
Abstract
Neuromuscular disorders (NMD) such as neuropathy or myopathy are rare and often severe inherited disorders, affecting muscle and/or nerves with neonatal, childhood or adulthood onset, with considerable burden for the patients, their families and public health systems. Genetic and clinical heterogeneity, unspecific clinical features, unidentified genes and the implication of large and/or several genes requiring complementary methods are the main drawbacks in routine molecular diagnosis, leading to increased turnaround time and delay in the molecular validation of the diagnosis. The application of massively parallel sequencing, also called next generation sequencing, as a routine diagnostic strategy could lead to a rapid screening and fast identification of mutations in rare genetic disorders like NMD. This review aims to summarize and to discuss recent advances in the genetic diagnosis of neuromuscular disorders, and more generally monogenic diseases, fostered by massively parallel sequencing. We remind the challenges and benefit of obtaining an accurate genetic diagnosis, introduce the massively parallel sequencing technology and its novel applications in diagnosis of patients, prenatal diagnosis and carrier detection, and discuss the limitations and necessary improvements. Massively parallel sequencing synergizes with clinical and pathological investigations into an integrated diagnosis approach. Clinicians and pathologists are crucial in patient selection and interpretation of data, and persons trained in data management and analysis need to be integrated to the diagnosis pipeline. Massively parallel sequencing for mutation identification is expected to greatly improve diagnosis, genetic counseling and patient management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Neuromuscular disorders
Inherited neuromuscular disorder (NMD) is a wide term covering different genetic disorders affecting muscles (different types of myopathies and dystrophies, ion channel muscle diseases and malignant hyperthermia), nerves (Charcot–Marie–Tooth neuropathies also called hereditary motor and sensory neuropathies, amyotrophic lateral sclerosis, hereditary ataxias and spinal muscular atrophies) and neuromuscular junctions (myasthenic syndromes [41]) (http://musclegenetable.fr/) [44]. Muscle weakness, twitching, cramps and numbness are common features in several NMDs. These disorders are rare and often severe, affecting children and adults with considerable burden to the patients, their families and public health systems [25].
Provisions in genetic diagnosis laboratories
The most important criterion for using a test in standard health care is whether the test will lead to a better outcome for the patient [97]. The clinical test should address the specific problem. The aim of a clinical genetic service is to make genetic diagnosis and estimation of transmission risk for an affected patient and to provide genetic counseling and preventive and therapeutic guidance for family members [31]. Anonymity and confidentiality are important standards in genetic diagnosis. Data should be reliable and diagnosis should be cost-effective. The turnaround time for test results is another important issue in genetics diagnosis.
Challenges in molecular diagnosis
Genetic heterogeneity
NMDs are one of the most genetically heterogeneous disorders class with more than 300 implicated genes. This genetic heterogeneity can be seen, for instance in Charcot–Marie–Tooth (CMT) neuropathies with more than 30 causative genes [83] and more than 40 loci (http://www.molgen.ua.ac.be/CMTMutations) or in autosomal recessive limb girdle muscular dystrophy with 15 genes implicated to date [49]. This high degree of genetic heterogeneity is problematic for molecular genetics diagnosis as it could be time-consuming and costly to test the different implicated genes in diagnostic laboratories. The rarity of mutations in some genes explains the fact that samples should be sent to several specialized laboratories to cover all candidate genes.
Implication of large genes
Several of the largest human genes are mutated in neuromuscular disorders such as DMD (MIM #300377) spanning more than 2.3 Mb with 79 exons [86], TTN (MIM #188840) with 363 exons with an open reading frame spanning more than 100 Kb, NEB (MIM #161650) with 183 exons [22] and RYR1 (MIM #180901) with 106 exons [73]. These genes are also mainly expressed in muscle, precluding cDNA analysis if such tissue is not available. While these genes are known to be implicated in diseases, they might not be fully tested or only the mutation hot spot regions may be analyzed at first [4].
Clinical heterogeneity
Clinical heterogeneity is another parameter in diagnosis of NMDs. For example, mutations in the Caveolin 3 gene (MIM #601253) have been detected in four different skeletal muscle disease phenotypes: rippling muscle disease, limb-girdle muscular dystrophy, hyperCKemia and distal myopathy [28]. Patients can have an overlap of these symptoms, or the same mutation can cause diverse clinical phenotypes with different severities. Thus, it is sometimes difficult to suggest the best candidate genes to direct molecular diagnosis.
Unspecific clinical features
Another reason precluding the suggestion of the best molecular diagnosis approach is the fact that a large number of patients display unspecific clinical and histopathological features. For instance, reviews of cases with congenital myopathies that are usually sub-classified into nemaline, core or centronuclear myopathies reveal no specific signs in almost half of them, whereas other patients have diverse but overlapping clinical and histopathological manifestations [64]. Moreover, when a sequence change is found in one of the prioritized genes in diagnostic laboratories, further investigation might be stopped, which may lead in some cases to missing the real disease-causing mutation. Conversely, when no change is found, patients undergo additional time-consuming, costly and sometimes painful tests to precise the clinical diagnosis and prioritize other genes [75].
Requirements for several approaches
Different types of mutations are detected in patients with NMDs. For example, 60–65 % of patients with Duchenne and Becker muscular dystrophies (DMD and BMD) have deletions in the dystrophin gene, DMD (MIM #300377), 5–15 % have duplications, and the rest have point mutations or small insertions–deletions [1, 63]. Thus, diagnostic laboratories should apply different techniques to detect disease-causing mutations in patients, which is laborious, expensive, necessitates different platforms and increases turnaround time [76].
