
Abstract
Favorable outcome after chemotherapy of glioblastomas cannot unequivocally be linked to promoter hypermethylation of the O 6-methylguanine-DNA methyltransferase (MGMT) gene encoding a DNA repair enzyme associated with resistance to alkylating agents. This indicates that molecular mechanisms determining MGMT expression have not yet been fully elucidated. We here show that glioblastomas are capable to downregulate MGMT expression independently of promoter methylation by elongation of the 3′-UTR of the mRNA, rendering the alternatively polyadenylated transcript susceptible to miRNA-mediated suppression. While the elongated transcript is poorly expressed in normal brain, its abundance in human glioblastoma specimens is inversely correlated with MGMT mRNA expression. Using a bioinformatically guided experimental approach, we identified miR-181d, miR-767-3p, and miR-648 as significant post-transcriptional regulators of MGMT in glioblastomas; the first two miRNAs induce MGMT mRNA degradation, the latter affects MGMT protein translation. A regression model including the two miRNAs influencing MGMT mRNA expression and the MGMT methylation status reliably predicts The Cancer Genome Atlas MGMT expression data. Responsivity of MGMT expressing T98G glioma cells to temozolomide was significantly enhanced after transfection of miR-181d, miR-767-3p, and miR-648. Taken together, our results uncovered alternative polyadenylation of the MGMT 3′-UTR and miRNA targeting as new mechanisms of MGMT silencing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is the most common and most aggressive primary brain tumor [32]. Research on epigenetic regulation has recently led to the discovery of an important biomarker that has gained translational relevance: Epigenetic silencing of the DNA repair enzyme O 6-methylguanine-DNA methyltransferase (MGMT) by promoter methylation has been shown to be associated with a substantial survival advantage after radiotherapy plus concomitant and adjuvant temozolomide (TMZ) in GBM patients. The 18-month survival rate was 62 % among patients with a methylated MGMT promoter compared with only 8 % in the absence of promoter methylation [5, 28]. The association between promoter methylation and favorable treatment response after chemotherapy with alkylating agents is explained by the assumption that methylation of a CpG island within the promoter of the MGMT gene silences its transcription. This subsequently reduces MGMT protein expression, thereby preventing the disadvantageous repair of chemotherapy-induced DNA damage [12].
However, discordant findings exist; a considerable number of GBM patients with unmethylated tumors experience an unexpected favorable outcome after radiochemotherapy, and some patients with a methylated promoter do not benefit from concomitant and adjuvant TMZ treatment. In these discordant cases, occurring in approximately 20 % of all patients, MGMT mRNA expression was found to be low (high) despite an unmethylated (methylated) MGMT promoter [4, 24] and was shown to determine outcome independently of MGMT promoter methylation: Patients with low MGMT expression did significantly better than those with high transcript levels [16]. These discordant findings point to the existence of so far unknown post-transcriptional regulation of MGMT, which currently impairs the use of this biomarker for treatment decision (in favor of chemotherapy or not) in GBM patients.
The current study aimed to identify additional pathways regulating the expression of MGMT. We particularly focused on post-transcriptional mechanisms involving the 3′-UTR of the MGMT gene and potential targeting by micro-RNAs (miRNAs), which have been recognized to essentially impact multiple biological characteristics of GBM [17].
Materials and methods
Human tissue samples
GBM tissue samples were obtained and processed as described previously [16]. All patients gave written informed consent, and the prospective study protocol was reviewed and approved by the institutional review board of the Ludwig Maximilians University, Munich, Germany (AZ 216/14).
Cell culture
HEK 293 cell lines were routinely maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplied with 10 % fetal bovine serum (FBS), 1 % penicillin/streptomycin/glutamine (v/v) and 1 % NEAA. HEK 293 Tet-Off Advanced cells require the addition of 100 μg/ml geneticin. U87 cells were grown in DMEM medium supplied with 10 % FBS, 2 % glutamine, 1 % penicillin/streptomycin, and 1 % sodium-pyruvate. T98G cell were maintained in DMEM (4.5 g/l Glucose) supplemented with 10 % FBS, 1 % penicillin/streptomycin/glutamine (v/v), 1 % sodium-pyruvate, and 1 % NEAA. A549 cells were cultured in RPMI medium supplied with 10 % FBS, 1 % penicillin/streptomycin/glutamine, and 1 % HEPES (10 mM).
