
Abstract
The formation of low-order oligomers of β-amyloid (Aβ) within the brain is widely believed to be a central component of Alzheimer’s disease (AD) pathogenesis. However, despite advances in high-throughput and high-resolution techniques such as xMAP and mass spectrometry (MS), investigations into these oligomeric species have remained reliant on low-resolution Western blots and enzyme-linked immunosorbent assays. The current investigation compared Aβ profiles within human cortical tissue using sodium dodecyl sulphate (SDS) polyacrylamide gel electrophoresis (PAGE), xMAP and surface enhanced laser desorption/ionization time-of-flight MS and found that whilst there was significant correlation across the techniques regarding levels of monomeric Aβ, only SDS-PAGE was capable of detecting dimeric isoforms of Aβ. The addition of synthetic di-tyrosine cross-linked Aβ1–40Met35(O) to the AD tissue demonstrated that the MS methodology was capable of observing dimeric Aβ at femto-molar concentrations, with no noticeable effect on monomeric Aβ levels. Focus turned to the association between SDS-PAGE and levels of observable dimeric Aβ within the AD brain tissue. These investigations revealed that increased levels of dimeric Aβ were observed with increasing concentrations of SDS in the sample buffer. This finding was subsequently confirmed using synthetic Aβ1–42 and suggests that SDS was inducing the formation of dimeric Aβ. The findings that SDS promotes Aβ dimerization have significant implications for the putative role of low-order oligomers in AD pathogenesis and draw into question the utility of oligomeric Aβ as a therapeutic target.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The formation of β-amyloid (Aβ) oligomers in the brain and the subsequent progressive deposition of Aβ are believed to play central roles in Alzheimer’s disease (AD) pathogenesis [12]. Initial investigations proposed that fibrillar Aβ was responsible for mediating neurotoxicity, however, it has long been recognized that fibrillar plaque density and number do not correlate with clinical severity [9, 45]. This finding, supported by recent imaging studies [27, 37], has led researchers to look elsewhere. More recent investigations have indicated that a strong correlation exists between cognitive impairment and soluble Aβ isoforms [26]. Further attempts to characterize these oligomers, primarily relying on Western blot methods, have implicated SDS-stable, low-order, oligomeric Aβ as the form of Aβ most likely to cause neuronal dysfunction [26, 36, 51, 54]. Not surprisingly the inhibition of Aβ and its oligomers have become attractive targets for therapeutic interventions [51], albeit with limited success [38]. Most recently, the humanized monoclonal antibodies bapineuzumab, directed at residues 1–5 of the Aβ peptide, and solanezumab, directed at residues 16–24 of the Aβ peptide, both failed to meet their primary measures of cognition and daily living [15]. These disappointing results have led to speculation within the field of whether targeting Aβ’s accumulation and aggregation remains a legitimate therapeutic strategy.
Initial support for the role of SDS-stable, low-order oligomers stemmed from investigations using Chinese hamster ovary (CHO) cells expressing mutant (V717F) amyloid precursor protein (APP), known as 7PA2 cells. In addition to monomeric Aβ, 7PA2 cells have been reported to naturally secrete SDS-stable, low-order Aβ oligomers which inhibit synaptic function and disrupt long-term potentiation (LTP), a correlate of memory formation [16, 32, 50, 52, 53]. Similar disruptions to synaptic function and LTP have recently been reported using low-order oligomeric Aβ isolated from AD-affected human cortical tissue [39].
Given the increasing focus on low-order oligomeric Aβ over the past years we have sought to develop techniques capable of analyzing these species using both synthetic Aβ and biological tissues. The use of SELDI-TOF MS, in conjunction with hydrophobic arrays, enabled the resolution of synthetic Aβ1–42 species ranging from monomeric to tetrameric bound to lipid rafts [13]. Furthermore, the use of Aβ-specific antibodies in conjunction with SELDI-TOF MS enabled the observation of trimeric Aβ in the brains of transgenic mouse models of AD [7] and the observation of dimeric Aβ in the blood cellular fraction of AD-affected patients [49].
To date despite the ability of high-resolution MS techniques to analyze low-order oligomeric Aβ species, investigations into AD-affected cortical tissue have remained reliant on the lower resolution western blotting techniques. The current investigation aimed to assess whether the ability to observe low-order Aβ oligomers using SELDI-TOF MS was transferable to AD-affected human cortical tissue. The investigation also aimed to evaluate whether the high-throughput xMAP multiplex immunoassay could be adapted to measure soluble levels of Aβ in AD-affected cortical tissue. xMAP uses bead-based antibody capture compared to ELISAs microplate wells which result in smaller sample volume requirements and a reduction in non-specific binding [5]. xMAP is fast becoming a widely used technique in the analysis of Aβ levels in plasma [10, 20, 48] and cerebrospinal fluid [30, 40].
Materials and methods
Collection and characterization of brain tissue
Human brain tissue was collected at autopsy. The sourcing and preparation of the tissue was conducted by the Australian Brain Bank Network (ABBN). Pathological diagnosis of AD was made in accordance with the standard criteria set out by the National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease [6, 28] and pathological diagnosis of frontotemporal lobar degeneration (FTLD) was made in accordance with the guidelines established by the Work Group on Frontotemporal Dementia and Pick’s Disease [21, 25]. All procedures described in this manuscript were conducted in accordance with the Florey Neurosciences Ethics Committee.
Preparation of human brain homogenate
Grey matter was isolated from the post-mortem frontal cortices of neurologically normal, FTLD, familial AD (fAD) and sporadic AD-affected individuals. The isolated tissue was subsequently added to 1× Dulbecco phosphate buffered saline (PBS), devoid of Mg and Ca, (Invitrogen) but containing ethylenediaminetetraacetic acid (EDTA)-free protease inhibitors (Roche) to yield 100 mg tissue per mL of buffer. The brain sample was then homogenized, using an ultrasonic cell disrupter (Virsonic 600; VirTis, Gardiner, NY), three times for 30 s each at 24,000 rpm. The protein concentrations of the samples were assessed using the Bradford Assay (BioRad) before the homogenates were aliquoted and stored at −80 °C until required. All sample preparation was undertaken on ice.
Western blot analysis of brain tissue
Brain homogenates were diluted in SDS-loading buffer, reduced in 10 % β-mercaptoethanol and heated at 90 °C for 5 min. Protein samples were then separated by 4–10 % SDS-PAGE (Invitrogen) and transferred onto nitrocellulose membranes. Non-specific immunoreactivity was controlled for by blocking the membranes with 5 % non-fat skim milk diluted in tris-buffered saline (TBS) before the membranes were probed overnight with the WO2 antibody [1:500], or for synaptophysin (1:75,000: Millipore). WO2 binds to the N terminus of Aβ at residues 5–8 [14] and was obtained from the WEHI Monoclonal Antibody Facility. Subsequent to the incubation, membranes underwent three 10-min washes with 0.1 % TBS/Tween before the immunocomplex was probed with horseradish peroxidise (HRP)-conjugated secondary antibodies.