Unidentified genes
The last obstacle is the unidentified genes in different NMDs. For instance, mutations in several genes, including SOD1 (MIM #147450) [81], SETX (MIM #608465) [14], ALS2 (MIM #606352) [35, 96], TARDBP (MIM #605078) [30], FUS (MIM #137070) [48], ANG (MIM #105850) [33, 39] and C9orf72 (MIM #614260) [19, 79] can cause amyotrophic lateral sclerosis (ALS), a neurodegenerative disorder characterized by the loss of motor neurons in brain and spinal cord. However, for a significant number of ALS patients, the causative mutation remains unknown, suggesting the involvement of other genes. More generally, about 40 % of patients with NMDs do not have a genetic diagnosis.
Benefits of definitive molecular diagnosis
Identification of related disease-causing mutations helps to confirm the clinical findings and provide an accurate diagnosis. The knowledge of the mutation and mutated gene usually improves disease management, and allows for inclusion into therapeutic trials. Genetic counseling becomes possible, as carrier status determination and prenatal diagnosis can decrease the risk of recurrence in affected families. In addition, finding the disease-causing mutation permits potential phenotype–genotype correlations and a better understanding of the underlying pathophysiological mechanisms, a pre-requisite for the development of specific therapeutic approaches.
Routine molecular techniques in genetics diagnostic laboratories
Today, several techniques are used in routine diagnostic to find causative mutations. The most common method is PCR amplification of coding/exonic sequences of candidate genes from genomic DNA, followed by Sanger sequencing of PCR products. This approach is the gold standard for detection of point mutations and small insertions–deletions. If a gene is large and mutations are spread throughout the entire gene, using this technique is extremely time-consuming, expensive and laborious. An alternative approach is reverse transcription PCR (RT-PCR) followed by Sanger sequencing of the entire cDNA [34, 59] which needs the access to specific tissues such as muscle for the muscle-specific genes. Such tissues may not be available all the times. Multiplex PCR and long-range PCR (LR-PCR) are other PCR-based techniques. Using multiplex PCR of only 19 exons of the DMD gene, about 98 % of deletions could be detected in patients with DMD/BMD [6, 13, 70]. With the MLPA technique in patients with sporadic amyotrophic lateral sclerosis, it was shown that SMN1 duplications are associated with ALS susceptibility whereas SMN1 deletions and SMN2 copy number status are not associated with ALS [10]. In patients with spinal muscular atrophy (SMA), a neuromuscular disorder characterized by degeneration and loss of alpha-motor neurons in the anterior horn of the spinal cord, LR-PCR was used to detect deletions of the SMN1 gene (MIM #600354) [3].
For detection of repeat expansions, a common type of mutation in NMDs, several methods are used, such as repeat primed PCR (RP-PCR) and Southern blot [32, 95]. In 56 out of 76 patients with myotonic dystrophy type 2 (DM2), a neuromuscular disorder characterized by myotonia and muscle dysfunction, and 25 out of 378 patients with spinocerebellar ataxia type 8, a slowly progressive neurodegenerative disorder, repeat expansions were detected using RP-PCR [46].
Comparative genomic hybridization (CGH) array is a method of choice for detection of large rearrangements and copy number variations. Using this technique, genomic rearrangements were detected in dysferlin (DYSF, MIM #603009) and calpain-3 (CAPN3, MIM # 114240) genes, implicated in two forms of limb-girdle muscular dystrophy types 2B and 2A, respectively [5].
Indirect diagnostic techniques such as monitoring the presence of a protein by Western blot [88] or enzymatic activity are other available approaches; however, they do not provide a specific knowledge of the mutation.
Massively parallel sequencing technology
As mentioned above, genetic and clinical heterogeneity, unspecific clinical features, implication of large genes and the necessity to apply multiple techniques are the main drawbacks in routine molecular diagnostic laboratories. Massively parallel sequencing (MPS), also called next generation (NGS) or high-throughput sequencing (HTS), allows to sequence target genes and regions, exome or whole genome, and is revolutionizing the molecular diagnosis as it now permits large-scale parallel sequencing and can be used to detect several types of mutations. The exome represents about 1–2 % of the genome but harbor 85 % of disease-causing mutations [15].
Template preparation and barcoding
The massively parallel sequencing technology is based on a combination of template preparation, sequencing, imaging/recording and data analysis [61]. DNA and library preparations are the first steps. Two to twenty microgram of high quality genomic DNA is randomly sheared into smaller size molecules by sonication or nebulization. Several DNA can also be pooled and sequenced together. The barcoding and pooling step can be done either before or after capture of specific targeted sequences, using a unique DNA tag sequence per sample [18]. In this way, it is possible to sequence a few genes in many patients or many genes from a few patients.
Targeted sequence enrichment
There is no enrichment step for whole genome sequencing [94]; however, if the aim is to sequence a portion of genomic DNA which can be the protein-coding part of the genome (exome) or regions of interest such as selected genes or a genomic region linked to a disease, such targeted sequencing includes the enrichment of the target sequences.
This enrichment or capture can be done on microarray or in solution [60]. In solid-phase enrichment, high-density primers targeting the sequences of interest are covalently attached to the slide array [25], while in solution-phase enrichment, primers are generated on beads [20]. Several solutions such as Illumina, Agilent and Nimblegen capture kits can be used; they have technical differences such as using RNA baits (Agilent) or DNA baits (Illumina and Nimblegen) with almost similar performance [17]. Other enrichment methods include RainStorm microdroplet-based technology from Raindance technologies which is a multiplex PCR method using microdroplets containing PCR components loaded on microfluidic chip to compartmentalize the PCRs by single primer pairs [87].