Vector construction
The psiCheck-2 Dual-Luciferase Vector (Promega) was used for generation of MGMT long and short 3′-UTR reporter constructs. Briefly, the long MGMT 3′-UTR was amplified by PCR from 100 ng of cDNA isolated from human glioma tissue with the primers given in Table 1 (synthesized by Metabion). The following cycling conditions were applied: 95 °C for 5 min denaturing; 36 cycles of 95 °C for 30 s, 62.9 °C for 30 s, 72 °C for 60 s; and a final extension at 72 °C for 5 min. PCR products were first ligated into the pSC-B amp/kan vector (UltraBlunt PCR Cloning Kit, Stratagene) according to the manufacturer’s protocol and subsequently subcloned into the PmeI and NotI restriction sites of the psiCheck-2 plasmid. To generate the short UTR reporter construct, a NotI restriction site was introduced into the long UTR reporter downstream of the first APA site using the QuikChange Lightning Mutagenesis Kit (Stratagene) and the primers listed in Table 1. For Tet-Off experiments, sequences encompassing the hRluc reporter gene coding region and the respective UTR were excised from the psiCheck-2 constructs using the restriction enzymes NheI and NotI and were subsequently ligated into the pTRE-Tight vector (Clontech). All constructs were verified by sequence analysis (MWG Biotech).
3′-Rapid amplification of cDNA ends (RACE)
3′-RACE reactions were performed with 1 μg total RNA starting material using the FirstChoice RLM-RACE kit (Ambion). Primer sequences are given in Table 1. PCR products were subcloned into the StrataClone Blunt Vectoramp/kan (Stratagene).
Cell transfections
Cell transfections were performed using the Neon™ Transfection System (Invitrogen). To determine MGMT 3′-UTR stability, HEK 293 Tet-Off Advanced cells were transfected with 1 μg of pTRE-Tight vector containing the MGMT 3′-UTR variants. After 30 h, doxycycline was added to a final concentration of 3 μg/ml. Cells were harvested 8 or 30 h later for qPCR analyses of Renilla luciferase mRNA or reporter gene assays, respectively.
Reporter gene assays
U87 cells were co-transfected with 1 μg of psiCheck-2 luciferase reporter vector containing MGMT 3′-UTR variants and either control or pre-miR microRNA precursor molecules (Ambion) at concentrations of 50 nM. 48 h later, luciferase activities were measured with the Dual-Glo Luciferase Assay System (Promega), and Renilla luciferase activities were normalized to Firefly luciferase activities. All experiments were performed at least in triplicates.
RNA isolation and synthesis of cDNA
Total RNA was isolated using either the RNAqueos® Micro Kit or the mirVana miRNA Isolation Kit followed by a DNase-digest with the Turbo DNA-free Kit (Ambion). RNA quantity was assessed using the NanoDrop® ND-1000 spectrophotometer (Peqlab). cDNA was synthesized from 1 μg of total RNA using the SuperScript III First Strand Synthesis System (Invitrogen) and random hexamers.
Quantitative real-time PCR (qPCR)
Analyses were performed on a Light Cycler 480 (Roche Diagnostics) using UPL probes and reference gene normalization to SDHA and TBP as described in [15]. Expression of mature miRNAs was performed using the TaqMan® MicroRNA Assay (Applied Biosystems); RNU6b served as endogenous control.
MTT assay
Cell growth and viability was determined by colorimetric MTT cell viability/proliferation assay. Briefly, T98G cells were transfected with scrambled control, premiR-767-3p, premiR-181d, premiR-648 or with all 3 premiRs simultaneously (20 nM each premiR) and seeded into 24-well plates. 12 h after electroporation, cells were treated with TMZ (Sigma) dissolved in DMSO. After incubation for further 12 h, cells were treated with 100 μl MTT reagent (Sigma; 5 mg/ml) for 1 h. Finally, optical density at 550 nm was determined.
Western blot analysis
Western blotting was performed with 30 μg of total protein extract on a 10 % SDS-gel. MGMT protein was detected using anti-MGMT mouse monoclonal antibody (GeneTex). Immunoreactive bands were detected using horseradish peroxidase-labelled horse anti-mouse antibody (Cell Signaling).
Bioinformatics and statistical analyses
MGMT mRNA expression data, miRNA expression data, and methylation data were downloaded from The Cancer Genome Atlas (TCGA) database (https://tcga-data.nci.nih.gov/tcga, Level 3 data) [2]. The Cancer Genome Atlas is a public repository that comprises tumor tissue from a diverse set of malignancies. The database aims at providing all levels of biomedical information available, such as genomic, transcriptomic, proteomic, as well as clinical data. At the time of writing, TCGA contains samples from a total of more than 600 GBM patients. In the current study, only samples with complete information were considered (n = 206), herein referred to as the training set. A set of n = 19 patients enrolled in our own study is referred to as validation set 1, and a second download of n = 71 newly added patients from the TCGA database is referred to as validation set 2. MiRNAs were identified using four different programs (TargetScan, PITA, RNA22, miRDB) [11, 19, 23, 31], implementing both prediction scores and agreement between the different programs.