Probed membranes were washed three times in 0.1 % TBS/Tween for 10 min each, and the immunoreactive proteins were detected using enhanced chemiluminescence upon reaction with ECL (Millipore). Relative optical density of the blots was undertaken using Image J (1.44p) and western blot densitometry data were normalized to the loading control, β-tubulin.
SELDI-TOF MS analysis of brain tissue
Mass spectrometric analysis of the brain tissue was carried out using ProteinChip® PS10 Arrays (Bio-Rad; CAT #C55-30044) loaded with the specific antibody, WO2 (2 μL at 0.25 mg/mL) or G2–10 (2 μL at 0.25 mg/mL). G2–10 binds to the C terminus of Aβ40 at residues 33–40 [14] and was made in-house. Chips were then incubated overnight at 4 °C in a humidity chamber before excess antibodies were removed and 10 μL blocking buffer (0.5 M ethanolamine in PBS) was added and incubated for 30 min. After the removal of the blocking buffer, arrays were washed three times for 5 min with 120 μL of 0.5 % Triton X-100/PBS (wash buffer) followed by three 5 min washes with 100 μL PBS.
Brain homogenates (40 μL) were prepared with 40 μL of urea 8 M and 240 μL of wash buffer and placed for 10 min in an ultra-sound bath with ice. One hundred and fifty microlitres of sample mix was added to each spot and incubated at room temperature for 3 h.
Subsequent to incubation, samples were removed and the arrays underwent three 10-s washes on a vigorous shaking table with 150 μL wash buffer, and 150 μL PBS, followed by two washes with 150 μL HEPES. The arrays were then dried before two 1 μL aliquots of a 50 % saturated solution of sinapinic acid (SPA) energy absorbing molecules (EAM) were applied, with air-drying between treatments. The 50 % saturated solution was prepared by suspending 5 mg ProteinChip SPA EAM (Bio-Rad; CAT # C30-00002) in 0.5 % trifluoroacetic acid (TFA; Sigma-Aldrich; St Louis, Missouri), 50 % acetonitrile [ACN; high performance liquid chromatography (HPLC) grade] and 15 % isopropyl alcohol (IPA; HPLC grade) and 34.5 % HPLC-grade H2O.
Each sample was analyzed in duplicate. All arrays were analyzed blind to diagnostic status using a ProteinChip SELDI System Enterprise Edition (BioRad). All spectra were internally normalized and peak intensities were normalized using total ion current. Peak detection was carried out using the inbuilt BioRad ProteinChip Data Manager Biomarker Wizard (Version 3.07.004). Peaks were auto-detected to clusters with both first pass signal-to-noise ratio and valley depth set at 5.0, and a minimum peak threshold of 20 % of all spectra. Cluster completion was achieved with a Cluster Window Mass of 1.0 peak width and a second pass signal-to-noise ratio and valley depth of 2.0. Aβ species, including potential oxidations, were matched to peaks within the resulting spectra using m/z.
INNO-BIA xMAP analysis of brain tissue
The INNO-BIA plasma Aβ forms assays (Innogenetics NV, Ghent, Belgium), a multiplex microsphere-based Luminex xMAP approach, were adapted to analyze Aβ levels in the PBS-soluble fraction of the brain homogenate, prepared by centrifuging the sample for 30 min at 15,000×g at 4 °C. Aβ1–40, Aβ1–42 and Aβ n–40, Aβ n–42 were measured using Module A and B of the immunoassay kit, respectively.
Assay day 1
All kit components and samples were brought to room temperature (temperature range 23–25 °C) on a laboratory bench. Samples (1 mg tissue/4 mL buffer) were centrifuged at 1,500×g for 5 min at room temperature to pellet any precipitates. The samples were then immediately diluted 1:3 in the kit diluent and left at room temperature for an average of 30 min while the plate and working solutions were prepared. The beads were vortexed and then incubated for 10 min in a sonicating water bath at room temperature. A mixture of a 1:100 bead stock solution was distributed to 96-well filter plates provided in the kit (Millipore Corporation, Bedford, Massachusetts), using calibrated manual multichannel pipettes. The wells were drained by vacuum manifold (Millipore Corporation, Bedford, Massachusetts). Then 75 μL of standards, blanks and samples were added to the wells in duplicate, followed by the addition of biotinylated detector MAb (25 μL/well) (MAb 3D6 for Module A and MAb 2G3 for Module B) and incubated overnight at 2–8 °C in the dark (plates covered with aluminium foil) on a plate shaker (600 rpm). In addition, on each plate a two-run validation control samples consisting of synthetic Aβ1–40, Aβ1–42 were also included.
Assay day 2
The plates were allowed to come back to room temperature on an orbital shaker, covered in foil and were then washed three times with diluted wash buffer provided in the kit. After the last wash step, the detection reagent comprising diluted phycoerythrine-labelled streptavidine was added (100 μL/well) and incubated for 1 h on an orbital shaker at room temperature, covered in foil. The plates were then washed 3× times with wash buffer and 100 μL of reading solution, provided in the kit, was added to each well and incubated for 10 min at room temperature on an orbital shaker and then analyzed. All assays were analyzed on a Bio-Rad Bioplex 200 instrument using v5 software (Bio-Rad Laboratories, USA) with 5PL regression type logistic. The data were fitted to a calibration curve constructed with the median fluorescence values for each replicate of the standards and concentrations were determined by sigmoidal curve fitting. The same batch number was used for all kits assayed in the study and the same algorithm was used for the four analytes.
Preparation of monomeric Aβ1–42 and Aβ1–40Met35(O) dimer
Synthetic Aβ1–42 was purchased from W. M. Keck Laboratory (Yale University, New Haven, CT). Continuous-flow Fmoc-SPPS (solid-phase peptide synthesis) was used for the synthesis of Aβ1–40 peptides. Peptides were synthesized on a 0.1-mmol scale using Fmoc-L-Ala-PEG-PS resin as a solid support on an Applied Biosystems Pioneer Synthesizer as described previously [47]. Aβ peptides were dissolved in HFIP at a concentration of 1 mg/ml (w/v) to induce a monomeric and helical conformation of the peptides [43]. Aliquots of 100 μL were dried under vacuum. Dried samples were stored at −80 °C.
