving CNS vascular breakdown and can enhance WNV neuroinvasion [8, 9, 34, 45]. LACV infection also induces the upregulation of multiple cytokines [44]. However, one of the distinctions between susceptible 3 week old mice and resistant 6 week old mice is the presence of a strong type I IFN response in adult mice [44]. This IFN response may help maintain the integrity of the BBB in adult animals by preventing vascular leak and inhibiting neuroinvasion. In contrast, the poor type I IFN response in 3 week old mice may contribute to the blood vessel permeability found in this study (Figs. 2, 3). The decrease in vascular permeability at 4 dpi (Fig. 2a) may be due to an initial burst of type I IFNs by brain cells as they recognize virus infection. Further investigation into BBB permeability in response to both peripheral and CNS cytokines during virus infection is necessary to fully piece together how virus infection influences the BBB.
The OB in humans is volumetrically smaller than in mice when compared as a relative ratio to the rest of the brain. However, virus infection has been observed in the OB and OSNs in human patients. For example, human herpes virus 6 was found at higher levels in the OB and olfactory tract than in other regions of the brain [16]. Despite these findings, it may be difficult to determine in human patients if capillaries in the OB or olfactory tract are more prone to vascular leak during virus infection or whether viruses primarily infect the OB by transport along OSNs. Similar to our mouse studies, virus neuroinvasion may be possible through both routes. The actual route contributing to neuroinvasion will likely depend on the site of virus replication. Infections within the respiratory system with easy access to OSNs will likely invade along OSNs while blood-borne viruses may preferentially enter via weakened blood vessels.
Although OSNs did not contribute to neuroinvasion following peripheral LACV infection (Fig. 6e), neuroinvasion along OSN projections is thought to be utilized by several neurotropic viruses [2, 11, 39]. The present study demonstrates that LACV utilizes transport along OSN pathways when administered i.n. (Fig. 6c, d). Specifically, adult mice, which are resistant to peripheral LACV infection, are highly susceptible when virus is administered through the nose (Supplemental Fig. 3b). Interestingly, OSNs are only infected when virus is administered by this route and not by i.p. infection (Fig. 5). This finding is novel in that it clearly establishes neuroinvasion occurring through distinct and exclusionary mechanisms depending on the route of inoculation. Furthermore, since the molecular mechanisms of OSN anterograde transport remain poorly understood [48], i.n. inoculation of LACV in adult mice may be an ideal model for studying viral OSN anterograde neuroinvasion.
OSNs were not infected in either adult or weanling mice when LACV was administered i.p. despite infection of other cells in the olfactory epithelium (Fig. 5b, d). However, OSNs were susceptible to infection when virus was applied directly to the olfactory epithelial surface (Fig. 5a, c). This observation suggests that the apical surface of OSNs, which reside at the epithelial surface near the mucus of the nasal turbinate, may express a LACV-specific receptor that facilitates infection. Additionally, LACV-infected OSNs were clustered together in small aggregates rather than being evenly dispersed throughout the epithelium (data not shown). This aggregation could be related to focal settling of virus to a small area of epithelium in which the covering mucus turned over slowly [17]. However, because LACV administered i.n. was diluted in an aqueous solution that bathes the entire nasal epithelium, it is possible that a large area of epithelium is exposed to virus and does not become infected. This would suggest that only a subset of OSNs is susceptible to LACV infection. As OSNs express only a single odorant receptor and these are clustered together zonally [35], it possible that OSN susceptibility to LACV may be related to odorant receptor expression or other proteins that are specifically expressed on the epithelial surface of these cells.
Hematogenous spread of LACV could be explained by direct LACV infection of BCECs, which would then allow virus entry into the CNS. Human cytomegalovirus and Hepatitis C virus directly infect BCECs and increase permeability of endothelial tight junctions [4, 13]. Others such as HIV and Japanese encephalitis virus utilize either clathrin-dependent or -independent transcytosis mechanism to cross the BBB [10, 25]. In one study, immunohistochemistry analysis of LACV-infected suckling mice detected LACV within brain capillaries [20] suggesting that these cells may be capable of transmitting virus to the CNS. However, another study did not observe endothelial cell infection [3]. Our immunohistochemical studies found virus only within neurons at any time and purified BCECs were not positive for virus (Fig. 3 and Supplemental Fig. 1). Furthermore, mass spectrometry comparison of OB BCECs from LACV-infected mice did not identify any viral proteins or any proteins that were only detectable in LACV-infected cells (Table 2, data not shown). These data suggest that LACV is not harbored by endothelial cells. Finally, LACV is internalized via a clathrin-mediated mechanism [18] while transcytosis through BCECs typically utilizes a caveolin-1 mediated mechanism [36]. Thus, LACV likely does not cross the BBB by mechanisms involving transcytosis or direct endothelial cell infection.