The training set was used for the selection of candidate regulatory miRNAs by considering correlation of miRNA expression with MGMT expression, significant negative correlation in bivariate linear regression analysis including MGMT methylation status (cg12981137) and the expression of the respective miRNA in glioma tissue, and selection in a linear regression stepwise forward algorithm. After identification and biomolecular validation of the two candidate miRNAs with the most significant impact on MGMT expression levels, a multivariate linear regression model was trained and evaluated on the training set. In order to apply the model to the validation sets, miRNA and mRNA expression values were discretized into 5 quantiles, whereas methylation status was binarized with the median value being the cut-off. The model was retrained on the discretized data (MGMT-expression = −1.57 × (methylation state) − 0.17 × (hsa-miR-181d expression) − 0.14 × (hsa-miR-767-3p expression) + 3.43). All data were analyzed using SigmaPlot 11.0 software (Systat Software) and the statistical software package R. Analysis was performed with Student’s t test with p < 0.05 considered as significant and p < 0.001 considered as highly significant.
Results
GBMs express two MGMT transcripts differing in their 3′-UTR lengths
Recent data indicate that MGMT expression in GBM is not only regulated by promoter methylation but also by further so far unexplored mechanisms [16]. MiRNAs have gained significant attention as potent regulators of gene expression [20]. To determine, whether MGMT is subject to post-transcriptional regulation by specific miRNAs, we performed bioinformatics analyses of the MGMT-3′-UTR. This suggested that MGMT may be subject to alternative polyadenylation, since two possible MGMT transcripts with 3′-UTR lengths of 105 and 522 nt were found, with the short transcript containing a canonical poly(A) signal upstream the putative cleavage site and the long transcript possessing an alternative poly(A) site (Fig. 1a). To determine which of these isoforms are expressed in human normal brain tissue and in GBMs, we performed 3′-RLM-RACE analyses (positions of RACE forward primers are depicted in Fig. 1a). As shown in Fig. 1b, in normal brain tissue, only one MGMT transcript was expressed, whereas in GBMs, we detected a second mRNA. Amplicon lengths of approximately 440 and 850 nt pointed at the expected 105 and 522 nt MGMT transcripts, which were verified by sequencing.

Malignant gliomas express two MGMT mRNA isoforms with different 3′-UTR lengths. a Schematic diagram of polyadenylation sites within the MGMT 3′-UTR and position of 3′-RACE forward primer with expected PCR product lengths. b 3′-RLM-RACE performed on total RNA from normal brain and GBM tissues. One experiment representative of n = 3 is shown (MM, molecular marker). c Expression of MGMT 3′-UTR variants was determined by qPCR in normal brain (n = 6) and in GBM tissues (n = 49). The correlation of MGMT mRNA expression and 3′-UTR long/short ratio in GBM samples relative to normal brain is illustrated; r = −0.86, p < 0.0001. d GBMs were categorized into 3 groups with respect to MGMT expression: (1) low MGMT expression (n = 13); (2) medium MGMT expression (n = 25); (3) high MGMT expression (n = 11). Values represent the 3′-UTR long/short ratio relative to normal brain; mean ± SEM; *p < 0.05
Low MGMT expression is accompanied by enhanced expression of the long 3′-UTR
We next systematically analyzed the expression patterns of the two MGMT isoforms in six normal brain samples and in 49 GBM samples using qPCR with specifically designed primer-probe sets. While the elongated MGMT 3′-UTR generally is poorly expressed in normal brain tissue with a median expression ratio long/short 3′-UTR of less than 0.1, GBM tissues showed a clear tendency towards 3′-UTR-elongation with decreasing MGMT expression (Fig. 1c). Thus, we categorized GBM samples into three groups with respect to MGMT expression based on the dataset described in [16]: low MGMT expression (relative expression level <0.35, encompassing 25 % of GBMs), medium MGMT expression (>0.35, <0.75, 50 % of GBMs), and high MGMT expression (>0.75, 25 % of GBMs). In tumors with high MGMT expression, 3′-UTR expression patterns did not significantly differ from normal brain, which is unmethylated and exhibits the highest MGMT expression levels [16]. In contrast, reduced MGMT expression levels were found to be associated with a higher abundance of the elongated 3′-UTR. As compared to normal brain, a 3.2- and 5.1-fold increase of the long 3′-UTR was detected in the medium and low expression group, respectively (Fig. 1d). These results were independent of the promoter methylation status, as the medium expression group exhibits homogenous 3′-UTR expression patterns although consisting of both unmethylated and methylated tumors (Summary of methylation status in Supplementary Table S1).