Resin-bound Aβ11–42 Met35(O) was prepared according to standard methods [1, 47]. Dityrosine was prepared according to the previously reported method [2, 42]. Fmoc protection of dityrosine and incorporation into SPPS of the Aβ1–40Met35(O) dimer were performed according to the previously reported method [18].
Peptide stock solutions were prepared by making a 250 μM concentration by weight, dissolving the peptide in 20 mM NaOH, and then diluting in 1× PBS (1:5). Solutions were sonicated in an ice-water bath for 5 min and spun for 5 min at 15,800×g at 4 °C. The supernatant was retained. The resulting Aβ concentration was determined using a molar extinction coefficient value of 75,887 L/mol/cm and measuring absorbance at 214 nm before adjusting the final concentration of the stock solution to 200 μM. The Aβ stock solution was subsequently diluted with the stock 20 mM NaOH in 1× PBS solution to a concentration of 2 μM for analysis using western blots. Synthetic Aβ analyzed by SELDI-TOF MS was prepared in the aforementioned urea/wash buffer solution. All chemicals in the preparation were purchased from Sigma unless otherwise stated.
Statistical analysis
Comparisons across the groups for Aβ levels and demographic data were conducted using Chi-square tests and one-way analysis of variance (ANOVA) with psost hoc Tukey’s multiple comparisons tests. Bivariate pairwise comparisons were undertaken using Pearson’s correlation. Data are presented as mean ± standard deviation (SD) unless otherwise stated. All analyses were performed using Graphpad Prism® for Windows (Version 5.03, 2009). Outliers deemed significant by the Extreme Studentized Deviate method (ESD) were removed. Statistical comparisons, other than correlations, did not include data from the fAD tissue.
Results
Subject characteristics
Clinical information pertaining to the 26 individuals from whom brain tissue was collected is outlined in Table 1. Comparative analysis across the groups revealed a significant difference regarding age at death (p = 0.0206) with post hoc analyses revealing that the fAD group were significantly younger than the AD group at time of death. Despite trends towards lower total brain weight in diseased tissue compared to control tissue no significant differences were observed. Significant differences were not observed across the group for gender ratios and PMI. Evidence of congophilic amyloid angiopathy (CAA) was evident in four of the AD cases, in addition to two of the fAD cases.
Western blot analysis of Aβ in human brain homogenate
WO2 western blot analysis of the frontal brain homogenate demonstrated that bands consistent with putative ‘monomeric’ and ‘dimeric’ Aβ species were clearly visible within all AD brain homogenates in addition to a small proportion of control cortical tissue (Fig. 1a), consistent with previously published reports [33]. Densitometric analysis of the blots revealed that significant differences were evident across the diagnostic groups regarding levels of both ‘monomeric’ (p = 0.0026) and ‘dimeric’ Aβ (p = 0.0161) with post hoc analyses revealing that elevated levels of both bands were observed in the AD tissue compared to both control and FTLD tissue (Fig. 1b, c).

WO2 western blot analysis of human brain homogenates. a Representative western blot showing bands consistent with ‘monomeric’ and ‘dimeric’ Aβ. Densitometric analysis of the b ‘monomeric’ Aβ and c ‘dimeric’ Aβ bands in conjunction with ANOVA revealed that both ‘monomeric’ and ‘dimeric’ bands were significantly elevated within the AD tissue. β-tubulin loading control was used to normalize data. d Pearson’s correlation revealed a significant positive correlation between the ‘monomeric’ and ‘dimeric’ Aβ bands visualized by the WO2 western blots. The ΔE9 fAD case was shown to be an outlier and was removed from this analysis so as not to drive the positive correlation
The association between the ‘monomeric’ and ‘dimeric’ bands was assessed and revealed a significant positive correlation between the ‘monomeric’ and ‘dimeric’ Aβ bands (r = 0.696, p = 0.0001) (Fig. 1d). Data pertaining to the ΔE9 fAD case, which showed high levels of both monomeric and dimeric Aβ, was shown to be an outlier and was removed from this analysis so as not to drive the positive correlation.
xMAP analysis of Aβ in human brain homogenate
The adaptation of the xMAP INNO-BIA plasma Aβ forms assays in the current investigation enabled analysis of the following Aβ isoforms within the PBS-soluble fraction of the brain homogenates: Aβ1–40, Aβ1–42 and Aβ n–40, Aβ n–42 (Table 2).
Comparative analysis of Aβ levels showed that significant differences across the groups were evident for levels of Aβ n–40 (p = 0.0443), Aβ1–42 (p < 0.0001), and Aβ n–42 (p = 0.0004). However, no significant difference was found for Aβ1–40. Post hoc analyses demonstrated that the AD brain fractions had significantly higher levels of Aβ n–40 compared to control tissue and significantly higher levels of Aβ1–42 and Aβ n–42 compared to both control and FTLD tissue.
SELDI-TOF MS analysis of Aβ in human brain homogenate
Profiling of the human brain homogenates using WO2 in conjunction with SELDI-TOF MS indicated that the monomeric band resolvable by western blot consisted of a number of n-terminally truncated Aβ peptides ranging from Aβ5–42 through Aβ1–42 (Fig. 2a). A comparison between the expected molecular weight and observed molecular weights of the Aβ peptides observed in the spectra is outlined in Table 3. As expected Aβ1–42 was the predominant species observed in the sporadic AD brains, however, Aβ4–42 was also observable at near equivalent levels, despite being largely overlooked in the literature whilst more widely studied species such as pEAβ3–42 were only observable at low levels (Fig. 2b). A peak observed with an average mass of 4329 Da could be attributed to either Aβ3–42 (4328 Da) or Aβ1–40 (4330 Da) as the resolution of the SELDI-TOF MS would not allow for a distinction to be made. However, subsequent analysis of the AD homogenate with G210, an Aβ40 specific antibody, showed no peaks consistent with any Aβ n–40 species unless the homogenate was spiked with synthetic Aβ1–40 prior to analysis (Fig. 2c). Whilst Aβ1–40 was not observed using G210, low levels of Aβ1–42 were observable in the AD tissue; likely the result of non-specific binding.