Sequencing
For some of the sequencing platforms such as Helicos BioSciences, single molecule template is used, whereas in others such as Illumina, SOLiD and Roche/454, clonally amplified DNA is required to detect the signal produced by the incorporation of nucleotides [57]. Thus, for sequencing platforms using single molecule templates, the amount of starting DNA is lower and there is no PCR amplification step that could create artificial mutations and AT or GC-rich amplification bias.
In all the MPS platforms, either templates, primers or polymerase enzyme are immobilized on a solid support before the sequencing reaction [61]. Sequencing and recording steps are different [54]. Illumina technology is based on clonally amplified templates coupled with cyclic reversible termination method with four fluorescent colors. First, one fluorescently modified nucleotide complementary to the template sequence is incorporated. After washing and imaging for detection of the incorporated nucleotide, a cleavage step removes the fluorescent dye and a novel incorporation step is performed. These steps are done in a cyclic manner, 72 or 100 times or more [9]. It is possible to sequence from both extremities of the DNA template (paired-end sequencing). HeliScope single-molecule sequencer of Helicos BioSciences works with one fluorescent color (Cy5) using a cyclic reversible termination approach [12]. SOLiD (Sequencing by Oligonucleotide Ligation and Detection) sequencer from Applied Biosystems is another MPS technology based on sequencing by ligation using DNA ligase and a cleavable two-base-encoded probe consisting of two nucleotides combined with a particular dye [91]. Roche/454 sequencing platform is based on pyrosequencing. In this sequencing by synthesis method, a pyrophosphate (PPi) is released after incorporation of one dNTP. This released pyrophosphate is converted into detectable light through a series of enzymatic reaction [80]. Ion Torrent commercializes semiconductor sequencing machines where the release of a proton after incorporation of a nucleotide leading to change in pH is detected by voltage change. If two nucleotides are incorporated, the voltage will be double [82]. For human whole genome sequencing, apart from the above mentioned sequencing platforms, Complete Genomics offers a non-commercialized solution. In that case, DNA nanoballs containing hundreds of copies of a short DNA fragment are sequenced using combinatorial probe anchor-ligation (cPAL) where fluorescent molecules are attached to each nucleotide by a ligase enzyme [23] (Table 1).
Data analysis
The final steps in massively parallel sequencing are genome alignment, variant calling and data analysis. The generated sequence reads are aligned to a human reference genome such as UCSC assembly hg18/NCBI 36 or hg19/GRCH 37, and the variations are detected using different programs including BWA [50, 89] for alignment and Samtools [51] for variant calling. For filtering and scoring the variants and detection of disease-causing mutations, there are several strategies depending on the mode of inheritance and number of affected/non-affected sequenced cases [29]. Sequencing several affected and eventually non-affected individuals from the same family improves the filtering; variations common in affected and absent in non-affected persons will be prioritized. Several public websites display polymorphisms and allele frequencies such as dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 genomes (http://www.1000genomes.org/) or the Exome Variant Server, NHLBI Exome Sequencing (http://evs.gs.washington.edu/EVS/) which covers SNPs but no indels. Many softwares are available to filter sequence variants, such as ALAMUT software (Interactive Biosoftware, Rouen, France), Cartagenia benchlab (http://www.cartagenia.com/); SIMPLEX (http://simplex.i-med.ac.at) [27] or ANNOVAR (http://www.openbioinformatics.org/annovar/) [93], and most integrate tools to predict the pathogenicity of the variations on RNA splicing and protein function such as SIFT [65], Polyphen [2, 77] or NNSPLICE (Fig. 1).

Workflow of massively parallel sequencing and analysis. a 2–20 μg of genomic DNA is sheared randomly and targeted sequences are enriched following capture protocols in solid or liquid phases. Targeted genes/regions or exome can be enriched, or the whole genome can be sequenced. Amplified or single molecules are prepared for sequencing according to each platform. Output sequences are used for mapping to a reference genome, variation calling and filtering, using different algorithms and softwares. The last step is validation, where the interesting variations are confirmed by Sanger sequencing from the starting DNA, segregation of the variation in the family investigated, and a healthy control population checked. Sequencing of additional patients with similar phenotypes to identify more mutated patients with variations in the same gene, and functional studies could be done to confirm the implication of the gene or the pathogenicity of the variation if not previously known. b Different steps of filtering are shown. From the list of called variations, the known non-pathogenic variations are removed by comparison with dbSNP, 1,000 genomes and Exome Variant Server databases. Based on disease inheritance mode, homozygous or heterozygous changes are selected. Next, in silico prediction of pathogenecity and effect on splicing can be tested using different programs such as PolyPhen, SIFT or NNSPLICE. Scoring and ranking of the different variations/genes based on expression profile and function using different databases such as GENATLAS or Genecards provide a list of prioritized genes and variations
Massively parallel sequencing for NMD diagnosis
In 2010, Lupski et al. [58] published the first paper using massively parallel sequencing technology in neuromuscular diseases, where they showed the result of whole genome sequencing of a patient with a recessive form of Charcot–Marie–Tooth neuropathy. They prioritized sequence variants in genes known to be implicated in Charcot–Marie–Tooth neuropathies and found the compound heterozygous mutations p.Arg954X and p.Tyr169His in the SH3TC2 gene as the causes of the neuropathy. Since then, several studies have been published using massively parallel sequencing for detection of causative mutations in different NMDs. Through exome sequencing, Montenegro et al. [62] found a missense variation, p.Val95Met, in the GJB1 gene, a known gene in CMT neuropathies, in a family with CMT. Sanger sequencing confirmed this change and validated the complete co-segregation within the family.