Expression of full-length MGMT 3′-UTR confers reduced protein expression due to a reduced mRNA stability
To determine how the different 3′-UTR isoforms influence protein expression, we fused the full-length and the shortened MGMT 3′-UTRs to the MCS of the psiCheck-2 vector downstream of the Renilla luciferase (hRluc) gene and performed transfection experiments in HEK 293 cells. As shown in Fig. 2a, the full-length UTR reporter yielded significantly lower luciferase activity than the construct containing the short UTR region (p = 0.008). Thus, expression of long MGMT 3′-UTR confers reduced protein expression, most likely due to an impaired mRNA stability and/or translation. To directly determine the stability of both mRNA isoforms, we performed experiments with a Tet-Off system. Therefore, both the short and the elongated MGMT 3′-UTR were subcloned downstream of the Renilla luciferase gene. These constructs were subsequently ligated into the pTRE-Tight vector, in which addition of doxycycline turns off transcription of the inserted luciferase-3′-UTR construct. The decay of luciferase mRNA and luciferase protein resulting from the vector constructs containing the long 3′-UTR was significantly faster than those derived from the short UTR constructs (Fig. 2b, c). These results indicate that the reduced protein expression of the long MGMT 3′-UTR is due to a reduced mRNA stability.

Expression of the long MGMT 3′-UTR isoform reduces MGMT protein levels and mRNA stability. a MGMT 3′-UTR isoforms were subcloned into the psiCheck-2 reporter vector and protein expression was determined by luciferase assay. Results are expressed as Rluc/Fluc ratio and represent the mean ± SEM; n = 6; *p < 0.05. b HEK 293 Tet-Off Advanced cells were transiently transfected with pTRE-Tight vector containing the MGMT 3′-UTR variants, together with pTRE-Tight Firefly Luciferase Vector as transfection efficiency control. 30 h post-transfection doxycycline was added. MGMT mRNA stability was analyzed by quantitative RT-PCR (n = 5) and by c determination of reporter activity (n = 5). Data are presented as hRluc expression values in doxycycline treated cells relative to untreated cells. Values are mean ± SEM; *p < 0.05
3′-UTR elongation renders MGMT prone to miRNA regulation
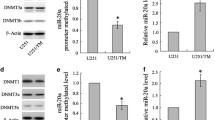
As the elongated 3′-UTR may harbor miRNA binding sites and/or AU-rich sequences (AREs) rendering it prone to an enhanced degradation, we next screened the long 3′-UTR in silico for the respective consensus sequences. As AREs were not detected, we focused on miRNA-mediated regulation. Using four different miRNA target prediction programs, 32 potential miRNAs target sites have been predicted within the 3′-UTR of the MGMT mRNA. Two of them were localized within the short UTR (miR-655 and miR-1197), 29 were exclusively situated in the long UTR (in the sequence between the two APA sites), and one site (miR-181d) was localized in both UTRs. To further restrict the number of the miRNAs potentially targeting the long 3′-UTR, we screened the TGCA database for miRNAs that are expressed in human GBMs. We additionally performed correlation analyses to identify miRNAs that show a negative correlation with MGMT expression (Supplementary Table S2). Taking into account prediction scores, correlation analyses, and seed match position, nine miRNAs were selected for experimental validation: those miRNAs potentially targeting the short UTR as well as seven miRNAs potentially targeting the long UTR (Fig. 3a). To analyze direct binding and targeting of the predicted miRNAs, we subcloned both 3′-UTRs into the MCS of the psiCheck-2 vector, which contains both Renilla reniformis luciferase (Rluc) and Photinus pyralis (Firefly) luciferase (Fluc) on a single plasmid with the MCS located downstream of the Renilla encoding region. The reporter vector was co-transfected with the respective pre-miRs (or with scrambled control), and Rluc/Fluc ratios were calculated. The three miRNA binding sites located in the short UTR—namely miR-181d, miR-655 and miR-1197—did not exhibit significant regulatory activity when transfected with the short-UTR luciferase construct, which indicates that the MGMT isoform containing the short UTR is not influenced by miRNA regulation (Fig. 3b). Out of the candidate miRNAs potentially regulating the long UTR, miR-661, miR-370, miR-181d, miR-767-3p, and miR-648 significantly repressed luciferase activity (inhibition by 14.4 ± 8 %, 15.2 ± 6 %, 29.2 ± 2 %, 31.6 ± 2 % and 36.5 ± 4 %, respectively) and thus turned out to be regulators of the long UTR-expressing MGMT isoform (Fig. 3c). To further confirm these findings, we systematically introduced two nucleotide mutations within the particular predicted seed region of the respective miRNA to prevent specific miRNA-target interactions. As shown in Supplementary Figure S1, mutations resulted in a complete reversal of the inhibitory effect of the three miRNAs. Western blotting experiments with A549 cells (expressing both MGMT isoforms) transfected with the respective pre-miRs were in line with these experiments, as a significant protein reduction was seen for miR-181d, miR-767-3p, and miR-648. Notably, the strongest reduction was achieved by miR-648 (Fig. 3d), whereas miR-661 and miR-370 did not exert visible effects on MGMT protein expression. Unexpectedly, the strong effect of miR-648 on MGMT protein could not be seen on mRNA level, as qPCR experiments revealed that MGMT transcripts were only significantly repressed by miR-181d and miR-767-3p (Fig. 3e). This indicates that miR-181d and miR-767-3p exert their effects by degradation of mRNA, whereas miR-648 inhibits protein translation.

