
Abstract
We examined the phosphorylated 43-kDa TAR DNA-binding protein (pTDP-43) inclusions as well as neuronal loss in full-length spinal cords and five selected regions of the central nervous system from 36 patients with amyotrophic lateral sclerosis (ALS) and 10 age-matched normal controls. The most severe neuronal loss and pTDP-43 lesions were seen in lamina IX motor nuclei columns 4, 6, and 8 of lower cervical segments and in columns 9–11 of lumbosacral segments. Severity of pTDP-43 pathology and neuronal loss correlated closely with gray and white matter oligodendroglial involvement and was linked to onset of disease, with severe involvement of columns 4, 6, and 8 of upper extremity onset cases and severe involvement of columns of 9, 10, and 11 in cases with lower extremity onset. Severe TDP-43 lesions and neuronal loss were observed in stage 4 cases and sometimes included Onuf’s nucleus. Notably, three cases displayed pTDP-43 aggregates in the midbrain oculomotor nucleus, which we had not seen previously even in cases with advanced (i.e., stage 4) pathology. pTDP-43 aggregates were observed in neurons of Clarke’s column in 30.6 % of cases but rarely in the intermediolateral nucleus (IML). Gray matter oligodendroglial pTDP-43 inclusions were present in areas devoid of neuronal pTDP-43 aggregates and neuronal loss. Taken together, our findings indicate that (1) the dorsolateral motor nuclei columns of the cervical and lumbosacral anterior horn may be the earliest foci of pTDP-43 pathology in the spinal cord, (2) gray matter oligodendroglial involvement is an early event in the ALS disease process that possibly heralds subsequent involvement of neurons by pTDP-43 pathology, and (3) in some very advanced cases, there is oculomotor nucleus involvement, which may constitute an additional neuropathological stage (designated here as stage 5) of pTDP-43 pathology in ALS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most frequent adult-onset motor neuron disease characterized by rapidly progressive paresis leading to death with a mean survival of approximately 3 years [31]. The phosphorylated 43-kDa TAR DNA-binding protein (pTDP-43) was identified as the major component in neuronal aggregates in sporadic ALS and the largest subset of frontotemporal lobar degeneration (FTLD) [41]. TDP-43 is a highly conserved and widely expressed 414 amino acid proteins that are involved in splicing of pre-mRNA, transcriptional repression, microRNA synthesis, and in maintaining mRNA stability and nuclear-cytoplasmic shuttling [34, 37]. Progressive accumulation of similar protein aggregates is recognized as a defining feature of many neurodegenerative diseases, including accumulation of abnormally phosphorylated tau in Alzheimer’s disease (AD) and α-synuclein in Parkinson’s disease (PD). Remarkably, many of these diseases show a characteristic spreading pattern of their underlying pathology across specific brain regions with disease progression [1, 2, 4–6, 58]. The cellular mechanisms underlying this spreading are incompletely understood, but increasing evidence indicates that misfolded protein aggregates can propagate by a self-perpetuating process that leads to amplification and neuron-to-neuron transmission of these pathologies [23, 28].
While the concept of “prion-like” propagation is now recognized for tau, amyloid-β (Aβ), and α-synuclein, evidence for cell-to-cell transmission of pTDP-43 is just emerging [44]. Recently, we presented evidence for a possible sequential dissemination of pTDP-43 pathology in a large cohort of autopsy cases with ALS, in which pTDP-43 lesions were proposed to spread initially from the agranular motor cortex, spinal cord α-motoneurons and brainstem motor nuclei, and then from premotor and prefrontal neocortical areas to brainstem precerebellar nuclei and striatum, and finally from postcentral neocortical areas to anteromedial portions of the temporal lobe [9]. However, in that study, the lesions were not examined in sections through all segments of the spinal cord for potential longitudinal and transverse dissemination of pTDP-43 within the spinal cord during the progression of ALS. In fact, although many studies have analyzed pTDP-43 pathology in selected segmental sections of the spinal cord [7, 9, 16, 21, 22, 30, 40–43, 49, 54, 55], none to date have evaluated the extent and severity of pTDP-43 pathology in full-length ALS spinal cords. Here, we examined the distribution of pTDP43 pathology and neuronal loss at all levels of the spinal cord from clinically well-characterized ALS cases, and we incorporated these findings into our previously described pTDP-43 staging algorithm and related this to the clinical presentations of ALS.
Materials and methods
Autopsy cohort
We included 36 patients with a clinical diagnosis of ALS (Table 1), in accordance with modified El Escorial Criteria [13], and a confirmed neuropathological diagnosis of ALS in subjects followed to autopsy in the Center for Neurodegenerative Disease Research (CNDR) at the University of Pennsylvania (n = 13; 5 females, 8 males; age range 51–85 years; mean age ± SD: 65.4 ± 10.8 years) and in the Department of Neuropathology, Chiba University, Japan (n = 23; 9 females, 14 males; age range 42–82 years; mean age ± SD: 64.6 ± 11.1 years), between 1985 and 2012. Informed written consent was obtained from all patients or from their next of kin. The study was approved by the University of Pennsylvania Institutional Review Board (Penn IRB) and by the University of Chiba Ethical Committee. Ten full-length spinal cords from controls age-matched to the ALS cases were collected by the Clinical Neuroanatomy Section of the Department of Neurology, University of Ulm, Germany for comparison with the ALS cases.
Detailed clinical characteristics (gender, age at onset, age at death, site of onset, disease duration) were ascertained from an integrated autopsy database, as described previously [59, 62] and by retrospective chart review of clinical visits within the University of Pennsylvania and University of Chiba Health System (Table 1). For University of Pennsylvania cases, ALS global disease severity data, as measured by a functional rating score (ALSFRS-R) [15], also were available.