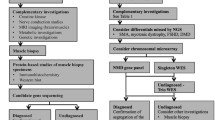
In our recent publication, we showed the efficacy of massively parallel sequencing in molecular diagnosis of patients with different NMDs by capturing and sequencing 267 genes implicated in NMDs [92]. We could retrieve successfully all known mutations, detect and precise a large deletion linked to DMD, and identify novel disease-causing mutations in patients awaiting molecular diagnosis since more than 15 years. Other targeted sequencing focused on subsets of NMD genes. 25 patients with Duchenne or Becker muscular dystrophies (DMD/BMD) with known or unknown mutations were sequenced with the Illumina genome analyzer using a capture kit targeting the whole genomic sequence of muscular dystrophy-related genes after DNA barcoding [53]. The authors could detect disease-causing mutations in 24 patients out of 25 and concluded that this technology is useful for diagnosis of patients with DMD/BMD. Hoischen et al. [42] reported the validation of an array based sequence capture of seven genes and two loci related to the autosomal recessive form of ataxias, by sequencing these genes in five patients with known mutations and two unaffected persons using a one-quarter Roche GS FLX Titanium sequencing run. Other examples were dedicated to other specific disease classes [36, 45] (Table 2).
Such targeted parallel sequencing of all candidate genes is especially appropriate for disorders with high genetic heterogeneity like NMDs, and should ease the identification of allelic diseases, i.e. different diseases caused by mutations of the same gene. In addition, for large genes, such as TTN, which are difficult to fully test by conventional Sanger sequencing routinely even if known to be implicated in disorders, different studies have shown the effectiveness of massively parallel sequencing in variant detection of such a large gene [68, 72, 92]. Although there are some limitations in massively parallel sequencing as described below, important issues in genetic diagnosis laboratories such as cost and turnaround time can be resolved using massively parallel sequencing. Transfer of these technologies to diagnosis laboratories will benefit from the constantly dropping costs and increasing output. Reliable data can be produced if the depth of sequence coverage is high enough. In Fig. 2, an example of MPS achieved data is shown.

An example of massively parallel sequencing data for a patient with myotubular myopathy. a The table shows three different variations: the first row is the disease-causing mutation which is a deletion of four nucleotides in the MTM1 gene. The second row is a synonymous change and the third is an intronic insertion. b Deletion of four nucleotides at the beginning of exon 4 in the MTM1 gene displayed with the integrative genomics viewer (IGV)
Possible applications of massively parallel sequencing in diagnosis
Diagnosis of affected individuals
Ideal strategies for diagnostic laboratories are methods that are simple with high accuracy and low error rate in a short run time; they should also be cost-effective with easy data analysis and interpretation [20]. Massively parallel sequencing is a useful alternative or complementary technique for molecular diagnosis. Routine use of massively parallel sequencing leads to a rapid screening and fast identification of mutations in rare genetic disorders through sequencing of either the exons or the genomic sequence of all genes or a subset of genes. Nowadays, with benchtop sequencers such as Ion Torrent PGM (Ion Torrent, Guilford, CT), MiSeq (Illumina Inc., San Diego, CA) and 454 GS Junior (454 Life Sciences, Roche, Branford, CT), these new technologies can be transferred easily to clinics for mutation identification in patients, carrier status determination and prenatal diagnosis. The cost of whole genome sequencing of a DNA without interpretation is about 6,500 USD and exome sequencing is offered for about 1,000 USD (an example: http://www.edgebio.com). High throughput screening at low cost and low complexity can be addressed by targeted sequencing rather than complete sequencing of exomes or genomes, combined with multiplexing of barcoded samples. The cost for this strategy depends on the number of mixed samples and the size of targeted regions and is generally less than 1,000 USD per sample. Moreover, targeted and exome sequencing are starting to be proposed on a routine diagnosis basis (for examples: http://www.bcm.edu/geneticlabs/ or http://genetics.emory.edu/egl/). Financing this cost depends on specific countries regulation and insurance policies.
Massively parallel sequencing will probably replace most of the genetic screening methods but will not substitute for clinical and histopathological investigations. While until now, clinical and pathological diagnoses were used to orient genetic screening, massively parallel sequencing may now be used on a first intention to better orient clinical tests that could be invasive, costly and necessitate patient travel. Although biopsies might be less needed for establishing the molecular diagnosis, they will be necessary for understanding the pathogenesis of a disease. Thus, massively parallel sequencing will promote a more efficient integrated diagnosis encompassing clinic, histopathology and molecular analyses.
A rapid and accurate molecular diagnosis will have important impacts on patients as it will improve disease management, may lead to inclusion into therapeutic trials, will help genetic counseling and will reduce further unhelpful investigations.
Prenatal diagnosis
This technology is also applicable to noninvasive prenatal diagnostic by massively parallel sequencing of fetal DNA present in the maternal plasma. The circulating cell-free fetal DNA is assessed using a set of differentially methylated markers [71] or by exome or genome sequencing compared to parents DNAs [26]. Currently, this approach is used for detection of severe monogenic disorders, fetal chromosomal aneuploidies and determination of blood groups [56]. Such approach will avoid the need for invasive and risky procedures such as chorionic villus sampling. Comprehensive diagnosis of any Mendelian disorders such as NMDs early in pregnancy might permit early termination of pregnancy in an ethically acceptable way. It can have a significant impact on the reproductive decision making and pregnancy management as it has been shown that families with increased risk of having a child with a severe disorder tend to have fewer children [78].