3′-UTR lengthening renders MGMT mRNA prone to miRNA-targeting. a Schematic diagram of selected predicted miRNA binding sites in the short and long MGMT 3′-UTR. b PsiCheck-2 reporter vector containing the short MGMT 3′-UTR variant was transfected with either scrambled control or pre-miR molecules as indicated. Luciferase activity was determined and data are presented as Rluc/Fluc activity relative to scrambled control; mean ± SEM; n = 6. c PsiCheck-2 reporter vector containing the long MGMT 3′-UTR variant was transfected with either scrambled control or pre-miR molecules as indicated. Luciferase activity was determined and data are presented as Rluc/Fluc activity relative to scrambled control; mean ± SEM; n = 6; *p < 0.05. d Western blot analysis of A549 cells transiently transfected with scrambled control or pre-miR molecules as indicated using a MGMT specific antibody (β-actin served as loading control; one experiment representative of n = 6 is shown). e MGMT mRNA expression was analyzed in the same samples by quantitative RT-PCR. Values are mean ± SEM; n = 6; *p < 0.05
We next studied the expression of the three miRNAs regulating the long 3′-UTR MGMT isoform in primary human GBM tissue specimen as compared to normal brain tissue. As shown in Fig. 4a, expression of miR-767-3p is strongly enhanced (10.1 ± 1.9-fold), and miR-648 is up-regulated 2.1 ± 0.4-fold, whereas miR-181d remains unchanged.

In human GBM, MGMT expression is influenced by miRNA-targeting a MiRNA expression in GBM samples was quantified by qPCR with TaqMan miRNA assays relative to RNU6b. Data are presented as relative miRNA expression in GBM samples relative to normal brain; mean ± SEM; n = 19; *p < 0.05. b Linear regression analysis of MGMT expression using TGCA data with observed MGMT expression plotted against predicted MGMT expression and calculated as: −1.03 × expression(miR-767-3p) − 0.23 × expression(miR-181d) − 2.1 × methylation status + 7.64. c Validation of the linear regression model using n = 19 GBM samples. Observed MGMT expression is plotted against predicted MGMT expression; correlation: 0.79, r 2: 0.60 (methylation: r 2 0.54, miR-767-3p: r 2 0.34, hsa-miR-181d: r 2 0.11), CI 0.52–0.91, p = 6.2e−05
After identification of candidate miRNAs that influence MGMT expression in vitro, we evaluated the predictive capabilities of these miRNAs in GBM data from The Cancer Genome Atlas (TCGA) database. Therefore, we constructed a linear regression model based on the expression of these two miRNAs. Methylation state was also included in the model, as it has repeatedly been shown to have great impact on MGMT expression. As expected, methylation status, miR-767-3p expression, and miR-181d expression are highly significant predictors of MGMT mRNA expression (Fig. 4b) and explain a large fraction of the observed variation (multiple r 2 0.45). In a linear regression model, each coefficient represents by how much the prediction is influenced by the covariate. Accordingly, methylation contributes to the model the most, followed by miR-767-3p and miR-181d, respectively (Table 2). The fact that all covariates in our model are negative implies an inverse relationship with MGMT expression. It is not surprising that miR-648 expression has no predictive value on MGMT mRNA expression levels, as according to our experimental analyses, its main effect appears to be in translational repression rather than mRNA degradation. We further tested whether additional inclusion of TGCA data on copy numbers of DNA regions affecting the MGMT gene would improve our model. Even though copy numbers were found to be significantly associated with MGMT expression values (coefficient: 0.41, p value: 0.02), the observed variance of MGMT expression explained by our model (r 2) increased only by 0.01, thereby not leading to significant enhancement of predictability.
For further validation, we have applied the model to our dataset of GBM patients (Fig. 4c) and to 71 patient samples newly added to TGCA database (Supplementary Fig. S2). Predictions showed high correlation with the observed MGMT expression data (p < 0.01) in both validation sets.