a Representative WO2 SELDI-TOF MS spectra of AD, FTLD and neurologically normal cortical tissue. b Spectra arising from the AD tissue displayed an array of N-terminally truncated Aβ species of which Aβ4–42 and Aβ1–42 were the predominant species. Data represent average peak intensity of Aβ species observed within AD tissue. c The peak observable at 4,329 Da could have been attributed to either Aβ3–42 (4,329 Da) or Aβ1–40 (4,330 Da). However, analysis of the AD homogenate with G210, an Aβ40-specific antibody, showed no peaks consistent with Aβ n –40 species unless the homogenate was spiked with synthetic Aβ1–40 prior to analysis. Low levels of Aβ1–42 likely resulting from non-specific binding with G210 were observable in the AD tissue
Comparative analysis of these Aβ species showed that there were significant differences across the groups for Aβ4–42 (p = 0.0313), pEAβ3–42 (p = 0.0154), Aβ3–42 (p = 0.0090), Aβ2–42 (p = 0.0014), and Aβ1–42 (p = 0.0028), however, no significant difference was observed across the groups for Aβ5–42 (Fig. 3). Post hoc analyses indicated that there were significantly higher levels of pEAβ3–42 and Aβ3–42 in the AD brain homogenate compared with control tissue and significantly higher Aβ2–42 and Aβ1–42 in the AD tissue compared to both FTLD and control homogenate.

Comparative analysis of the Aβ species observed within the WO2 SELDI-TOF MS spectra revealed that whilst no significant difference was found across the diagnostic groups with regard to a Aβ5–42, significant differences were observed for peaks corresponding to b Aβ4–42 (p = 0.0313), c pEAβ3–42 (p = 0.0154), d Aβ3–42 (p = 0.0090), e Aβ2–42 (p = 0.0014), and f Aβ1–42 (p = 0.0028). Post hoc analyses indicated that there were significantly higher levels of pEAβ3–42 and Aβ3–42 in the AD brain homogenate compared with control tissue and significantly higher Aβ2–42 and Aβ1–42 in the AD tissue compared to both FTLD and control homogenates
Analysis of fAD brain homogenates
Specific demographic information pertaining to the fAD brain homogenates is outlined in Table 4. Analysis of the fAD homogenates using western blot and SELDI-TOF MS showed that monomeric Aβ profiles in the cortical tissue of those individuals with presenilin point mutations were indistinguishable from those observed in sporadic cases (Fig. 4a, b). The deletion of Exon nine (ΔE9), however, resulted in an Aβ profile distinct from the sporadic cases. Like sporadic AD, the ΔE9 homogenate showed an array of N-terminally truncated Aβ isoforms, however, unlike sporadic AD, the isoforms in the ΔE9 homogenate were n–40 species (Aβ4–40, Aβ2–40, Aβ1–40) (Fig. 4c). Furthermore the ΔE9 homogenate showed Aβ1–38, a species not observed in other fAD or AD spectra. A comparison between the expected molecular weight and observed molecular weights of the Aβ peptides observed in the spectra is outlined in Table 5. Moreover, the levels of Aβ observed in the ΔE9 homogenate were higher than those observed in the other fAD and sAD cases, when analyzed by western, xMAP and SELDI-TOF MS.

a Representative WO2 western blots of the three fAD cases showed that there were elevated levels of observable Aβ/APP fragments within the ΔE9 tissue compared to the two PS1 mutations. b Representative WO2 SELDI-TOF MS spectra showed that the PS1 mutations resulted in Aβ profiles indistinguishable from those observed in the sporadic AD tissue. The Aβ profile arising from the ΔE9 tissue, however, showed a distinct pattern of N-terminally truncated Aβ n–40 species alongside Aβ1–38, pEAβ3–42 and Aβ1–42. The peak visible before pEAβ3–42 in the C263T spectra (4,270 Da) did not match any known Aβ species. c These N terminal species were also readily observable when analyzed with the Aβ40 specific antibody G2–10
Western blot analysis of synaptophysin levels
Synaptophysin levels in the brain homogenates were assessed, using western blots, as a general measure of neuronal health. Comparative analysis of these blots indicated that brain homogenates affected by AD, FTLD and fAD all had significantly lower levels of synaptophysin than the control tissue (p < 0.0001) (Fig. 5a, b).

a Representative western blot analysis of synaptophysin levels within the AD and control tissue. β-tubulin loading control was used to normalize data. b Densitometric analysis in conjunction with one-way ANOVA indicated that brain homogenates affected by AD, FTLD and FAD all had significantly lower levels of synaptophysin than the control tissue (p < 0.0001)
The association between synaptophysin levels and Aβ levels as measured by western blot, xMAP and SELDI-TOF MS was assessed using Pearson’s correlations (Table 6). These analyses revealed that there were significant negative correlations between synaptophysin levels and both ‘monomeric’ Aβ (r = −0.46, p = 0.0168) and ‘dimeric’ Aβ (r = −0.51, p = 0.0083) as measured by western blot. Significant negative correlations were also observed between synaptophysin levels and xMAP measures of Aβ1–40 (r = −0.59, p = 0.0015), Aβ n–40 (r = −0.62, p = 0.0007) and Aβ n–42 (r = −0.41, p = 0.0380), but not between Aβ1–42 and synaptophysin levels. Analyses of the relationships between the individual Aβ species, as measured by SELDI-TOF MS, and synaptophysin levels showed that there was a significant negative correlation between Aβ3–42 and synaptophysin (r = −0.54, p = 0.0040). However, significant correlations with synaptophysin were not observed for Aβ5–42, Aβ4–42, pEAβ3–42, Aβ2–42, nor Aβ1–42.
Comparisons across techniques
Comparisons across the three Aβ measuring techniques were made using Pearson’s correlations (Table 7). ‘Monomeric’ Aβ measured by western blot was found to be positively correlated with xMAP measures of Aβ1–40 (r = 0.72, p < 0.0001), Aβ n–40 (r = 0.52, p = 0.0060) and Aβ n–42 (r = 0.43, p = 0.0277). No significant association between ‘monomeric’ Aβ measured by western and xMAP measure of Aβ1–42 was observed.
Comparisons between the Aβ species measured by xMAP and those measured by SELDI-TOF MS showed that there were significant positive correlations between levels of Aβ1–40 (r = 0.85, p < 0.0001) and Aβ1–42 (r = 0.55, p = 0.0033). Comparisons between the xMAP measure of Aβ n–42 and SELDI-TOF MS Aβ n–42 species, including Aβ1–42, were made by summing the peak intensities of the N-terminally truncated Aβ species observable by SELDI. This analysis revealed that there was also a significant positive correlation between the two Aβ n–42 measures (r = 0.55, p = 0.0033).
Finally comparisons were also undertaken between levels of Aβ as measured by western blot and SELDI-TOF MS, respectively. Peak intensities of the various Aβ isoforms observable using SELDI-TOF MS were summed to account for the less specific separation of individual Aβ isoforms via western blot. Pearson’s correlation showed that there was a significant positive correlation between the summed Aβ peak intensities and the ‘monomeric’ Aβ observable with western blot (r = 0.53, p = 0.0052).