Tissue preparation, staining, and immunohistochemistry
The spinal cord was dissected and removed in toto from the level of C2 to the level of the caudal sacral segments. After fixation, the dura mater was removed, preserving the dorsal and ventral roots. The nerve roots were marked with indelible ink at each spinal cord segment. After placement of the marks and evaluation of the completeness of the four major divisions, tissue blocks from each segment were placed in standard embedding cassettes with the ventral limit of the spinal cord facing the lower part of the cassette and the left side of the block facing the right side of the cassette (from the dissector’s perspective). All tissue samples were embedded in paraffin, and the left dorsal horn of the spinal cord was marked with a pinprick and indelible black ink to permit analysis of potential disease laterality. The tissue blocks were cut in the transverse plane on a giant sliding microtome (Jung, Heidelberg, Germany). Two sets of 70-µm sections were made from the paraffin blocks, as previously described [20]. This technique is performed on free-floating sections to enable recognition of specific cell types and cytoarchitectonic units and the accurate assessment of pathological changes, e.g., those associated with synucleinopathies, tauopathies, and pTDP-43 proteinopathies. The first set of sections was stained for topographical overview and neuronal loss using a pigment-Nissl stain for lipofuscin pigment (aldehyde fuchsin) and basophilic Nissl material (Darrow red). In a second set of slides, pTDP-43 pathology was analyzed by immunohistochemistry (IHC) using a commercially available 409/410 rabbit polyclonal antibody (1:10.000, Cosmo Bio, Carlsbad, CA, USA) and assessed according to a semiquantitative rating scale (0, not detectable, ≤2 aggregates per region; +, mild; ++, moderate; +++, severe/numerous) [9]. Severity of pTDP-43 pathology was throughout adjusted for neuronal loss. In assigning cases to a given neuropathological stage, the extent (topographical distribution pattern) was assigned more weight than the abundance of pTDP-43 pathology as previously described [9, 10]. Neuronal loss was also evaluated using a semiquantitative scale (0, absent or not detectable; +, mild; ++, moderate; +++, severe).
Within lamina IX of the spinal cord anterior horn, pTDP-43 pathology and neuronal loss were evaluated separately for each of 11 motor nuclei columns according to the classification of Routal and Pal [50]. These motor columns were identified in control cases first (Fig. 1) and employed as templates for the analysis of the corresponding sections from ALS cases.

Motor nuclei columns of the spinal cord anterior horn in controls. Pigment-Nissl staining (aldehyde fuchsin and Darrow red) of motor nuclei columns of lamina IX in different segments of spinal cord from C2 to S3 in control cases using the organizational scheme proposed by [50]. The right anterior horn is shown in all sections. IML intermediolateral nucleus, C Clarke’s column (posterior thoracic nucleus), O Onuf’s nucleus. Scale bar in C2 is valid for all segments
In addition to the spinal cord pathology, pTDP-43 lesions were evaluated in 22 selected regions of the brain as previously described [9] and staged based on our previously published ALS staging protocol. These regions included the agranular motor cortex (Brodmann areas 4 and 6), medulla oblongata at level of the hypoglossal nucleus (XII), the middle frontal gyrus, striatum, and the hippocampal formation.
IHC was performed with a monoclonal antibody against phosphorylated tau (PHF-Tau; 1:2,000; Clone A8; Pierce Biotechnology, Rockford, IL, USA), α-synuclein (Syn303; 1:4,000, generated in CNDR [18], and amyloid-β (NAB228; 1:15 000; generated in CNDR) [35].
Double-labeling IHC was also performed on selected 70-μm sections using the pTDP-43 antibody above together with the Vector SG (SK-4700, Vector) blue chromogen and the SMI-311 monoclonal antibody (1:1,000, Covance, Princeton, NJ, USA), the rabbit polyclonal anti-oligodendrocyte-specific protein (OSP) antibody (1:400, Abcam, Cambridge, UK), and the anti-oligo2 antibody (Millipore, Billerica, MA, 1:400). Neurofibrillary tangle (NFT) stages [1, 2] and phases of Aβ deposition [58] are shown in Table 1.
Genetic analysis
Genomic DNA was obtained for all 13 University of Pennsylvania cases, but was not available from the University of Chiba cases. Genomic DNA was extracted from brain tissue following the manufacturer’s protocols [QIAsymphony DNA Mini Kit (Qiagen)]. Genotyping for a C9orf72 repeat expansion was performed as previously described in detail [12]. In the 7 cases lacking a C9orf72 expansion, the coding regions of other genes associated with ALS/motor neuron disease or FTLD-TDP (GRN, TARDBP, CHMP2B, ALS2, SETX, SQSTM1, FUS, VAPB, ANG, FIG4, OPTN, VCP, UBQLN2, DCTN1, PRPH, SOD1, NEFH, DAO, SPG11, TAF15, EWSR1, PFN1) were sequenced using a custom-designed targeted next generation sequencing panel to screen for known pathogenic missense mutations. The regions covered included the five prime and three prime untranslated regions and the whole-coding and flanking intronic regions. The genomic libraries were constructed using the Haloplex target enrichment kit according to the manufacturer’s instruction (Agilent, CA). Briefly, genomic DNA was fragmented using eight different restriction enzymes and denatured. The DNA was hybridized with a biotinylated custom probe library and sample barcodes, and the target fragments were retrieved with magnetic streptavidin beads and circularized by ligation. Captured DNA targets were amplified by PCR, validated, and quantified. Equimolar amounts of differentially indexed samples were pooled for sequencing on the MiSeq platform (Illumina). Sequence reads were aligned and analyzed by SureCall software (Agilent). No pathogenic mutation was identified; however, three variants of unknown significance were identified in three cases.
Statistical analysis
Data analysis was performed using SPSS (Version 17.0 SPSS Inc., Chicago, IL, USA). The average (and range) of data on patient characteristics was estimated by calculating the median (and 25–75th percentiles). Differences between two clinical subgroups were compared using Wilcoxon–Mann–Whitney Test. To compare raw data of multiple subgroups, Kruskal–Wallis analysis of variance on ranks was applied and in case of significance, by Dunn’s Method. Trend analysis was conducted using the Mantel–Haenszel Chi-square test. All correlations were studied using Spearman’s rank-order correlation coefficient. Bonferroni correction for multiple testing was applied when contrasts were not driven by a specific hypothesis. For all other tests, p values <0.05 were considered significant. All statistical tests were two-sided.