Carrier detection
The massively parallel sequencing approach might also apply to families without previous cases of severe genetic disorders. It would be technically possible to determine the carrier risks of individuals for any Mendelian disorders and predict for example, for which recessive diseases both parents are carriers of a heterozygous mutation [8]. Based on this knowledge, parents would have the possibility to test the fetus by sequencing the gene(s) with heterozygous mutations, or performing in vitro fertilization (IVF) and selecting non-affected zygotes. This approach will not predict de novo mutation, another source of disease-causing mutations.
Massively parallel sequencing limitations and improvements for clinical use
Although massively parallel sequencing is an appropriate alternative technique to use in diagnostic laboratories, some issues need to be addressed.
False negatives
One challenge in the use of massively parallel sequencing for diagnosis is the detection of different types of mutations, especially repeat expansions or structural variations. Some of the most common NMDs are due to repeat expansions; these mutations may be missed leading to false negative results. Increasing sequence coverage [40], improving bioinformatics algorithms and softwares and novel sequencing technologies may solve this problem in order to propose an exclusion diagnosis. Alternatively, a combination of different methods can be proposed. Detection of structural variations will benefit from whole genome sequencing. Another obstacle is the incomplete coverage of commercial exome capture libraries. Although new versions of capture kits are released consistently, none of them captures all coding parts of all genes [47]. This issue can be addressed by targeted sequencing of genes and regions of interest and improving the capturing process.
False positives
Another drawback is the high error rates in massively parallel sequencing compared to Sanger sequencing. Artificial mutations can be produced during templates amplification or sequencing, leading to false positive results. Thus, Sanger sequencing of interesting variants detected by massively parallel sequencing is an essential validation step which is increasing the cost and turnaround time. This issue can be addressed by improving capturing and sequencing approaches to increase variant coverage, and thus leads to achieve reliable data. In addition, better data filtering protocols can reduce the pool of false positives.
Volume of data
Massively parallel sequencing generates a high volume of data which becomes problematic for data management, analysis and storage in diagnostic laboratories [74]. As the genetic test results should be kept at least 5 years or even 10–20 years [85], substantial investment in infrastructure and informatics is needed. Cloud computing can be a solution for reducing the cost of expensive computing infrastructure [20, 84]. Importantly, decreasing costs in massively parallel sequencing outpaces the increase in calculation power and storage capacity of computers. As sequencing becomes cheaper than data storage of the corresponding sequence output, re-sequencing of a patient DNA might be more cost-effective than saving original data [38]. In other words, DNA is becoming the cheapest storage support.
Variants filtering and mutation identification
As a high number of variations are detected by massively parallel sequencing, it is difficult to distinguish between individual, rare and non-pathogenic variations without clinical significance versus disease-causing mutations. Defining the complete list of polymorphisms in different populations will require sequencing a large part of the world populations [52]. A recent study hypothesized that 27 % of published mutations appear to be sequencing errors, common polymorphisms, or have a lack of pathogenicity evidence [8]. This issue can be addressed by further analysis and validation such as in silico predictions of pathogenicity, detection of mutations in the same gene in unrelated individuals with the same disorder and absence in a control population, co-segregation in affected families and finally functional studies [90]. Multiple criteria should be combined in order to prove the pathogenicity of the variation.
Incidental findings
With massively parallel sequencing, incidental findings are an issue and unsolicited/unexpected information can be problematic, especially for unreported diseases. Diagnostic laboratories should prepare informative, complete and precise consent and result reports to use massively parallel sequencing for diagnosis application. Guidelines should be established and implemented by national committees in order to determine whether the analysis and reports should be selective or complete and what is the best way to cope with the ethical issues [16, 31].
Trained people
Having enough trained people for data analysis and information transfer to patients will be a key point for the future. Specific trainings are needed for interpreting genetic data for rare and common diseases and eventually genetic predispositions, and how to transfer this information to patients. Ideally a team of bioinformaticians, scientists, geneticists and clinicians will collaborate together in order to provide an accurate and accessible results to patients. Development of robust, easy to use and practical softwares in the clinical setting and comprehensive mutations and polymorphisms databases are necessary. For a full understanding of genetic variations, considerable amount of genetic data from various populations should be gathered and interpreted. Recurrent re-analysis of the sequencing data will be needed while our understanding of genetic variations improves. Well-trained clinicians and pathologists in neuromuscular disorders and bioinformaticians will have crucial roles in the selection of patients and interpretation of the obtained data.
Conclusion
Although there are some limitations and challenges using massively parallel sequencing for diagnosis, this technology appears mature enough for a routine approach in clinics. As the cost of massively parallel sequencing declines consistently and the technology improves continually leading to obtaining faster and more accurate data, it is strongly anticipated that this recent technology will complement clinical and pathological investigations and will greatly improve diagnosis and management of Mendelian disorders.