Observing dimeric Aβ using SELDI-TOF MS
Attempts were also made to compare the levels of ‘dimeric’ Aβ observed using western blot with ‘dimeric’ Aβ observable within the MS spectra. However, analysis of the MS spectral range in which ‘dimeric’ Aβ should be visible revealed no observable peaks consistent with any ‘dimeric’ Aβ isoforms (Fig. 6a). The inability to observe the ‘dimeric’ Aβ species, observable by western, within the SELDI spectra raised the possibility that the confirmation of the ‘dimeric’ isoforms may have blocked the WO2 epitope and thus prevented binding from occurring or alternatively that the high abundance of monomeric isoforms were saturating the detector, thus precluding dimeric species from being readily observable. This idea was tested by analyzing the AD cortical tissue spiked with 100 fmol of di-tyrosine cross-linked Aβ1–40Met35(O) (Fig. 6b). The ability of this methodology to analyze dimeric Aβ1–42 has been reported previously [49]. The femto-molar concentrations of synthetic dimeric Aβ1–40 were readily observable within the spectra. Moreover, the concentrations of synthetic dimer observable by SELDI MS were orders of magnitude lower than those observable by western blots (Fig. 6c). These findings raised the possibility that the lack of ‘dimeric’ species observed was not an aberration of the SELDI spectra, but rather that the presence of ‘dimeric’ species within the western blots were confounding these results.

a Representative WO2 SELDI-TOF MS spectra of AD and control tissue demonstrated that whilst monomeric Aβ species were readily observable, peaks consistent with dimeric Aβ were not seen within the spectra. b Analysis of AD homogenate spiked with 100 fM of di-tyrosine cross-linked Aβ1–40Met35(O) demonstrated that the SELDI-TOF MS methodology was capable of observing dimeric Aβ at low concentrations. c WO2 western blot of increasing concentrations of synthetic di-tyrosine cross-linked Aβ1–40Met35(O): A 100 fM, B 100 pM, C 100 nM, D 500 nM, E 1 μM, F 5 μM, and G 10 μM
‘Dimeric’ Aβ and SDS-PAGE
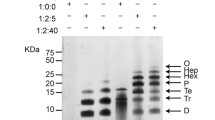
Previous reports in the literature have reported that SDS induces β-sheet formation by Aβ which increases Aβ toxicity [46], promotes the formation of Aβ globulomers [11], and prompts the formation of Aβ trimers [3]. Given these previous reports, the role of SDS in the formation of ‘dimeric’ Aβ was tested. AD brain homogenates were prepared in sample buffer, with concentrations of SDS ranging from 0.5 to 4 %, before being analyzed by western blot (Fig. 7a). Densitometric analysis of the blots showed that there was a significant difference in the levels of observable ‘dimeric’ Aβ across the groups (Fig. 7b). Whilst there was a readily observable trend towards more intense ‘dimeric’ bands in the higher SDS conditions post hoc analyses showed that there were no significant differences across the concentrations. However, when this experiment was repeated using synthetic Aβ1–42 not only was there a significant difference across the range of SDS concentrations, but there was also a significant difference between specific concentrations; there was significantly greater dimeric Aβ in 2 % SDS than 0.5 %, there was significantly greater dimeric Aβ in 3 % SDS than 0.5 % SDS, and 1 % SDS and there was significantly greater dimeric Aβ in 4 % SDS than 0.5 % SDS, 1 % SDS and 2 % SDS (Fig. 7c, d). No significant differences were observed in monomeric Aβ levels (Supplementary Fig. 1). Attempts were made to analyze whether SDS-induced ‘dimeric Aβ’ could be observed using SELDI MS, however, interactions between SDS and the array surface prevented this line of investigation (data not shown).

a WO2 western blots of an AD brain homogenate prepared in sample buffer, with concentrations of SDS ranging from 0.5 to 4 %. a Densitometric analysis of the blots showed that there was a significant difference in the levels of observable ‘dimeric’ Aβ across the groups (p = 0.0462) (n = 3). c WO2 western blots of synthetic Aβ1–42 prepared in sample buffer with concentrations of SDS ranging from 0.5 to 4 %. d Densitometric analysis of the blots showed that there was a significant increase in the ‘dimeric’ Aβ bands observed as the concentration of SDS in the sample buffer was increased (p < 0.0001) (n = 4). Data normalized to normal (2 %) SDS concentration
Discussion
Consistent with the previous reports [19, 33, 44], the current MS investigation demonstrated that the AD-affected cortical tissue is laden with a wide spectrum of N-terminally truncated Aβ n–42 peptides, including Aβ1–42, Aβ2–42, pEAβ3–42, Aβ3–42, Aβ4–42 and Aβ5–42. Of these, Aβ1–42 and Aβ4–42 were the predominant species detected; a finding which was consistent with earlier reports [19, 33, 44]. To date, Aβ1–42 has received the lion’s share of the attention whilst Aβ4–42 has largely been overlooked, despite both peptides being isolated from the AD-affected brain over 25 years ago [23]. The tendency for the scientific community to overlook Aβ4–42 potentially stems from the faster aggregation kinetics and higher β-sheet formation associated with N-terminally truncated Aβ isoforms, making Aβ4–42 a difficult peptide to synthesize [24, 31]; a fact that the current authors can anecdotally confirm.
Familial forms of the disease, resulting from point mutations in PSEN1, resulted in cortical monomeric Aβ profiles which were indistinguishable from those observed in sporadic AD, albeit occurring some 10 years earlier in the subject’s life. However, the brain tissue arising from the carrier of a ΔE9 exhibited pEAβ3–42 and Aβ1–42 alongside a predominant Aβ1–40 peak and a series of N-terminally truncated Aβ n–40 isoforms. The ΔE9 mutation has been reported as having an unusual but characteristic pattern of Aβ deposition known as ‘cotton wool’ plaques and has been shown to result in higher ratios between Aβ1–40 and Aβ1–42 in human cortical tissue than those observed in sporadic AD and other PS-1 mutations [22]. Higher ratios between Aβ1–40 and Aβ1–42 have also been observed in mouse embryonic fibroblasts with the ΔE9 mutation compared to other PS1 variants [4]. A majority of histopathological investigations conducted to date have reported a significant increase in Aβ1–42 observable within the ‘cotton wool plaques’ [17, 55]; however, in some individual cases Aβ1–40 has been shown to be the predominant species [55]. On the surface, these reports appear to contradict the current findings of abundant Aβ n–40 species within ΔE9-affected tissue, however, the methodologies utilized in the current study would not have disrupted the abundant Aβ1–42 sequestered in plaques and raise the possibility that the ΔE9 mutation results in increased levels of soluble Aβ n–40 species.