Transfection of MGMT expressing human GBM cells with miR-181d, miR-767-3p, and miR-648 sensitizes cells to alkylating agents
MGMT expression levels have been shown to be a major predictor of TMZ sensitivity in human glioma cells [7, 13, 29]. We therefore assumed that miRNA-induced down-regulation of MGMT would sensitize MGMT-expressing glioma cells to TMZ. As expected, transfection of T98G cells (exhibiting MGMT expression patterns resembling those of unmethylated GBM with high MGMT expression, data not shown) with miR-181d, miR-767-3p, and miR-648 resulted in significant reduction of MGMT protein expression (Fig. 5a). We next examined the cytotoxic effects of TMZ on both miRNA- and negative control-transfected T98G cells with the aid of an MTT assay. As shown in Fig. 5b, T98G cells transfected with all three miRNAs showed a decrease in viable cells of approximately 40 % as compared to control cells (transfected with scrambled control) after treatment with 100 and 500 μM TMZ; all three miRNAs contribute to this effect (Fig. 5c). Thus, transfection of the three miRNAs significantly increased TMZ cytotoxicity of T98G glioma cells.

MiR-targeting of MGMT reduces cell viability upon TMZ treatment. T98G cells were transiently transfected with premiR-767-3p, premiR-181d and premiR-648 (either individually or altogether) or with scrambled control. a Western blot analysis of cells transfected with all three premiRs simultaneously using a MGMT specific antibody and β-actin as loading control; one experiment representative of n = 3 is shown. b 12 h after electroporation of all three premiRs simultaneously, cells were treated with TMZ (or with DMSO only) for further 12 h at final concentrations as indicated. The viability of the cells as determined by MTT assay is shown; mean ± SEM; n = 6; *p < 0.01. c 12 h after transfection of individual premiRs, TMZ at a final concentration of 500 nM was added. After 12 h, cell viability was determined. Data are presented as % viability of premiR-transfected cells relative to control (transfected with premiR scrambled control); mean ± SEM; n = 6; *p < 0.05
Discussion
In gliomas, promoter hypermethylation status of MGMT has emerged as an important predictor of responses to chemotherapy with alkylating agents [30]. However, an incomplete understanding of the mechanisms regulating the expression of MGMT still impairs its clinical use as a valid biomarker. In the current study, we provide evidence that the expression of MGMT is not only controlled by promoter methylation, but also by post-transcriptional processes involving alternative polyadenylation (APA) of the MGMT 3′-UTR and miRNA targeting. We show that in addition to gene silencing by promoter methylation, human glioma cells down-regulate MGMT expression via elongation of the 3′-UTR of the mRNA, thereby rendering the alternatively polyadenylated transcript susceptible to miRNA-mediated suppression.
In the last few years, APA has increasingly gained attention as a critical post-transcriptional regulator of gene expression [3]. Several array analyses pointed out that in states of increased proliferation and malignant transformation, a global shortening of 3′-UTRs occurs which, by removal of regulatory sequence elements, affects expression quantitatively [22, 25]. Shorter transcripts have been shown to possess less miRNA binding sites and AU-rich elements (AREs), resulting in increased transcript stability with production of higher levels of protein [6, 27]. In contrast, 3′-UTR lengthening has been described to occur during developmental differentiation processes and has been associated with down-regulation of genes involved in DNA replication and cell cycle regulation [9]. Although in cancer, truncation of messages is more prevalent, elongation of selected transcripts may also occur, and recent reports presumed that this might enhance tumorigenesis as well [26]. Consistent with this notion, we have identified MGMT as such a candidate gene in human gliomas. In normal brain tissue, MGMT is expressed as a transcript with a relatively short 3′-UTR that apparently is not post-transcriptionally regulated. In GBM, a second mRNA appears which is 417 nt longer, exhibits a significantly reduced stability, and produces reduced protein levels. Accordingly, MGMT expression in tumor samples is inversely correlated with the abundance of the long 3′-UTR, irrespective of the promoter methylation status. The exact mechanisms regulating APA are not fully elucidated, yet. Here, the predominating short transcript possesses a canonical poly(A) signal, while elongation of the UTR is accomplished by utilization of the “weaker” distally localized alternative polyadenylation site [3]. Taking into account recent studies that have identified a large number of 3′-processing factors and co-factors determining the shifts in poly(A) site utilization [8, 10], one can speculate that tumor-specific changes in the stoichiometry of these factors or of RNA binding proteins may account for this phenomenon.