One final mechanism of virus entry is the Trojan horse method, where an infected immune cell during normal neurosurveillance is the initial source of neuroinvasive virus. Leukocyte infiltration into the CNS during LACV infection is observed in both mice and humans [3, 27]. However, using immunohistochemistry and flow cytometry, we did not observe cells in the CNS, other than neurons infected by LACV (data not shown). Also, immune cells isolated from the brains of infected mice were negative for LACV by both flow cytometry and real-time PCR analysis (data not shown). Furthermore, transfer studies of either splenocytes or blood cells from LACV-infected mice to naïve controls did not result in virus infection in recipient mice (Fig. 1). Thus, although we cannot completely rule out a Trojan horse method, it is unlikely that LACV spreads to the CNS via this mechanism. Instead, the ability of free virus from plasma to induce neurological disease (Fig. 1) and the breakdown of the BBB to allow the crossing of virus-sized particles into the CNS (Fig. 4) indicate that the most likely mechanism for neuroinvasion is hematogenous spread across capillaries in susceptible areas such as the OB where BCECs are activated and undergoing cytoskeletal rearrangement.
References
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7:41–53
Allavena RE, Desai B, Goodwin D, Khodai T, Bright H (2011) Pathologic and virologic characterization of neuroinvasion by HSV-2 in a mouse encephalitis model. J Neuropathol Exp Neurol 70:724–734
Bennett RS, Cress CM, Ward JM, Firestone CY, Murphy BR, Whitehead SS (2008) La Crosse virus infectivity, pathogenesis, and immunogenicity in mice and monkeys. Virol J 5:25
Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD (2006) Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol 80:11539–11555
Brandt I, Brittebo EB, Feil VJ, Bakke JE (1990) Irreversible binding and toxicity of the herbicide dichlobenil (2,6-dichlorobenzonitrile) in the olfactory mucosa of mice. Toxicol Appl Pharmacol 103:491–501
Butchi NB, Woods T, Du M, Morgan TW, Peterson KE (2011) TLR7 and TLR9 trigger distinct neuroinflammatory responses in the CNS. Am J Pathol 179:783–794
Curmi PA, Andersen SS, Lachkar S, Gavet O, Karsenti E, Knossow M, Sobel A (1997) The stathmin/tubulin interaction in vitro. J Biol Chem 272:25029–25036
Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS (2014) Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. MBio 5:e01476–e014714
de Vries HE, Blom-Roosemalen MC, Van OM, de Boer AG, van Berkel TJ, Breimer DD, Kuiper J (1996) The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol 64:37–43
Dohgu S, Ryerse JS, Robinson SM, Banks WA (2012) Human immunodeficiency virus-1 uses the mannose-6-phosphate receptor to cross the blood-brain barrier. PLoS ONE 7:e39565
Dups J, Middleton D, Yamada M, Monaghan P, Long F, Robinson R, Marsh GA, Wang LF (2012) A new model for Hendra virus encephalitis in the mouse. PLoS ONE 7:e40308
Ferrari G, Langen H, Naito M, Pieters J (1999) A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell 97:435–447
Fletcher NF, Wilson GK, Murray J, Hu K, Lewis A, Reynolds GM, Stamataki Z, Meredith LW, Rowe IA, Luo G, Lopez-Ramirez MA, Baumert TF, Weksler B, Couraud PO, Kim KS, Romero IA, Jopling C, Morgello S, Balfe P, McKeating JA (2012) Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology 142:634–643
Gaensbauer JT, Lindsey NP, Messacar K, Staples JE, Fischer M (2014) Neuroinvasive arboviral disease in the United States: 2003 to 2012. Pediatrics 134:e642–e650
Haddow AD, Odoi A (2009) The incidence risk, clustering, and clinical presentation of La Crosse virus infections in the eastern United States, 2003–2007. PLoS ONE 4:e6145
Harberts E, Yao K, Wohler JE, Maric D, Ohayon J, Henkin R, Jacobson S (2011) Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc Natl Acad Sci USA 108:13734–13739
Harkema JR, Carey SA, Wagner JG (2006) The nose revisited: a brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol Pathol 34:252–269
Hollidge BS, Nedelsky NB, Salzano MV, Fraser JW, Gonzalez-Scarano F, Soldan SS (2012) Orthobunyavirus entry into neurons and other mammalian cells occurs via clathrin-mediated endocytosis and requires trafficking into early endosomes. J Virol 86:7988–8001