The adaptation of the xMAP INNO-BIA assays in the current study demonstrated that such high-throughput techniques can successfully be used to assess Aβ levels in human cortical tissue. Significant correlations between Aβ levels as measured by xMAP, Western blot or SELDI-TOF MS suggest that these techniques appear to be measuring overlapping pools of Aβ, at least with regard to Aβ n–42 species. The significantly higher levels of Aβ n–40 species observed in the AD fractions using the xMAP assays were somewhat surprising as they did not correspond to the Aβ profiles observed by SELDI-TOF MS. This discrepancy could be due to the differences in the antibodies used in the two methods; however, the observation of Aβ n–40 species in the familial tissue suggests that this was unlikely. Alternatively the discrepancy might arise from the xMAP antibodies either binding larger aggregates of Aβ/APP, that fall outside the range of MS detection, or Aβ species other than the Aβ n–40 species that the antibodies are reported to be specific for. These notions are, however, somewhat difficult to evaluate, due to the fact that xMAP works in a proverbial “black box”, measuring a simple immunoresponse that gives no indication to the possible range of species being detected; although, future investigations that can adapt the xMAP antibodies for analysis using SELDI-TOF MS may provide a better approach.
At this stage, it is important to note the differences between SELDI-TOF MS and more traditional MS investigations into Aβ levels in human brain. Traditional MS studies rely on serial high-speed centrifugations of the human samples in buffers such as TBS/PBS and formic acid before the ‘soluble’ Aβ is collected and analyzed using immunoprecipitation (IP) with an Aβ-specific antibody [29, 33]. The captured Aβ is subsequently eluted from the IP beads and freeze-dried before being resuspended and analyzed using MS to determine the proteomic profile of Aβ in the human brain. Unlike traditional MS techniques, SELDI-TOF MS involves an on-array IP which eradicates the need for samples to undergo centrifugation, elution, freeze-drying and re-suspension. Instead the Aβ in the samples binds directly to the Aβ-specific antibodies bound to the array surface, thus allowing a more direct analysis of Aβ levels within the human brain homogenates.
Investigations into Aβ1–40 levels in sporadic AD tissue resulted in a number of contradictory observations. Analysis by SELDI-TOF MS with the Aβ40-specific antibody (G2–10) suggested that the peak observed at 4329 Da in the AD tissue belonged to Aβ3–42. Analysis by xMAP, however, suggested that there was more than 100 pg/mL of Aβ1–40 and more than 200 pg/mL of Aβ n–40 within the AD tissue, levels which the SELDI-TOF MS was more than capable of measuring. The lack of Aβ n–40 species observable within the MS spectra raises the question of whether the xMAP system is measuring aggregated Aβ n–40 species which lie outside the dynamic range of SELDI-TOF MS or whether the signals are the result of non-specific binding occurring with other proteins in the samples. Previous MS studies have reported that Aβ1–40 is readily observable when formic acid extractions are used to disrupt Aβ plaques, a finding which has been confirmed by MS/MS techniques [29, 33]. In addition, a number of MS studies report observing Aβ1–40 within the soluble fractions of brain often in the midst of a range of Aβ n–42 isoforms [19, 44]; however, like the current study, these studies invariably relied on m/z for the identification of these peaks, raising the possibility that soluble levels of Aβ3–42 have erroneously been reported as Aβ1–40 in the literature.
The formation of small-order Aβ oligomers has long been thought of as a fundamental stage in the pathogenesis of AD and over the past decade these oligomeric species have become an attractive target for AD therapeutics [38, 51]. Consistent with the previously published reports, bands consistent with ‘dimeric’ Aβ were readily observable in western blots of both synthetic Aβ and AD-affected cortical tissue. However, peaks consistent with ‘dimeric’ Aβ were not observed in the MS spectra arising from AD tissue, despite the abundance of monomeric Aβ isoforms. It is possible that differences in the sample buffer used for western blots and SELDI-TOF MS, namely the presence of urea in the MS sample buffer, may have been a limiting factor in the observation of small-order Aβ oligomers in the MS spectra. However, as the methodology had previously been utilized to observe Aβ oligomers in mouse brain homogenates [7] and human blood [49], in addition to the capture of synthetically derived dimeric Aβ in the present study, it is unlikely that the presence of urea precluded the observation of dimeric Aβ in the MS spectra. With this in mind, the lack of observable dimeric Aβ in the current MS analysis means that a MS spectrum of brain-derived, low-order oligomeric Aβ remains unreported in the scientific literature.
Interactions between SDS and Aβ have been widely reported in the literature with a number of studies reporting that SDS induces Aβ β-sheet formation [35, 46], increases Aβ toxicity [46], Aβ globulomer formation [11], and Aβ trimer formation [3]. The findings of the current study indicate that SDS is also capable of inducing the dimerization of Aβ, both in its synthetic form and in isoforms derived from AD-affected cortical tissue. It is noted that whilst significantly different, the SDS induction of dimeric Aβ appeared to be less responsive in the brain homogenates when compared to experiments using synthetic Aβ. This difference is likely to be due to the variations in the complexity of the proteomic environments and the availability of additional Aβ interactions in the two experimental settings. The current findings are in line with a recent investigation by Rangachari et al. [35] who reported that Aβ1–42 undergoes rapid conformational changes in the presence of SDS leading to conversion of monomeric Aβ1–42 to bands consistent with low-order oligomeric Aβ species, ranging from dimer through tetramer. In addition, studies using 7PA2 cells which have shown that bands previously reported as corresponding to ‘dimeric’ and ‘trimeric’ Aβ can also be increased through the inhibition of BACE1 [34]. However, rather than increasing the rate of Aβ oligomerization, inhibition of BACE1 in the cells reportedly led to differential processing of APP, resulting in a number of larger APP/Aβ fragments spanning the β-secretase site.