We further provide clear evidence that only the elongated MGMT transcript is significantly regulated by miRNAs, and that the altered properties of the long transcript are due to the influence of a set of specific miRNAs. Using a combined bioinformatic and experimental approach, we show that out of the various miRNAs, potentially targeting the long MGMT 3′-UTR, miR-181d, miR767-3p, and miR-648 are significant regulators of MGMT. Importantly, the first two miRNAs induce degradation of the MGMT transcript, whereas miR-648 negatively affects MGMT protein translation. Of note, miR-767-3p and miR-648 turned out to be significantly up-regulated in GBMs thus indicating that APA processes and altered miRNA expression patterns are strongly interrelated. The here described post-transcriptional regulation may provide an explanation for recently reported findings. A considerable number of patients with unmethylated tumors exhibit low MGMT expression levels and experience unexpected favorable clinical courses. Also, in methylated tumors, post-transcriptional regulation pathways may support MGMT suppression achieved by promoter methylation leading to extremely low MGMT expression levels. This could result in exceptionally good responses to radiochemotherapy. This assumption is supported by experiments with MGMT expressing glioma cell lines. Transfection of the three miRNAs resulted in significant reduction of MGMT expression and sensitization to temozolomide treatment.
Our findings were confirmed by a linear regression analysis on the TGCA expression data: a model containing both the methylation state and the expression of the two miRNAs (influencing MGMT transcripts) proved reliable in predicting the MGMT expression. This model also applied to our own data set consisting of 19 GBM patient samples. As the existence of additional miRNAs and/or factors that also impact MGMT expression is highly probable, this model still needs improvement. Accordingly, we could not confirm a correlation with survival using TGCA data as suggested by others for miR-181d [33]. This is not surprising, as prediction of a multidimensional measurement like patients’ survival requires the development of a complex predictive model including the majority of influencing parameters. So far, at least three different layers of epigenetic regulation influencing the expression of MGMT have been elucidated: (1) promoter methylation, (2) histone modifications [14, 34], and (3) the here described alternative polyadenylation with consecutive miRNA targeting. The complexity of this network is further enhanced by the fact that individual CpGs within the MGMT promoter differ with respect to their expression-inhibiting properties [1, 21], and that interactions between the layers of regulation have not yet been investigated. Moreover, the supposed influence of specific transcription factors still is elusive [18]. Complete elucidation of networks determining MGMT expression requires further research.
References
Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, Dietrich PY, Weller M, Mariani L, Heppner FL, McDonald DR, Lacombe D, Stupp R, Delorenzi M, Hegi ME (2012) MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 124(4):547–560. doi:10.1007/s00401-012-1016-2
McLendon, Friedman A, Bigner D et al (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216):1061–1068. doi:10.1038/nature07385
Di Giammartino DC, Nishida K, Manley JL (2011) Mechanisms and consequences of alternative polyadenylation. Mol Cell 43(6):853–866. doi:10.1016/j.molcel.2011.08.017
Everhard S, Tost J, El Abdalaoui H, Criniere E, Busato F, Marie Y, Gut IG, Sanson M, Mokhtari K, Laigle-Donadey F, Hoang-Xuan K, Delattre JY, Thillet J (2009) Identification of regions correlating MGMT promoter methylation and gene expression in glioblastomas. Neuro Oncol 11(4):348–356. doi:10.1215/15228517-2009-001
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352(10):997–1003. doi:10.1056/NEJMoa043331
Helfer S, Schott J, Stoecklin G, Forstemann K (2012) AU-rich element-mediated mRNA decay can occur independently of the miRNA machinery in mouse embryonic fibroblasts and Drosophila S2-cells. PLoS One 7(1):e28907. doi:10.1371/journal.pone.0028907
Hermisson M, Klumpp A, Wick W, Wischhusen J, Nagel G, Roos W, Kaina B, Weller M (2006) O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J Neurochem 96(3):766–776. doi:10.1111/j.1471-4159.2005.03583.x
Hu J, Lutz CS, Wilusz J, Tian B (2005) Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 11(10):1485–1493. doi:10.1261/rna.2107305
Ji Z, Lee JY, Pan Z, Jiang B, Tian B (2009) Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci USA 106(17):7028–7033. doi:10.1073/pnas.0900028106
Ji Z, Tian B (2009) Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS One 4(12):e8419. doi:10.1371/journal.pone.0008419
John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS (2004) Human MicroRNA targets. PLoS Biol 2(11):e363. doi:10.1371/journal.pbio.0020363