Results
Aggregated pTDP-43 in motor nuclei columns of lamina IX
We analyzed pTDP-43 lesions and neuronal loss in full-length spinal cords from 10 control cases age-matched to patients with ALS. No pTDP-43 aggregates and no loss of α-motoneurons were observed in any of the controls.
Generally, in the motor nuclei columns of the anterior horn of ALS cases, pTDP-43 pathology presented in the form of intraneuronal dash-like, skein-like, or roundish dot-like inclusions in the cytoplasm of α-motoneurons (Fig. 2a, b). Widespread pTDP-43 pathology was seen across the entire length of the spinal cord, with the most severe pTDP-43 aggregates occurring in columns 6, 7, and 8, and at levels C6-Th1, L5, and S1 (Table 2). Similarly, the heaviest neuronal loss was observed in motor nuclei columns 6 and 8, and at levels of C5-Th1 as well as L5. Neuronal loss correlated strongly with TDP-43 pathology across all motor nuclei columns (p < 0.001, rho > 0.4 for each correlation). Six cases (16.7 %) with widespread and severe pTDP-43 pathology showed mild pTDP-43 aggregates in the motoneurons of Onuf’s nucleus (Fig. 2l), although there was no evidence of neuronal loss (Fig. 2m).

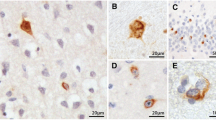
pTDP-43 pathology in motor and non-motor areas of the spinal cord of ALS. a, b Detailed view of skein-like lesions and large rounded aggregates in α-motoneurons using double-labeling IHC with the pTDP-43 antibody (dark blue) and an antibody to the heavy neurofilament subunit (brown) c Skein-like neuronal aggregates (arrow). d Example of pTDP-43 pathology in motor nuclei columns of ALS showing the spinal cord anterior horn at segment L4; numbers indicate the respective motor nuclei columns at this level. e–i, k Images show involvement by pTDP-43 pathology of non-motor areas in the spinal cord of ALS. e Bilateral involvement of Clarke’s column (arrows) at segment L1. f and g show pTDP-43 positive aggregates in neurons of Clarke’s column from segment L2 at higher resolution. h While pTDP-43 aggregates were observed in Clarke’s column of some ALS cases, there was no neuronal loss there: pigment-Nissl staining shows intact neurons of Clarke’s columns at segment L2 in the same ALS case as in g. i, k Involvement of the IML (arrow) at segment Th6 (i) where aggregates are mild in pTDP-43 IHC without detectable neuronal loss in pigment-Nissl staining (k). m, n Mild pTDP-43 IHC (arrow) in Onuf’s nucleus at segment S2 without neuronal loss (arrow) as shown by pigment-Nissl staining (n). Scale bar in (a) is valid for (b). Scale bar in (f) also applies to (g–m). Scale bar in d is valid for e
We also looked to see if different types of ALS onset might influence patterns of spinal cord TDP-43 pathology. Both pTDP-43 lesions and loss of lamina IX α-motoneurons tended to be more severe in ALS cases that had an extremity onset of disease as compared with bulbar onset cases (Fig. 3a, b). In cases with upper extremity onset of disease, pTDP-43 pathology was significantly more severe in motor nuclei columns 1–6 and 8 in comparison to cases with a lower extremity or a bulbar onset of disease (Fig. 3a, b). Similarly, neuronal loss was more severe in motor nuclei columns 1–6 and 8 of upper extremity onset cases as opposed to cases with a lower extremity or a bulbar onset of disease. In cases with a lower extremity onset of disease, pTDP-43 pathology was more severe in columns 9, 10, and 11 as compared with other types of onset (Fig. 3a, b).

Graphic showing burden of pTDP-43 pathology and severity of neuronal loss in 11 motor nuclei columns at different segments of the spinal cord anterior horn in ALS. a Barplots show severity of neuronal loss and b burden of pTDP-43 pathology in 11 motor nuclei columns of lamina IX anterior horn for bulbar (white bar), cervical (black bar), and lumbar (blue bar) types of ALS onset. c Barplots show severity of neuronal loss and d burden of pTDP-43 pathology from segments C2 to S3 for bulbar (white), cervical upper extremity (black), and lumbar lower extremity (blue) types of ALS onset. e Barplots show the burden of pTDP-43 pathology and f severity of neuronal loss in bulbar (white), cervical (black), and lumbar (blue) bulbar (white), cervical (black), and lumbar (blue) motor nuclei columns of lamina IX of the anterior horn depending on the site of clinical onset of paresis. e, f Blue bars show extent of pathology on the side contralateral to that of the first clinical manifestation of paresis; black bars indicate the extent of pathology on the side ipsilateral to first clinical manifestation. Bars indicate mean and 95 % confidence interval throughout. Significant difference in Wilcoxon Mann–Whitney Test (p < 0.05) is indicated by an asterisk
The severity of pTDP-43 pathology in various spinal cord segments was then compared among different onset types (Fig. 3d). In cases with an upper extremity onset of disease, pTDP-43 aggregates and neuronal loss were more severe at levels C3-Th1 than in the other onset types, whereas neuronal loss was more severe at levels L1-S1 in cases with a lower extremity onset of disease than in other types of onset (Fig. 3c, d). Assessment of a possible influence of the side of disease onset on pTDP-43 pathology in the ALS spinal cords revealed that the severity of both the pTDP-43 pathology and neuronal loss was laterality associated, i.e., with the side of the first clinical manifestation (Fig. 3e, f). pTDP-43 lesions were more severe in motor nuclei columns 2 and 8 of the side ipsilateral to first manifestation of paresis in comparison to the corresponding columns of the contralateral side (Fig. 3e). Furthermore, neuronal loss was more severe in ipsilateral motor nuclei columns 2, 4, 5, and 8 than in the corresponding contralateral columns (Fig. 3f). Finally, the severity of neuronal loss increased in nearly all motor nuclei columns with increasing duration of ALS, although no significant correlation with age at onset, age at death, or the ALSFRS-R could be found (Suppl. Table 1). Our cohort included 6 cases with a C9orf72 hexanucleotide repeat expansion. Severity of pTDP-43 pathology in different motor nuclei columns of these cases did not show a significant difference to cases without this expansion (data not shown).