It has been widely established that Aβ is a pleomorphic peptide whose structure has been found to be dependent on its environmental context, with metals, lipid membranes, salts, detergents, and pH levels all significantly affecting Aβ’s structure. In a biological context, protein/peptide structure and function are intimately linked, consequently not only does the environment of the peptide modify its structure but it will also impact on its subsequent toxicity [3, 8, 11, 35, 41, 46]. Such environmental effects make isolating and analyzing small-order oligomers of Aβ problematic as each proteomic technique exerts a different set of environmental constraints on the Aβ peptide, some resulting in the formation of species that may not always be observed in a native setting. Therefore, ideally it would be best if methods could be developed to identify various forms of Aβ in situ as any manipulation has the potential to introduce artefacts. However, given that these methods do not yet exist, in this work we have striven to restrict manipulation of the samples using simple brain homogenates and the limited sample preparation required by SELDI-TOF MS. With this in mind, the lack of small order oligomers observed using SELDI-TOF MS suggests that prevailing notions regarding the importance of dimeric Aβ may be resulting from an over interpretation of the published literature rather than an accurate depiction of Aβ’s structure and role in AD pathogenesis. The current findings do, however, reaffirm that given the right context Aβ, from human tissue, is capable of forming small-order oligomeric structures. However, whether these structures are stable and isolatable or whether the structural plasticity of these Aβ assemblies makes attempts to isolate them futile remains to be seen.
The reliance on low-resolution analytical techniques in the formation of the Aβ-centric theory of AD has potentially led to an overly simplistic understanding of APP processing and Aβ oligomerization. For over a decade, this knowledge has been used as the cornerstone of AD therapeutics which has repeatedly attempted to regulate Aβ oligomerization and aggregation within the AD brain; however, to date these attempts have met with limited success. The advent and advances of high-resolution techniques, such as MS, provide researchers with the tools to re-examine a number of the now widely held assumptions of AD pathogenesis. These techniques can now be utilized to dispel common misconceptions in the field and gain a more thorough understanding of Aβ’s role in AD pathology, a knowledge that could potentially be translated into a more rational and effective approach to the treatment of this devastating disease.
References
Barnham KJ, Ciccotosto GD, Tickler AK, Ali FE, Smith DG, Williamson NA, Lam YH, Carrington D, Tew D, Kocak G, Volitakis I, Separovic F, Barrow CJ, Wade JD, Masters CL, Cherny RA, Curtain CC, Bush AI, Cappai R (2003) Neurotoxic, redox-competent Alzheimer’s b-amyloid is released from lipid membrane by methionine oxidation. J Biol Chem 278(44):42959–42965
Barnham KJ, Haeffner F, Ciccotosto GD, Curtain CC, Tew D, Mavros C, Beyreuther K, Carrington D, Masters CL, Cherny RA, Cappai R, Bush AI (2004) Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease b-amyloid. Faseb J 18(12):1427. doi:10.1096/fj.04-1890fje
Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB (2003) Amyloid b-protein (Ab) assembly: Ab40 and Ab42 oligomerize through distinct pathways. Proc Natl Acad Sci USA 100(1):330–335
Cacquevel M, Aeschbach L, Houacine J, Fraering PC (2012) Alzheimer’s disease-linked mutations in presenilin-1 result in a drastic loss of activity in purified γ-secretase complexes. PLoS ONE 7(4):e35133. doi:10.1371/journal.pone.0035133
Carson RT, Vignali DAA (1999) Simultaneous quantitation of 15 cytokines using a multiplexed flow cytometric assay. J Immunol Methods 227(1–2):41–52. doi:10.1016/s0022-1759(99)00069-1
The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease Neurobiol Aging 18(4 Suppl):S1–2
Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, Filiz G, Perez KA, Nurjono M, Caragounis A, Du T, Laughton K, Volitakis I, Bush AI, Li QX, Masters CL, Cappal R, Cherny RA, Donnelly PS, White AR, Barnham KJ (2009) Increasing Cu bioavailability inhibits Ab oligomers and tau phosphorylation. Proc Natl Acad Sci USA 106(2):381–386. doi:10.1073/pnas.0809057106
Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ (2001) Alzheimer’s disease amyloid-b binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem 276:20466–20473
Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 16(3):285–298
Figurski MJ, Waligórska T, Toledo J, Vanderstichele H, Korecka M, Lee VMY, Trojanowski JQ, Shaw LM (2012) Improved protocol for measurement of plasma β-amyloid in longitudinal evaluation of Alzheimer’s Disease Neuroimaging Initiative study patients. Alzheimer Dement 8(4):250–260
Gellermann GP, Byrnes H, Striebinger A, Ullrich K, Mueller R, Hillen H, Barghorn S (2008) Ab-globulomers are formed independently of the fibril pathway. Neurobiol Dis 30(2):212–220
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356. doi:10.1126/science.1072994
Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ (2008) Amyloid-b peptide (Ab) neurotoxicity is modulated by the rate of peptide aggregation: Abeta dimers and trimers correlate with neurotoxicity. J Neurosci 28(46):11950–11958
Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters CL, Beyreuther K (1996) Analysis of heterogeneous b-A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive western blot assay. J Biol Chem 271:22908–22914
Karran E (2012) Current status of vaccination therapies in Alzheimer’s disease. J Neurochem 123(5):647–651. doi:10.1111/jnc.12009
Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ (2005) Amyloid-b protein immunotherapy neutralizes Ab oligomers that disrupt synaptic plasticity in vivo. Nat Med 11(5):556–561
Koivunen J, Verkkoniemi A, Aalto S, Paetau A, Ahonen J-P, Viitanen M, Någren K, Rokka J, Haaparanta M, Kalimo H, Rinne JO (2008) PET amyloid ligand [11C]PIB uptake shows predominantly striatal increase in variant Alzheimer’s disease. Brain 131(7):1845–1853. doi:10.1093/brain/awn107
Kok WM, Scanlon DB, Karas JA, Miles LA, Tew DJ, Parker MW, Barnham KJ, Hutton CA (2009) Solid-phase synthesis of homodimeric peptides: preparation of covalently-linked dimers of amyloid-b peptide. Chem Commun (Camb) 41:6228–6230. doi:10.1039/b912784d
Lewis H, Beher D, Cookson N, Oakley A, Piggott M, Morris CM, Jaros E, Perry R, Ince P, Kenny RA, Ballard CG, Shearman MS, Kalaria RN (2006) Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-β(42) peptide in vascular dementia. Neuropathol Appl Neurobiol 32(2):103–118. doi:10.1111/j.1365-2990.2006.00696.x