References
Abbs S, Bobrow M (1992) Analysis of quantitative PCR for the diagnosis of deletion and duplication carriers in the dystrophin gene. J Med Genet 29(3):191–196
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. doi:10.1038/nmeth0410-248
Alias L, Bernal S, Fuentes-Prior P, Barcelo MJ, Also E, Martinez-Hernandez R, Rodriguez-Alvarez FJ, Martin Y, Aller E, Grau E, Pecina A, Antinolo G, Galan E, Rosa AL, Fernandez-Burriel M, Borrego S, Millan JM, Hernandez-Chico C, Baiget M, Tizzano EF (2009) Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum Genet 125(1):29–39. doi:10.1007/s00439-008-0598-1
Andersen PS, Havndrup O, Hougs L, Sorensen KM, Jensen M, Larsen LA, Hedley P, Thomsen AR, Moolman-Smook J, Christiansen M, Bundgaard H (2009) Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat 30(3):363–370. doi:10.1002/humu.20862
Bartoli M, Negre P, Wein N, Bourgeois P, Pecheux C, Levy N, Krahn M (2012) Validation of comparative genomic hybridization arrays for the detection of genomic rearrangements of the calpain-3 and dysferlin genes. Clin Genet 81(1):99–101. doi:10.1111/j.1399-0004.2011.01708.x
Beggs AH, Koenig M, Boyce FM, Kunkel LM (1990) Detection of 98 % of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 86(1):45–48
Belaya K, Finlayson S, Slater CR, Cossins J, Liu WW, Maxwell S, McGowan SJ, Maslau S, Twigg SR, Walls TJ, Pascual Pascual SI, Palace J, Beeson D (2012) Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am J Hum Genet 91(1):193–201. doi:10.1016/j.ajhg.2012.05.022
Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, Sheth V, Woodward JE, Peckham HE, Schroth GP, Kim RW, Kingsmore SF (2011) Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 3(65):65ra64. doi:10.1126/scitranslmed.3001756
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O’Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456(7218):53–59. doi:10.1038/nature07517
Blauw HM, Barnes CP, van Vught PW, van Rheenen W, Verheul M, Cuppen E, Veldink JH, van den Berg LH (2012) SMN1 gene duplications are associated with sporadic ALS. Neurology 78(11):776–780. doi:10.1212/WNL.0b013e318249f697
Bohm J, Leshinsky-Silver E, Vassilopoulos S, Le Gras S, Lerman-Sagie T, Ginzberg M, Jost B, Lev D, Laporte J (2012) Samaritan myopathy, an ultimately benign congenital myopathy, is caused by a RYR1 mutation. Acta Neuropathol 124(4):575–581. doi:10.1007/s00401-012-1007-3
Braslavsky I, Hebert B, Kartalov E, Quake SR (2003) Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci USA 100(7):3960–3964. doi:10.1073/pnas.0230489100
Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT (1988) Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 16(23):11141–11156
Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74(6):1128–1135. doi:10.1086/421054
Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP (2009) Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA 106(45):19096–19101. doi:10.1073/pnas.0910672106
Christenhusz GM, Devriendt K, Dierickx K (2012) To tell or not to tell? A systematic review of ethical reflections on incidental findings arising in genetics contexts. Eur J Hum Genet. doi:10.1038/ejhg.2012.130
Clark MJ, Chen R, Lam HY, Karczewski KJ, Euskirchen G, Butte AJ, Snyder M (2011) Performance comparison of exome DNA sequencing technologies. Nat Biotechnol 29(10):908–914. doi:10.1038/nbt.1975
Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, Pawlowski TL, Laub T, Nunn G, Stephan DA, Homer N, Huentelman MJ (2008) Identification of genetic variants using bar-coded multiplexed sequencing. Nat Methods 5(10):887–893. doi:10.1038/nmeth.1251
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2):245–256. doi:10.1016/j.neuron.2011.09.011
Desai AN, Jere A (2012) Next-generation sequencing: ready for the clinics? Clin Genet 81(6):503–510. doi:10.1111/j.1399-0004.2012.01865.x
Dias C, Sincan M, Cherukuri PF, Rupps R, Huang Y, Briemberg H, Selby K, Mullikin JC, Markello TC, Adams DR, Gahl WA, Boerkoel CF (2012) An analysis of exome sequencing for diagnostic testing of the genes associated with muscle disease and spastic paraplegia. Hum Mutat 33(4):614–626. doi:10.1002/humu.22032
Donner K, Sandbacka M, Lehtokari VL, Wallgren-Pettersson C, Pelin K (2004) Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur J Hum Genet 12(9):744–751. doi:10.1038/sj.ejhg.5201242
Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, Dahl F, Fernandez A, Staker B, Pant KP, Baccash J, Borcherding AP, Brownley A, Cedeno R, Chen L, Chernikoff D, Cheung A, Chirita R, Curson B, Ebert JC, Hacker CR, Hartlage R, Hauser B, Huang S, Jiang Y, Karpinchyk V, Koenig M, Kong C, Landers T, Le C, Liu J, McBride CE, Morenzoni M, Morey RE, Mutch K, Perazich H, Perry K, Peters BA, Peterson J, Pethiyagoda CL, Pothuraju K, Richter C, Rosenbaum AM, Roy S, Shafto J, Sharanhovich U, Shannon KW, Sheppy CG, Sun M, Thakuria JV, Tran A, Vu D, Zaranek AW, Wu X, Drmanac S, Oliphant AR, Banyai WC, Martin B, Ballinger DG, Church GM, Reid CA (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327(5961):78–81. doi:10.1126/science.1181498
Edvardson S, Cinnamon Y, Jalas C, Shaag A, Maayan C, Axelrod FB, Elpeleg O (2012) Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann Neurol 71(4):569–572. doi:10.1002/ana.23524
Emery AE (1991) Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord 1(1):19–29
Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR (2012) Non-invasive prenatal measurement of the fetal genome. Nature 487(7407):320–324. doi:10.1038/nature11251
Fischer M, Snajder R, Pabinger S, Dander A, Schossig A, Zschocke J, Trajanoski Z, Stocker G (2012) SIMPLEX: cloud-enabled pipeline for the comprehensive analysis of exome sequencing data. PLoS ONE 7(8):e41948. doi:10.1371/journal.pone.0041948
Gazzerro E, Bonetto A, Minetti C (2011) Caveolinopathies: translational implications of caveolin-3 in skeletal and cardiac muscle disorders. Handb Clin Neurol 101:135–142. doi:10.1016/B978-0-08-045031-5.00010-4
Gilissen C, Hoischen A, Brunner HG, Veltman JA (2012) Disease gene identification strategies for exome sequencing. Eur J Hum Genet 20(5):490–497. doi:10.1038/ejhg.2011.258
Gitcho MA, Bigio EH, Mishra M, Johnson N, Weintraub S, Mesulam M, Rademakers R, Chakraverty S, Cruchaga C, Morris JC, Goate AM, Cairns NJ (2009) TARDBP 3′-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol 118(5):633–645. doi:10.1007/s00401-009-0571-7
Godard B, Kaariainen H, Kristoffersson U, Tranebjaerg L, Coviello D, Ayme S (2003) Provision of genetic services in Europe: current practices and issues. Eur J Hum Genet 11(Suppl 2):S13–S48. doi:10.1038/sj.ejhg.5201111
Goto K, Nishino I, Hayashi YK (2006) Rapid and accurate diagnosis of facioscapulohumeral muscular dystrophy. Neuromuscul Disord 16(4):256–261. doi:10.1016/j.nmd.2006.01.008
Greenway MJ, Alexander MD, Ennis S, Traynor BJ, Corr B, Frost E, Green A, Hardiman O (2004) A novel candidate region for ALS on chromosome 14q11.2. Neurology 63(10):1936–1938. pii:63/10/1936
Guis S, Figarella-Branger D, Monnier N, Bendahan D, Kozak-Ribbens G, Mattei JP, Lunardi J, Cozzone PJ, Pellissier JF (2004) Multiminicore disease in a family susceptible to malignant hyperthermia: histology, in vitro contracture tests, and genetic characterization. Arch Neurol 61(1):106–113. doi:10.1001/archneur.61.1.106
Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, Miyamoto N, Showguchi-Miyata J, Okada Y, Singaraja R, Figlewicz DA, Kwiatkowski T, Hosler BA, Sagie T, Skaug J, Nasir J, Brown RH Jr, Scherer SW, Rouleau GA, Hayden MR, Ikeda JE (2001) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 29(2):166–173. doi:10.1038/ng1001-166
Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, Ma D, Masi S, Allred P, Al-Lozi M, Reilly MM, Miller LJ, Jani-Acsadi A, Pestronk A, Shy ME, Muntoni F, Vallee RB, Baloh RH (2012) Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 78(22):1714–1720. doi:10.1212/WNL.0b013e3182556c05
Harms MB, Sommerville RB, Allred P, Bell S, Ma D, Cooper P, Lopate G, Pestronk A, Weihl CC, Baloh RH (2012) Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann Neurol 71(3):407–416. doi:10.1002/ana.22683
Hastings R, de Wert G, Fowler B, Krawczak M, Vermeulen E, Bakker E, Borry P, Dondorp W, Nijsingh N, Barton D, Schmidtke J, van El CG, Vermeesch J, Stol Y, Carmen Howard H, Cornel MC (2012) The changing landscape of genetic testing and its impact on clinical and laboratory services and research in Europe. Eur J Hum Genet 20(9):911–916. doi:10.1038/ejhg.2012.56
Hayward C, Colville S, Swingler RJ, Brock DJ (1999) Molecular genetic analysis of the APEX nuclease gene in amyotrophic lateral sclerosis. Neurology 52(9):1899–1901
Herdewyn S, Zhao H, Moisse M, Race V, Matthijs G, Reumers J, Kusters B, Schelhaas HJ, van den Berg LH, Goris A, Robberecht W, Lambrechts D, Van Damme P (2012) Whole-genome sequencing reveals a coding non-pathogenic variant tagging a non-coding pathogenic hexanucleotide repeat expansion in C9orf72 as cause of amyotrophic lateral sclerosis. Hum Mol Genet 21(11):2412–2419. doi:10.1093/hmg/dds055
Hill M (2003) The neuromuscular junction disorders. J Neurol Neurosurg Psychiatry 74(Suppl 2):ii32–ii37
Hoischen A, Gilissen C, Arts P, Wieskamp N, van der Vliet W, Vermeer S, Steehouwer M, de Vries P, Meijer R, Seiqueros J, Knoers NV, Buckley MF, Scheffer H, Veltman JA (2010) Massively parallel sequencing of ataxia genes after array-based enrichment. Hum Mutat 31(4):494–499. doi:10.1002/humu.21221
Jimenez-Escrig A, Gobernado I, Garcia-Villanueva M, Sanchez-Herranz A (2012) Autosomal recessive Emery-Dreifuss muscular dystrophy caused by a novel mutation (R225Q) in the lamin A/C gene identified by exome sequencing. Muscle Nerve 45(4):605–610. doi:10.1002/mus.22324
Kaplan JC (2011) The 2012 version of the gene table of monogenic neuromuscular disorders. Neuromuscul Disord 21(12):833–861
Kondo E, Nishimura T, Kosho T, Inaba Y, Mitsuhashi S, Ishida T, Baba A, Koike K, Nishino I, Nonaka I, Furukawa T, Saito K (2012) Recessive RYR1 mutations in a patient with severe congenital nemaline myopathy with ophthalomoplegia identified through massively parallel sequencing. Am J Med Genet A 158A(4):772–778. doi:10.1002/ajmg.a.35243
Krysa W, Rajkiewicz M, Sulek A (2012) Rapid detection of large expansions in progressive myoclonus epilepsy type 1, myotonic dystrophy type 2 and spinocerebellar ataxia type 8. Neurol Neurochir Pol 46(2):113–120. doi:18490
Ku CS, Cooper DN, Polychronakos C, Naidoo N, Wu M, Soong R (2012) Exome sequencing: dual role as a discovery and diagnostic tool. Ann Neurol 71(1):5–14. doi:10.1002/ana.22647
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918):1205–1208. doi:10.1126/science.1166066