Kaina B, Christmann M, Naumann S, Roos WP (2007) MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 6(8):1079–1099. doi:10.1016/j.dnarep.2007.03.008
Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, Wu W, James CD, Sarkaria JN (2009) Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol 11(3):281–291. doi:10.1215/15228517-2008-090
Kitange GJ, Mladek AC, Carlson BL, Schroeder MA, Pokorny JL, Cen L, Decker PA, Wu W, Lomberk GA, Gupta SK, Urrutia RA, Sarkaria JN (2012) Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res 18(15):4070–4079. doi:10.1158/1078-0432.CCR-12-0560
Kreth S, Heyn J, Grau S, Kretzschmar HA, Egensperger R, Kreth FW (2010) Identification of valid endogenous control genes for determining gene expression in human glioma. Neuro Oncol 12(6):570–579. doi:10.1093/neuonc/nop072
Kreth S, Thon N, Eigenbrod S, Lutz J, Ledderose C, Egensperger R, Tonn JC, Kretzschmar HA, Hinske LC, Kreth FW (2011) O-methylguanine-DNA methyltransferase (MGMT) mRNA expression predicts outcome in malignant glioma independent of MGMT promoter methylation. PLoS One 6(2):e17156. doi:10.1371/journal.pone.0017156
Kreth S, Thon N, Kreth FW (2012) Epigenetics in human gliomas. Cancer Lett. doi:10.1016/j.canlet.2012.04.008
Lavon I, Fuchs D, Zrihan D, Efroni G, Zelikovitch B, Fellig Y, Siegal T (2007) Novel mechanism whereby nuclear factor kappaB mediates DNA damage repair through regulation of O(6)-methylguanine-DNA-methyltransferase. Cancer Res 67(18):8952–8959. doi:10.1158/0008-5472.CAN-06-3820
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120(1):15–20. doi:10.1016/j.cell.2004.12.035
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR (2005) MicroRNA expression profiles classify human cancers. Nature 435(7043):834–838. doi:10.1038/nature03702
Malley DS, Hamoudi RA, Kocialkowski S, Pearson DM, Collins VP, Ichimura K (2011) A distinct region of the MGMT CpG island critical for transcriptional regulation is preferentially methylated in glioblastoma cells and xenografts. Acta Neuropathol 121(5):651–661. doi:10.1007/s00401-011-0803-5
Mayr C, Bartel DP (2009) Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138(4):673–684. doi:10.1016/j.cell.2009.06.016
Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I (2006) A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 126(6):1203–1217. doi:10.1016/j.cell.2006.07.031
Ramakrishnan V, Kushwaha D, Koay DC, Reddy H, Mao Y, Zhou L, Ng K, Zinn P, Carter B, Chen CC (2011) Post-transcriptional regulation of O(6)-methylguanine-DNA methyltransferase MGMT in glioblastomas. Cancer Biomark 10(3–4):185–193. doi:10.3233/CBM-2012-0245
Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB (2008) Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320(5883):1643–1647. doi:10.1126/science.1155390
Singh P, Alley TL, Wright SM, Kamdar S, Schott W, Wilpan RY, Mills KD, Graber JH (2009) Global changes in processing of mRNA 3′ untranslated regions characterize clinically distinct cancer subtypes. Cancer Res 69(24):9422–9430. doi:10.1158/0008-5472.CAN-09-2236
Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM (2005) Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123(6):1133–1146. doi:10.1016/j.cell.2005.11.023
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10(5):459–466. doi:10.1016/S1470-2045(09)70025-7
Szeliga M, Zgrzywa A, Obara-Michlewska M, Albrecht J (2012) Transfection of a human glioblastoma cell line with liver-type glutaminase (LGA) down-regulates the expression of DNA-repair gene MGMT and sensitizes the cells to alkylating agents. J Neurochem 123(3):428–436. doi:10.1111/j.1471-4159.2012.07917.x
von Deimling A, Korshunov A, Hartmann C (2011) The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol 21(1):74–87. doi:10.1111/j.1750-3639.2010.00454.x
Wang X, El Naqa IM (2008) Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics 24(3):325–332. doi:10.1093/bioinformatics/btm595
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359(5):492–507. doi:10.1056/NEJMra0708126
Zhang W, Zhang J, Hoadley K, Kushwaha D, Ramakrishnan V, Li S, Kang C, You Y, Jiang C, Song SW, Jiang T, Chen CC (2012) miR-181d: a predictive glioblastoma biomarker that downregulates MGMT expression. Neuro Oncol 14(6):712–719. doi:10.1093/neuonc/nos089
Zhao W, Soejima H, Higashimoto K, Nakagawachi T, Urano T, Kudo S, Matsukura S, Matsuo S, Joh K, Mukai T (2005) The essential role of histone H3 Lys9 di-methylation and MeCP2 binding in MGMT silencing with poor DNA methylation of the promoter CpG island. J Biochem 137(3):431–440. doi:10.1093/jb/mvi048
Acknowledgments
We would like to thank J. Rink and Gaby Groeger for expert technical assistance.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Kreth and E. Limbeck contributed equally.
Rights and permissions
About this article
Cite this article
Kreth, S., Limbeck, E., Hinske, L.C. et al. In human glioblastomas transcript elongation by alternative polyadenylation and miRNA targeting is a potent mechanism of MGMT silencing. Acta Neuropathol 125, 671–681 (2013). https://doi.org/10.1007/s00401-013-1081-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1081-1