Janssen R, Gonzalez-Scarano F, Nathanson N (1984) Mechanisms of bunyavirus virulence. Comparative pathogenesis of a virulent strain of La Crosse and an avirulent strain of Tahyna virus. Lab Invest 50:447–455
Johnson RT (1983) Pathogenesis of La Crosse virus in mice. Prog Clin Biol Res 123:139–144
Kalinke U, Bechmann I, Detje CN (2011) Host strategies against virus entry via the olfactory system. Virulence 2:367–370
Kraus J, Oschmann P (2006) The impact of interferon-beta treatment on the blood-brain barrier. Drug Discov Today 11:755–762
Lamalice L, Le BF, Huot J (2007) Endothelial cell migration during angiogenesis. Circ Res 100:782–794
Lebrun L, Junter GA (1994) Diffusion of dextran through microporous membrane filters. J Membrane Sciences 88:253–261
Liou ML, Hsu CY (1998) Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res 293:389–394
Liu P, Woda M, Ennis FA, Libraty DH (2009) Dengue virus infection differentially regulates endothelial barrier function over time through type I interferon effects. J Infect Dis 200:191–201
McJunkin JE, de los Reyes EC, Irazuzta JE, Caceres MJ, Khan RR, Minnich LL, Fu KD, Lovett GD, Tsai T, Thompson A (2001) La Crosse encephalitis in children. N Engl J Med 344:801–807
Miller F, Afonso PV, Gessain A, Ceccaldi PE (2012) Blood-brain barrier and retroviral infections. Virulence 3:222–229
Mukherjee P, Woods TA, Moore RA, Peterson KE (2013) Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity 38:705–716
Neal JW (2014) Flaviviruses are neurotropic, but how do they invade the CNS? J Infect 69:203–215
Olofsson B (1999) Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal 11:545–554
Pate M, Damarla V, Chi DS, Negi S, Krishnaswamy G (2010) Endothelial cell biology: role in the inflammatory response. Adv Clin Chem 52:109–130
Pekosz A, Phillips J, Pleasure D, Merry D, Gonzalez-Scarano F (1996) Induction of apoptosis by La Crosse virus infection and role of neuronal differentiation and human bcl-2 expression in its prevention. J Virol 70:5329–5335
Phares TW, Kean RB, Mikheeva T, Hooper DC (2006) Regional differences in blood-brain barrier permeability changes and inflammation in the apathogenic clearance of virus from the central nervous system. J Immunol 176:7666–7675
Ressler KJ, Sullivan SL, Buck LB (1993) A zonal organization of odorant receptor gene expression in the olfactory epithelium. Cell 73:597–609
Rippe B, Rosengren BI, Carlsson O, Venturoli D (2002) Transendothelial transport: the vesicle controversy. J Vasc Res 39:375–390
Roe K, Kumar M, Lum S, Orillo B, Nerurkar VR, Verma S (2012) West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J Gen Virol 93:1193–1203
Salinas S, Schiavo G, Kremer EJ (2010) A hitchhiker’s guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol 8:645–655
Schafer A, Brooke CB, Whitmore AC, Johnston RE (2011) The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J Virol 85:10682–10690
Sotir MJ, Glaser LC, Fox PE, Doering M, Geske DA, Warshauer DM, Davis JP (2007) Endemic human mosquito-borne disease in Wisconsin residents, 2002-2006. WMJ 106:185–190
Stamatovic SM, Keep RF, Andjelkovic AV (2008) Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol 6:179–192
Talmon Y, Prasad BV, Clerx JP, Wang GJ, Chiu W, Hewlett MJ (1987) Electron microscopy of vitrified-hydrated La Crosse virus. J Virol 61:2319–2321
Taylor KG, Peterson KE (2014) Innate immune response to La Crosse virus infection. J Neurovirol 20:150–156
Taylor KG, Woods TA, Winkler CW, Carmody AB, Peterson KE (2014) Age-dependent myeloid dendritic cell responses mediate resistance to la crosse virus-induced neurological disease. J Virol 88:11070–11079
Tsao N, Hsu HP, Wu CM, Liu CC, Lei HY (2001) Tumour necrosis factor-alpha causes an increase in blood-brain barrier permeability during sepsis. J Med Microbiol 50:812–821
Tuszynski JA, Brown JA, Sept D (2003) Models of the collective behavior of proteins in cells: tubulin, actin and motor proteins. J Biol Phys 29:401–428
van den Pol AN, Ding S, Robek MD (2014) Long-distance interferon signaling within the brain blocks virus spread. J Virol 88:3695–3704