Spinal cord pTDP-43 pathology at different disease stages
Spinal cord involvement by TDP-43 pathology and neuronal loss were previously evaluated at different ALS stages albeit not in the same level of detail reported here [9]. While stages of pTDP-43 pathology did not correlate with disease duration across all ALS cases (rho = 0.25, p = 0.15), we did show a significant correlation with disease duration in the subgroup of cases with extremity onset (rho = 0.46, p = 0.01). No significant correlation of ALS stages of pTDP-43 pathology with age of onset, age of death, or the ALSFRS-R was detectable (rho < 0.4, p > 0.05 each).
The abundance of pTDP-43 pathology increased significantly from ALS stage 1 to stage 4 in motor nuclei columns 6 and 2 (p = 0.04 each) as well as 7 (p = 0.02) and showed a tendency to increase with higher ALS stages in column 1 and 8 but did not reach statistical significance (p = 0.07).
Our cohort included four ALS cases at stage 1, and in three of these cases (cases 8, 12, and 20 in Table 1) relevant neuronal loss was absent along the entire length of the spinal cord, whereas mild pTDP-43 lesions were observed in the motor nuclei columns of all spinal cord segments. In a single stage 1 case with lower extremity onset of disease (case 8 in Table 1), pTDP-43 lesions were present only between L3 and S3 in motor nuclei columns 2, 9, and 10 but not in the remainder of the spinal cord, but there also was no neuronal loss throughout the spinal cord. In contrast, stage 4 cases displayed both widespread and severe pTDP-43 pathology and neuronal loss, including all six cases in which involvement of Onuf’s nucleus was seen.
Remarkably, we observed three stage 5 cases (cases 16, 19, and 22 in Table 1) with severe pTDP-43 pathology that did not correspond to the any of the regional distribution patterns we reported earlier for ALS [9]: In addition to the regions involved in ALS stage 4, all three cases also showed pTDP-43 inclusions and neuronal loss in the oculomotor nucleus of the midbrain (Table 1) (Fig. 4a–c). These cases were furthermore characterized by severe pTDP-43 aggregates and widespread neuronal loss in the substantia nigra (Fig. 4g, compare 4 h), severe involvement of the neurons of the inferior olivary complex (Fig. 4d, e), and severe pTDP-43 aggregates in the dentate nucleus of the cerebellum (Fig. 4f). Disease duration (median 70 months, interquartile range 59–145 months) was significantly longer than all other ALS cases (median 24 months, interquartile range 17–36 months) (p = 0.009) in the present cohort (Table 1).

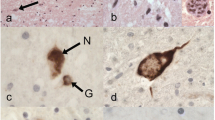
Late-stage pTDP-43 pathology in ALS. Protein aggregation of pTDP-43 and extensive neuronal loss of late-stage ALS (possible ALS stage 5). a Involvement of the oculomotor nucleus in the midbrain. b Pigment-Nissl staining of the same area as shown in a. c Bilateral involvement of the oculomotor nucleus (III), aq—cerebral aqueduct. d Section through the medulla oblongata at the level of the hypoglossal nucleus and inferior olivary complex showing, at higher resolution in e, severe involvement of the inferior olivary complex. f pTDP-43 pathology in the dentate gyrus of the cerebellum. g, h Midbrain sections containing the red nucleus (rn) and substantia nigra (sn). g Substantia nigra from an ALS stage 5 case with severe loss of melanized neurons. h Pigment-Nissl-stained section with intact substantia nigra from an ALS stage 1 case for purposes of comparison. i pTDP-43 IHC shows markedly atrophic anterior horn (ah) with a complete loss of α-motoneurons and widespread gray as well as white matter oligodendroglial lesions from a late-stage ALS case. Framed insert shows gray matter oligodendroglial with pTDP-43-positive aggregates at higher resolution. Scale bar in a applies to b, c, e and framed area in i. Scale bar in d applies to g and h, and scale bar in f is also valid for i
pTDP-43 pathology in neurons of lamina VII
The analysis of pTDP-43 pathology in lamina IX of the anterior horn was followed by evaluation of pathology in other gray matter areas of the spinal cord. In general, pTDP-43 lesions were rare outside the α-motoneurons of lamina IX. However, in 30.6 % of the ALS cases (n = 11) studied here, pTDP-43 aggregates were observed in the neurons of Clarke’s column (posterior thoracic nucleus) accompanied in 13.9 % of ALS cases by neuronal loss (Table 2) (Fig. 2e–h). The involvement of Clarke’s column and the neuronal loss therein were more prominent at lower thoracic and upper lumbar spinal cord levels (Table 2). Both parameters (neuronal loss, pTDP-43 lesions) in Clarke’s column were more severe in extremity onset cases (upper or lower extremity onset of disease) than in bulbar onset cases (p < 0.01 each). The majority of cases with involvement of Clarke’s column showed stage 4 pTDP-43 pathology (n = 9) and two cases showed stage 3 pathology. Rare or mild pTDP-43 inclusions without neuronal loss were also observed in the intermediolateral nucleus (IML) of five ALS cases (cases 14, 16, 17, 22, 35 in Table 1), four of which showed an extremity onset of disease (Fig. 2). All of these cases showed stage 3 pTDP-43 pathology or higher.