Lui JK, Laws SM, Li QX, Villemagne VL, Ames D, Brown B, Bush AI, De Ruyck K, Dromey J, Ellis KA, Faux NG, Foster J, Fowler C, Gupta V, Hudson P, Laughton K, Masters CL, Pertile K, Rembach A, Rimajova M, Rodrigues M, Rowe CC, Rumble R, Szoeke C, Taddei K, Taddei T, Trounson B, Ward V, Martins RN, Grp AR (2010) Plasma amyloid-b as a biomarker in Alzheimer’s disease: the AIBL study of aging. J Alzheimers Dis 20(4):1233–1242. doi:10.3233/jad-2010-090249
Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122(1):111–113. doi:10.1007/s00401-011-0845-8
Mann DM, Takeuchi A, Sato S, Cairns NJ, Lantos PL, Rossor MN, Haltia M, Kalimo H, Iwatsubo T (2001) Cases of Alzheimer’s disease due to deletion of exon 9 of the presenilin-1 gene show an unusual but characteristic b-amyloid pathology known as ‘cotton wool’ plaques. Neuropathol Appl Neurobiol 27(3):189–196
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82:4245–4249
McColl G, Roberts BR, Gunn AP, Perez KA, Tew DJ, Masters CL, Barnham KJ, Cherny RA, Bush AI (2009) The Caenorhabditis elegans Ab 1–42 model of Alzheimer disease predominantly expresses Ab 3–42. J Biol Chem 284(34):22697–22702. doi:10.1074/jbc.C109.028514
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58(11):1803–1809. pii:nsa10000
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Ab amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46:860–866
Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC (2006) [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 67(3):446–452. doi:10.1212/01.wnl.0000228230.26044.a4
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123(1):1–11. doi:10.1007/s00401-011-0910-3
Moore B, Chakrabarty P, Levites Y, Kukar T, Baine A-M, Moroni T, Ladd T, Das P, Dickson D, Golde T (2012) Overlapping profiles of Ab peptides in the Alzheimer’s disease and pathological aging brains. Alzheimer’s Res Ther 4(3):18
Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K (2005) Simultaneous measurement of β-amyloid(1–42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem 51(2):336–345. doi:10.1373/clinchem.2004.039347
Pike CJ, Overman MJ, Cotman CW (1995) Amino-terminal deletions enhance aggregation of b-amyloid peptides in vitro. J Biol Chem 270:23895–23898
Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ (1995) Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem 270(16):9564–9570
Portelius E, Bogdanovic N, Gustavsson MK, Volkmann I, Brinkmalm G, Zetterberg H, Winblad B, Blennow K (2010) Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol 120(2):185–193. doi:10.1007/s00401-010-0690-1
Portelius E, Olsson M, Brinkmalm G, Ruetschi U, Mattsson N, Andreasson U, Gobom J, Brinkmalm A, Holtta M, Blennow K, Zetterberg H (2013) Mass spectrometric characterization of amyloid-beta species in the 7PA2 cell model of Alzheimer’s disease. J Alzheimer Dis 33(1):85–93. doi:10.3233/jad-2012-120994
Rangachari V, Moore BD, Reed DK, Sonoda LK, Bridges AW, Conboy E, Hartigan D, Rosenberry TL (2007) Amyloid-beta(1–42) rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfatet. Biochemistry 46(43):12451–12462
Roher AE, Chaney MO, Kuo YM, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, Bonnell BS, Emmerling MR (1996) Morphology and toxicity of Ab-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J Biol Chem 271:20631–20635
Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O’Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL (2007) Imaging beta-amyloid burden in aging and dementia. Neurology 68(20):1718–1725
Selkoe DJ (2011) Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med 17(9):1060–1065
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14(8):837–842
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ (2009) Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 65(4):403–413. doi:10.1002/ana.21610
Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S, Lieberburg I, Becker GW, Brems DN et al (1994) Secondary structure of amyloid b peptide correlates with neurotoxic activity in vitro. Mol Pharmacol 45:373–379
Skaff O, Jolliffe KA, Hutton CA (2005) Synthesis of the side chain cross-linked tyrosine oligomers dityrosine, trityrosine, and pulcherosine. J Org Chem 70(18):7353–7363. doi:10.1021/jo051076m
Smith DP, Smith DG, Curtain CC, Boas JF, Pilbrow JR, Ciccotosto GD, Lau TL, Tew DJ, Perez K, Wade JD, Bush AI, Drew SC, Separovic F, Masters CL, Cappai R, Barnham KJ (2006) Copper-mediated amyloid-beta toxicity is associated with an intermolecular histidine bridge. J Biol Chem 281(22):15145–15154
Tabaton M, Piccini A (2005) Role of water-soluble amyloid-beta in the pathogenesis of Alzheimer’s disease. Int J Exp Pathol 86(3):139–145. doi:10.1111/j.0959-9673.2005.00428.x
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580
Tew DJ, Bottomley SP, Smith DP, Ciccotosto GD, Babon J, Hinds MG, Masters CL, Cappai R, Barnham KJ (2008) Stabilization of neurotoxic soluble beta-sheet-rich conformations of the Alzheimer’s disease amyloid-beta peptide. Biophys J 94(7):2752–2766
Tickler AK, Barrow CJ, Wade JD (2001) Improved preparation of amyloid-b peptides using DBU as Na-Fmoc deprotection reagent. J Pept Sci 7:488–494
Toledo J, Vanderstichele H, Figurski M, Aisen P, Petersen R, Weiner M, Jack C, Jagust W, Decarli C, Toga A, Toledo E, Xie S, Lee V, Trojanowski J, Shaw L (2011) Factors affecting Aβ plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol 122(4):401–413. doi:10.1007/s00401-011-0861-8
Villemagne VL, Perez KA, Pike KE, Kok WM, Rowe CC, White AR, Bourgeat P, Salvado O, Bedo J, Hutton CA, Faux NG, Masters CL, Barnham KJ (2010) Blood borne amyloid-beta dimer correlates with clinical markers of Alzheimer’s disease. J Neurosci 30(18):6315–6322
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416(6880):535–539
Walsh DM, Selkoe DJ (2007) A beta oligomers—a decade of discovery. J Neurochem 101(5):1172–1184
Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ (2005) Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci 25(10):2455–2462
Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ (2000) The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry 39(35):10831–10839
Wang J, Dickson DW, Trojanowski JQ, Lee VM (1999) The levels of soluble versus insoluble brain Ab distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol 158:328–337
Yokota O, Terada S, Ishizu H, Ujike H, Ishihara T, Namba M, Hayashi Y, Nishinaka T, Namba R, Nakashima H, Ueda K, Checler F, Kuroda S (2003) Variability and heterogeneity in Alzheimer’s disease with cotton wool plaques: a clinicopathological study of four autopsy cases. Acta Neuropathol 106(4):348–356. doi:10.1007/s00401-003-0737-7
Acknowledgment
This work was funded by National Health and Medical Research Council of Australia.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Watt, A.D., Perez, K.A., Rembach, A. et al. Oligomers, fact or artefact? SDS-PAGE induces dimerization of β-amyloid in human brain samples. Acta Neuropathol 125, 549–564 (2013). https://doi.org/10.1007/s00401-013-1083-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1083-z