Van RD, Verdijk R, Kuiken T (2015) The olfactory nerve: a shortcut for influenza and other viral diseases into the central nervous system. J Pathol 235:277–287
Winkler CW, Foster SC, Matsumoto SG, Preston MA, Xing R, Bebo BF, Banine F, Berny-Lang MA, Itakura A, McCarty OJ, Sherman LS (2012) Hyaluronan anchored to activated CD44 on central nervous system vascular endothelial cells promotes lymphocyte extravasation in experimental autoimmune encephalomyelitis. J Biol Chem 287:33237–33251
Wojciak-Stothard B, Tsang LY, Paleolog E, Hall SM, Haworth SG (2006) Rac1 and RhoA as regulators of endothelial phenotype and barrier function in hypoxia-induced neonatal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 290:L1173–L1182
Acknowledgments
This study was performed at Rocky Mountain Laboratories (RML) and funded by the Division of Intramural Research (DIR), as part of the Nation Institute of Allergy and Infectious Disease (NIAID) within the National Institutes of Health (NIH). We thank Suzette A. Priola, Byron Caughey, Lara M. Myers, Sonja M. Best, Roger A. Moore, Burhan A. Khan, Tyson A. Woods and Paul F. Policastro for critical reading of the manuscript. Also, we thank Dan Long, Vinod Nair, Nancy Kurtz and Aaron B. Carmody for technical assistance with experiments. Figure preparation and image presentation assistance were provided by Anita Mora and Ryan Kissinger.
Conflict of interest
All authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2015_1433_MOESM1_ESM.tif
Supplementary material 1 (TIFF 3513 kb). Supplemental Fig. 1. LACV does not infect BCECs. Representative images from the OB (a) and cerebellum (b) of weanling mice infected i.p. with 103 PFU LACV at 6 dpi demonstrate BCECs (CD31, red) are not infected with LACV (green) despite significant infection of neighboring neurons (See Fig. 2c for verification of neuronal infection). These observations are consistent with findings from i.n. infected animals (data not shown). Scale bars represent 200 μm
401_2015_1433_MOESM2_ESM.tif
Supplementary material 2 (TIFF 10119 kb). Supplemental Fig. 2. Peripheral LACV infection alters expression of BBB integrity specific proteins in the OB. CyDye labeled 2D-DIGE gels containing protein lysates from mock infected olfactory bulb (left column), LACV infected OB (middle column) and LACV infected cortex (right column). Spots for top seven proteins (A-G) within each gel are indicated by the red outline, with the center dot representing peak of signal intensity. Directly below each gel are 3D signal intensity plots obtained using DeCyder software (see Methods and Materials) for each spot demonstrating the area of each outline and the peak height and volume within the outline. Numerical representations of each plot are shown directly below
401_2015_1433_MOESM3_ESM.tif
Supplementary material 3 (TIFF 653 kb). Supplemental Fig. 3. LACV neuroinvasion is mechanistically distinct between i.n. and i.p. routes of inoculation. Weanling (a) and adult (b) C57/B6 mice were infected i.n. (filled circles) or i.p. (filled boxes) with 103 PFU of LACV and monitored for the development of terminal neurological symptoms. Resulting survival curves were compared using a Log-rank test (n = 7-18 mice per group,*p < 0.05, ***p < 0.005). A portion of mice included in the i.p. survival curves were previously published by our lab [44]
401_2015_1433_MOESM4_ESM.tif
Supplementary material 4 (TIFF 287 kb). Supplemental Fig. 4. Bulbectomized mice are resistant to neuroinvasion when inoculated i.n. Weanling (a) or adult (b) C57BL/6 mice were given BulbX or Sham surgery (see Materials and Methods) to remove the olfactory bulb. Three days post-surgery, 103 PFU LACV was administered i.n. to Sham (filled circles) and BulbX (filled boxes) animals and resulting survival curves compared using a Long-rank test (n = 7-14 mice per group, **p < 0.01)
Rights and permissions
About this article

Cite this article
Winkler, C.W., Race, B., Phillips, K. et al. Capillaries in the olfactory bulb but not the cortex are highly susceptible to virus-induced vascular leak and promote viral neuroinvasion. Acta Neuropathol 130, 233–245 (2015). https://doi.org/10.1007/s00401-015-1433-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1433-0