Aggregated pTDP-43 in ALS spinal cord oligodendrocytes
Neuronal pTDP-43 aggregates usually were accompanied by similar changes in oligodendrocytes (Fig. 5), but the pTDP-43 pathology did not develop in non-myelinating oligodendrocytes located along the somata of involved α-motoneurons of lamina IX (satellite cells). Severity of neuronal pTDP-43 inclusions and neuronal loss in different motor nuclei columns correlated closely with the severity of oligodendroglial pTDP-43 aggregates in the anterior horn gray matter and in corticospinal tract white matter (Suppl. Table 1). In addition, spinal cord gray matter oligodendroglial pTDP-43 inclusions correlated with the stage of pTDP-43 pathology (rho = 0.37, p = 0.025). Remarkably, oligodendroglial pTDP-43 inclusions were observed frequently in the anterior horn gray matter of cases that did not show (or only to a very mild extent) neuronal pTDP-43 aggregates and in which no loss of α-motoneurons was yet detectable (Fig. 5a–d). In addition, no oligodendroglial pTDP-43 aggregates were seen in the spinal cord white matter. With growing severity of neuronal TDP-43 lesions and increasing loss of α-motoneurons (Fig. 5e, f), oligodendroglial pTDP-43 lesions could be detected in the spinal cord white matter chiefly in the anterolateral funiculus and especially within corticospinal projections. Finally, in cases with the highest burden of neuronal pTDP-43 pathology and the most severe α-motoneuron loss, oligodendroglial TDP-43 lesions were widespread across both gray and white matter and even extended into non-motor areal of the spinal cord white matter, including the posterior funiculi (Fig. 5g–i) as schematically summarized in panels I–IV of Fig. 5k.

Oligodendroglial pTDP-43 pathology in ALS spinal cord. a, b Early involvement of gray matter oligodendroglia (small dot-like/rounded aggregates) at the level of S2 indicated by arrowheads, whereas α-motoneurons (arrow) and white matter oligodendroglia are largely unaffected, and no neuronal loss is detectable in b (pigment-Nissl staining). c Double-labeling IHC of segment C6 anterior horn (asterisk marks a normal α-motoneuron) using an OSP antibody (brown) and pTDP-43 antibody (dark blue). Arrowheads indicate oligodendrocytes containing aggregated pTDP-43, arrows point to oligodendrocytes without pTDP-43 aggregates. d Double-labeling IHC of segment L5 anterior horn using an oligo2 antibody (brown) and pTDP-43 antibody (dark blue). Arrowheads show oligodendrocytes with pTDP-43 aggregates, arrows point to intact oligodendrocyte devoid of pTDP-43 pathology. Micrographs in (e) and (f) show aggregates both in α-motoneurons (arrows) and gray matter oligodendrocytes (arrowheads) demonstrated by pTDP-43 IHC in segments L4 (e) and C7 (f). g C7 anterior horn with massive gray and white matter oligodendroglial pTDP-43 involvement. h Pigment-Nissl staining of same region as in (g) with severe loss of α-motoneurons. i White matter oligodendroglial pTDP-43 pathology in dorsal (sensory) white matter areas at the level of segment Th8. Framed inset shows dorsal white matter oligodendroglia at higher resolution. Scale bar in a also applies to b, scale bar in f is valid for framed area in i, and scale bar in g applies to h, i. k Schematic drawing illustrating the hypothetical sequence of oligodendroglial and neuronal pTDP-43 pathology in gray (upper half of each panel) or white (lower half of each panel) matter of the spinal cord: I—oligodendroglial pTDP-43 inclusions (indicated by brown dots in each panel) are present in the anterior horn gray matter of cases lacking neuronal pTDP-43 aggregates and without evident loss of α-motoneurons or detectable white matter oligodendroglial involvement. II, III—With increasing severity of neuronal pTDP-43 lesions (II) and progressive loss of α-motoneurons (III), oligodendroglial pTDP-43 aggregates are increasingly observed in the spinal cord white matter. IV—In cases with severe neuronal pTDP-43 pathology and extensive α-motoneuron loss, extensive oligodendroglial pTDP-43 pathology is observed across both gray and white matter
Discussion
Spinal cord involvement is at the very core of ALS pathology and is almost implicit in the standard definition of the disease [31, 51]; yet, there are no studies to date of how pTDP-43 pathology affects the different groups of neurons within the anterior horn, which thereby could provide a detailed analysis of pTDP-43 pathology along all segments of the spinal cord. However, this information is essential for understanding the progression of ALS, inasmuch as the spinal cord anterior horn is not a uniform en bloc entity but, rather, a complex anatomical system that shows a transversal and longitudinal somatotopic differentiation similar to the homunculus of the cerebral cortex [52, 53].
Somatotopic distribution of pTDP-43 pathology in the anterior horn of ALS
Within the spinal cord anterior horn, the neurons of lamina IX [48] are organized into a series of longitudinal motor columns, the numbers of which vary within different divisions of the spinal cord (Fig. 1). It is difficult to differentiate these motor nuclei columns in transverse 6- to 10-µm-thick sections because their borders are not always sharply delineated and the nuclei are often closely juxtaposed together. Consequently, the descriptions of these columns vary somewhat from author to author [27, 50, 52, 53]. Moreover, the problem of delineating different motor nuclei columns in ALS is aggravated by neuronal loss. Thus, here we used 70-µm-thick sections and a simplified version of the organizational scheme of the 11 motor nuclei columns as proposed by [50] supplemented by templates from control spinal cord sections (Fig. 1) to analyze pTDP-43 pathology in all corresponding segments of the anterior horn of ALS.
Not surprisingly, we found no pTDP-43 aggregates in motor nuclei columns of control cases, while pTDP-43 pathology was widespread across various motor nuclei columns and along different divisions (i.e., cervical, thoracic, lumbar, sacral) of the ALS spinal cord, and it was closely associated with α-motoneuron loss. This observation is in line with previous studies reporting the presence of severe pTDP-43 pathology in α-motoneurons of the anterior horn [7, 9, 16, 21, 22, 30, 40–43, 54, 55].
As a rule, neuronal loss in the motor nuclei columns only became apparent when moderate to severe pTDP-43 aggregation was present (Fig. 3c, d). As such, and consistent with earlier studies [10, 21, 22, 41], pTDP-43 lesions most likely precede neuronal loss, presumably reaching a given threshold before it causes neuronal dysfunction and death.
Other groups have reported a somatotopic differentiation of the cervical and lumbosacral enlargements of the spinal cord anterior horn, in which groups of α-motoneurons located in medial areas mainly innervate axial (trunk) muscles, whereas ventral groups project to proximal extremity muscles and lateral and dorsal areas innervate the more distal muscles of the extremities [27, 52, 53]. This transverse somatotopic division is augmented by a longitudinal differentiation of muscle innervation: motor neurons controlling hand movements are located primarily in segments C8 and Th1, and the distal lower extremity muscles are innervated mainly from the lower lumbar and upper sacral segments [27].
Here, both pTDP-43 pathology and neuronal loss were most severe in the dorsolateral motor nuclei columns of the lower cervical and lower lumbar/upper sacral segments (e.g., columns 4, 6, and 8 of the lower cervical segments, and columns 9–11 of the lumbosacral segments; see Fig. 3). Since the severity of pTDP-43 aggregates and neuronal loss are likely to increase in a specific region with progressing disease [40], our findings indicate that the dorsolateral motor nuclei columns of the lower cervical and lumbar anterior horn are possibly the earliest foci of pTDP-43 pathology in the ALS spinal cord. In addition, the remarkably severe involvement of dorsolateral motor nuclei columns coincides well with clinical observations of the early involvement of distal and extensor extremity muscles in limb onset ALS [31].
It should also be pointed out that the severity of motor nuclei column involvement was closely associated with the clinical onset of disease, with massive involvement of columns 4, 6, and 8 observed in cases with an upper extremity onset of disease, and with severe pTDP-43 pathology and neuronal loss in columns of 9, 10, and 11 of ALS cases with a lower extremity onset of disease (Fig. 3). In contrast, spinal cord neuronal loss was generally much less severe in cases with a bulbar onset of disease. Longitudinally, neuronal loss (and, to a lesser extent TDP-43 pathology) were accentuated in cervical segments in cases with upper extremity disease onset and decreased in a rostro-caudal gradient, whereas neuronal loss increased to lumbar and upper sacral segments in cases with lower extremity onset of disease.
Spinal cord involvement at different stages of TDP-43 pathology
We next evaluated how spinal cord pTDP-43 pathology might relate to stages of pTDP-43 pathology. As in our initial staging study [9], very few stage 1 cases (n = 4) were available. Here, all four stage 1 cases showed involvement of the motor cortex and brainstem somatomotor nuclei in addition to pTDP-43 lesions in the spinal cord. However, although neuronal loss was absent, pTDP-43 lesions in stage 1 cases were detectable along the entire length of the spinal cord in all but a single case, where pTDP-43 aggregates were confined to lower lumbar/upper spinal segments. Thus, these findings support the idea that spinal pTDP-43 pathology in ALS disseminates after a focal onset [46, 47]. However, it remains unclear if the pathology spreads between the α-motoneurons of different motor nuclei columns and across different segments or, alternatively, if it reaches the neurons of lamina IX by axonal connections of cortical projection neurons as recently hypothesized by our group [3]. We could not determine whether spinal cord lesions constitute the earliest foci of pTDP-43 pathology in ALS or whether such spinal pathology results from propagation of cortical pTDP-43 lesions because selected cortical areas rather than complete hemisphere series were available for this study.
We also analyzed spinal cord pathology in cases with the highest burden of pTDP-43 pathology (ALS stage 4) [9]. Neuropathological staging attempts are, by definition, cross-sectional, but we observed a correlation between the stages of pTDP-43 pathology and disease duration in the subgroup of extremity onset cases. However, previous studies have not unequivocally correlated severity of pTDP-43 pathology with disease duration, i.e. there are reports of mild pTDP-43 aggregates in ALS patients who survived for a long time, reflecting a relatively benign course of disease [26, 42]. In contrast, we saw severe pTDP-43 lesions and neuronal loss in three ALS patients with rapidly progressive disease who were kept alive over a median of 70 months by invasive ventilation via tracheostomy (Table 1). Furthermore, in some of these cases, the distribution of the aggregates did not resemble the regional distribution pattern observed by us previously in stage 4 cases [9]. We postulated that such cases with oculomotor involvement may constitute an additional advanced neuropathological stage (designated here as stage 5) of TDP-43 pathology in ALS. To assess the presence or absence of pTDP-43 pathology in the nuclei of the extrinsic eye muscles, frontal sections cut perpendicularly to the brainstem axis should be made as follows: a central section cut through the largest extent of the superior colliculus (N. III), a section cut at the latitude of the lower border of the inferior colliculus (N. IV), and a section cut at the latitude of the motor nucleus of the facial nerve (N. VI).
Involvement of the oculomotor nucleus (N. III) has been reported before in ALS [39], including cases with long-term survival on a respirator [24, 32, 33]. Moreover, our proposed stage 5 cases displayed massive and widespread neuronal loss that resulted in a marked bilateral atrophy of spinal cord anterior horns (Fig. 4i). In addition, neuronal loss extended beyond the motor areas, and abundant pTDP-43 aggregates were present in the cerebellar dentate nucleus, which occurs from stage 2 onwards but normally remains patchy [9], and there was nearly a complete loss of pigmented dopaminergic neurons in the substantia nigra reminiscent of PD [6] (Fig. 4g) but in the absence of Lewy body pathology.
Given previous clinical and neuropathological findings of non-motor involvement in ALS [10, 17, 21, 38, 45, 63], it is conceivable that with ongoing disease progression, ALS increasingly shifts from a disease of mainly motor neurons to a multi-system disease, a notion supported by our observation that the involvement of Clarke’s column and IML only was observed in ALS stages 3 and 4 (Table 2). There are no previous reports of pTDP-43 pathology in neurons of Clarke’s column, although mild neuronal loss in the posterior thoracic nucleus has been noted in cases with a long duration of disease [26]. Clarke’s column neurons receive proprioceptive afferences from skeletal muscles and project to the cerebellum via the posterior spinocerebellar tract [57]. However, all neurons of lamina VII are also targeted by brain regions involved in motor functioning, including the motor cortex, red nucleus, and periaqueductal gray matter [14, 61]. Whereas pTDP-43 pathology in the neurons of Clarke’s column underlines the concept of ALS as a multi-system disease [22], the clinical relevance of the pathology, there is difficult to interpret because neuronal loss was discrete in comparison to the very severe changes in lamina IX. The same applies to Onuf’s nucleus, where pTDP-43 lesions, which have been reported previously, were not accompanied by neuronal loss [56].
Stages of pTDP-43 pathology were related to disease duration in the subgroup of cases with extremity onset, and severity of neuronal loss increased with duration of disease, supporting the notion that accumulating pTDP-43 neuronal aggregation results in neuronal dysfunction and death, although the underlying molecular mechanisms of TDP-43-mediated neurodegeneration so far remain enigmatic (reviewed in [34]). Our cohort included 6 cases with a C9orf72 hexanucleotide repeat expansion, which has been linked to a younger age of onset of ALS [60], though the number of C9orf72 cases in our study was too small to exert a relevant influence here. Spinal cord pTDP-43 pathology and neuronal loss did not correlate with the ALSFRS-R, which is in line with our previous studies of spinal cord pTDP-43 pathology [9, 11].
Gray matter oligodendroglia as harbingers of neuronal pTDP-43 pathology
In addition to involvement of different neurons, we consistently observed widespread oligodendroglial pTDP-43 inclusions in the spinal cord gray and white matter of ALS (Fig. 4). In addition, severity of gray and white matter oligodendroglial pTDP-43 inclusions correlated closely with neuronal involvement and loss (Supp. Table 1). Oligodendrocytes form the myelin sheaths of the CNS and enable the rapid propagation of axonal action potentials with minimal energetic output. There is increasing evidence that, beyond their role as mere structural elements, oligodendrocytes also provide metabolic support to neurons by transferring glycolytic intermediates through the monocarboxylic transporter MCT1 [25, 36]. Motor neurons in the spinal cord have been shown to depend on this transporter for survival, which indicates that oligodendroglia play a key role in the preservation of motor neuron integrity [36]. Recently, extensive degeneration of gray matter oligodendrocytes was observed in SOD1 ALS mice that began prior to the onset of clinical disease symptoms [29]. Here, we repeatedly observed gray matter oligodendroglial pTDP-43 inclusions in areas without evident neuronal pTDP-43 aggregates, and in which no neuronal loss or white matter oligodendroglial pTDP-43 pathology were detected (Fig. 5a–d). Consequently, gray matter oligodendroglial involvement may be an early event in the disease process that heralds subsequent involvement of neuronal cells by pTDP-43 pathology (Fig. 5k). Based on their anatomical distribution, these gray matter oligodendrocytes are not satellite cells but are located in close proximity to axonal connections of α-motoneurons, e.g., corticospinal projections. TDP-43 can be transported along axons of cultured motor neurons [19], and we recently hypothesized that TDP-43 could propagate via axonal connections [8, 9]. Since gray matter oligodendroglial pTDP-43 inclusions occurred here in cases that did not yet show white matter oligodendroglial involvement, they are likely to reflect selective changes at the distal parts of axonal connections to α-motoneurons rather than changes along the entire length of the axon. Accordingly, we speculate that early gray matter oligodendroglial involvement may reflect an increasing propensity of TDP-43 to aggregate in distal portions of axons that could lead to aggregate formation in perineuronal oligodendrocytes before reaching α-motoneurons.
Based on the findings presented here and in a previous staging paper [9], we infer that the progression of ALS from focal sites of onset in the agranular motor cortex, brainstem motor nuclei of cranial nerves V, VII, and X–XII, as well as in spinal cord motor neurons to more widespread areas of the CNS could reflect the cell-to-cell spread of pathological TDP-43, and we speculate that this could have significant implications for the development of disease modifying therapies for ALS and related TDP-43 proteinopathies. Furthermore, early oligodendroglial involvement by pTDP-43 pathology as well as the close relation between neuronal and oligodendroglial pTDP-43 aggregation supports the notion that oligodendroglia could play an important role in ALS pathology.
References
Braak H, Alafuzoff I, Arzberger T et al (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Brettschneider J, Ludolph AC et al (2013) Amyotrophic lateral sclerosis-a model of corticofugal axonal spread. Nat Rev Neurol 9:708–714
Braak H, Del Tredici K (2011) The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 121:171–181
Braak H, Del Tredici K (2012) Where, when, and in what form does sporadic Alzheimer’s disease begin? Curr Opin Neurol 25:708–714
Braak H, Del Tredici K, Rüb U et al (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Braak H, Ludolph A, Thal DR et al (2010) Amyotrophic lateral sclerosis: dash-like accumulation of phosphorylated TDP-43 in somatodendritic and axonal compartments of somatomotor neurons of the lower brainstem and spinal cord. Acta Neuropathol 120:67–74
Brettschneider J, Del Tredici K, Irwin DJ et al (2014) Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol 127:423–439
Brettschneider J, Del Tredici K, Toledo JB et al (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38
Brettschneider J, Libon DJ, Toledo JB et al (2012) Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol 123:395–407
Brettschneider J, Toledo JB, Van Deerlin VM et al (2012) Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One 7:e39216
Brettschneider J, Van Deerlin VM, Robinson JL et al (2012) Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol 123:825–839
Brooks BR, Miller RG, Swash M et al (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299
Brown LT (1974) Rubrospinal projections in the rat. J Comp Neurol 154:169–187
Cedarbaum JM, Stambler N, Malta E et al (1999) The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 169:13–21
Cooper-Knock J, Hewitt C, Highley JR et al (2012) Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135:751–764
Donaghy C, Thurtell MJ, Pioro EP et al (2011) Eye movements in amyotrophic lateral sclerosis and its mimics: a review with illustrative cases. J Neurol Neurosurg Psychiatry 82:110–116
Duda JE, Giasson BI, Mabon ME et al (2002) Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol 52:205–210
Fallini C, Bassell GJ, Rossoll W (2012) The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet 21:3703–3718
Feldengut S, Del Tredici K, Braak H (2013) Paraffin sections of 70–100 mum: a novel technique and its benefits for studying the nervous system. J Neurosci Methods 215:241–244
Geser F, Brandmeir NJ, Kwong LK et al (2008) Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol 65:636–641
Geser F, Martinez-Lage M, Robinson J et al (2009) Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol 66:180–189
Guo JL, Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20:130–138
Harvey DG, Torack RM, Rosenbaum HE (1979) Amyotrophic lateral sclerosis with ophthalmoplegia. A clinicopathologic study. Arch Neurol 36:615–617
Horner PJ, Thallmair M, Gage FH (2002) Defining the NG2-expressing cell of the adult CNS. J Neurocytol 31:469–480
Iwanaga K, Hayashi S, Oyake M et al (1997) Neuropathology of sporadic amyotrophic lateral sclerosis of long duration. J Neurol Sci 146:139–143
Jenny AB, Inukai J (1983) Principles of motor organization of the monkey cervical spinal cord. J Neurosci 3:567–575
Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501:45–51
Kang SH, Li Y, Fukaya M et al (2013) Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16:571–579
Keller BA, Volkening K, Droppelmann CA et al (2012) Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol 124:733–747
Kiernan MC, Vucic S, Cheah BC et al (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955
Kimura T, Jiang H, Konno T et al. (2014) Bunina bodies in motor and non-motor neurons revisited: a pathological study of an ALS patient after long-term survival on a respirator. Neuropathology
Komachi H, Okeda R, Ishii N et al (1994) Motor neuron disease with dementia and ophthalmoplegia. A clinical and pathological study. J Neurol 241:592–596
Lee EB, Lee VM, Trojanowski JQ (2012) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13:38–50
Lee EB, Leng LZ, Zhang B et al (2006) Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem 281:4292–4299
Lee Y, Morrison BM, Li Y et al (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448
Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438
Miki Y, Mori F, Nunomura J et al (2010) Sporadic amyotrophic lateral sclerosis with pallido-nigro-luysian degeneration: a TDP-43 immunohistochemical study. Neuropathology 30:149–153
Mizuno Y, Fujita Y, Takatama M et al (2012) Comparison of phosphorylated TDP-43-positive inclusions in oculomotor neurons in patients with non-ALS and ALS disorders. J Neurol Sci 315:20–25
Mori F, Tanji K, Zhang HX et al (2008) Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 116:193–203
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Nishihira Y, Tan CF, Hoshi Y et al (2009) Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol 117:45–53
Nishihira Y, Tan CF, Onodera O et al (2008) Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182
Nonaka T, Masuda-Suzukake M, Arai T et al (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 4:124–134
Phukan J, Pender NP, Hardiman O (2007) Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol 6:994–1003
Ravits J, Paul P, Jorg C (2007) Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68:1571–1575
Ravits JM, La Spada AR (2009) ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 73:805–811
Rexed B (1954) A cytoarchitectonic atlas of the spinal cord in the cat. J Comp Neurol 100:297–379
Riku Y, Watanabe H, Yoshida M et al (2014) Lower motor neuron involvement in TAR DNA-binding protein of 43 kDa-related frontotemporal lobar degeneration and amyotrophic lateral sclerosis. JAMA Neurol 71:172–179
Routal RV, Pal GP (1999) A study of motoneuron groups and motor columns of the human spinal cord. J Anat 195(Pt 2):211–224
Rowland LP (2001) How amyotrophic lateral sclerosis got its name: the clinical-pathologic genius of Jean-Martin Charcot. Arch Neurol 58:512–515
Schoenen J (1982) Dendritic organization of the human spinal cord: the motoneurons. J Comp Neurol 211:226–247
Sharrard WJ (1955) The distribution of the permanent paralysis in the lower limb in poliomyelitis: a clinical and pathological study. J Bone Jt Surg Br 37-B:540–558
Stewart H, Rutherford NJ, Briemberg H et al (2012) Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 123:409–417
Sumi H, Kato S, Mochimaru Y et al (2009) Nuclear TAR DNA binding protein 43 expression in spinal cord neurons correlates with the clinical course in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 68:37–47
Takeda T, Uchihara T, Nakayama Y et al (2014) Dendritic retraction, but not atrophy, is consistent in amyotrophic lateral sclerosis-comparison between Onuf’s neurons and other sacral motor neurons. Acta Neuropathol Commun 2:11
Terman JR, Wang XM, Martin GF (1998) Origin, course, and laterality of spinocerebellar axons in the North American opossum, Didelphis virginiana. Anat Rec 251:528–547
Thal DR, Rub U, Orantes M et al (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Toledo JB, Van Deerlin VM, Lee EB et al (2013) A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement
Van Rheenen W, Van Blitterswijk M, Huisman MH et al (2012) Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology 79:878–882
Waldron HA, Gwyn DG (1969) Descending nerve tracts in the spinal cord of the rat. I. Fibers from the midbrain. J Comp Neurol 137:143–153
Xie SX, Baek Y, Grossman M et al (2011) Building an integrated neurodegenerative disease database at an academic health center. Alzheimers Dement 7:e84–e93
Zhang H, Tan CF, Mori F et al (2008) TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 115:115–122
Acknowledgments
We thank the patients who contributed to this study. We are also grateful to Kevin Raible, Terry Schuck, Sigrid Baumann, Gabriele Ehmke, Simone Feldengut, Julia Straub, and Thi Phuong Thu Brettschneider for technical support, together with David Ewert (University of Ulm) for assistance with the graphics. This study was supported by the NIH (AG033101, AG017586, AG010124, AG032953, AG039510, NS044266), the Wyncote Foundation, and the Koller Family Foundation, as well as by the German Motor Neuron Disease Network. VM-YL is the John H. Ware, 3rd, Professor of Alzheimer’s Disease Research. JQT is the William Maul Measey-Truman G. Schnabel, Jr. Professor of Geriatric Medicine and Gerontology. DJI is supported by the Penn Institute for Translational Medicine & Therapeutics (ITMAT). HB and KDT are supported by the Michael J. Fox Foundation for Parkinson’s Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Brettschneider, K. Arai, and K. Del Tredici contributed equally.
H. Braak and J. Q. Trojanowski are Senior authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Brettschneider, J., Arai, K., Del Tredici, K. et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol 128, 423–437 (2014). https://doi.org/10.1007/s00401-014-1299-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-014-1299-6