Laing NG (2012) Genetics of neuromuscular disorders. Crit Rev Clin Lab Sci 49(2):33–48. doi:10.3109/10408363.2012.658906
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. doi:10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. doi:10.1093/bioinformatics/btp352
Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T, Grarup N, Guo Y, Hellman I, Jin X, Li Q, Liu J, Liu X, Sparso T, Tang M, Wu H, Wu R, Yu C, Zheng H, Astrup A, Bolund L, Holmkvist J, Jorgensen T, Kristiansen K, Schmitz O, Schwartz TW, Zhang X, Li R, Yang H, Wang J, Hansen T, Pedersen O, Nielsen R (2010) Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet 42(11):969–972. doi:10.1038/ng.680
Lim BC, Lee S, Shin JY, Kim JI, Hwang H, Kim KJ, Hwang YS, Seo JS, Chae JH (2011) Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology: comprehensive mutational search in a single platform. J Med Genet 48(11):731–736. doi:10.1136/jmedgenet-2011-100133
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M (2012) Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012:251364. doi:10.1155/2012/251364
Liu MJ, Xie M, Mao J, Li H, Yan WH, Chen Y (2012) Application of next-generation sequencing technology for genetic diagnosis of Duchenne muscular dystrophy. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 29(3):249–254. doi:10.3760/cma.j.issn.1003-9406.2012.03.001
Lo YM, Chiu RW (2012) Genomic analysis of fetal nucleic acids in maternal blood. Annu Rev Genomics Hum Genet. doi:10.1146/annurev-genom-090711-163806
Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ (2012) Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 30(5):434–439. doi:10.1038/nbt.2198
Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, McGuire AL, Zhang F, Stankiewicz P, Halperin JJ, Yang C, Gehman C, Guo D, Irikat RK, Tom W, Fantin NJ, Muzny DM, Gibbs RA (2010) Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N Engl J Med 362(13):1181–1191. doi:10.1056/NEJMoa0908094
Lynch PJ, Krivosic-Horber R, Reyford H, Monnier N, Quane K, Adnet P, Haudecoeur G, Krivosic I, McCarthy T, Lunardi J (1997) Identification of heterozygous and homozygous individuals with the novel RYR1 mutation Cys35Arg in a large kindred. Anesthesiology 86(3):620–626
Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ (2010) Target-enrichment strategies for next-generation sequencing. Nat Methods 7(2):111–118. doi:10.1038/nmeth.1419
Metzker ML (2010) Sequencing technologies - the next generation. Nat Rev Genet 11(1):31–46. doi:10.1038/nrg2626
Montenegro G, Powell E, Huang J, Speziani F, Edwards YJ, Beecham G, Hulme W, Siskind C, Vance J, Shy M, Zuchner S (2011) Exome sequencing allows for rapid gene identification in a Charcot–Marie–Tooth family. Ann Neurol 69(3):464–470. doi:10.1002/ana.22235
Muntoni F, Torelli S, Ferlini A (2003) Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2(12):731–740 pii:S1474442203005854
Nance JR, Dowling JJ, Gibbs EM, Bonnemann CG (2012) Congenital myopathies: an update. Curr Neurol Neurosci Rep 12(2):165–174. doi:10.1007/s11910-012-0255-x
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ (2010) Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet 42(1):30–35. doi:10.1038/ng.499
Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461(7261):272–276. doi:10.1038/nature08250
Nowak KJ (2012) Trusting new age weapons to tackle titin. Brain 135(Pt 6):1665–1667. doi:10.1093/brain/aws123
Ohlsson M, Hedberg C, Bradvik B, Lindberg C, Tajsharghi H, Danielsson O, Melberg A, Udd B, Martinsson T, Oldfors A (2012) Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain 135(Pt 6):1682–1694. doi:10.1093/brain/aws103
Oudet C, Hanauer A, Clemens P, Caskey T, Mandel JL (1992) Two hot spots of recombination in the DMD gene correlate with the deletion prone regions. Hum Mol Genet 1(8):599–603
Palomaki GE, Deciu C, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, van den Boom D, Bombard AT, Grody WW, Nelson SF, Canick JA (2012) DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: an international collaborative study. Genet Med 14(3):296–305. doi:10.1038/gim.2011.73
Pfeffer G, Elliott HR, Griffin H, Barresi R, Miller J, Marsh J, Evila A, Vihola A, Hackman P, Straub V, Dick DJ, Horvath R, Santibanez-Koref M, Udd B, Chinnery PF (2012) Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 135(Pt 6):1695–1713. doi:10.1093/brain/aws102
Phillips MS, Fujii J, Khanna VK, DeLeon S, Yokobata K, de Jong PJ, MacLennan DH (1996) The structural organization of the human skeletal muscle ryanodine receptor (RYR1) gene. Genomics 34(1):24–41. doi:10.1006/geno.1996.0238
Pop M, Salzberg SL (2008) Bioinformatics challenges of new sequencing technology. Trends Genet 24(3):142–149. doi:10.1016/j.tig.2007.12.006
Prior TW (2010) Perspectives and diagnostic considerations in spinal muscular atrophy. Genet Med 12(3):145–152. doi:10.1097/GIM.0b013e3181c5e713
Prior TW, Bridgeman SJ (2005) Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J Mol Diagn 7(3):317–326. doi:10.1016/S1525-1578(10)60560-0
Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30(17):3894–3900
Raymond FL, Whittaker J, Jenkins L, Lench N, Chitty LS (2010) Molecular prenatal diagnosis: the impact of modern technologies. Prenat Diagn 30(7):674–681. doi:10.1002/pd.2575
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72(2):257–268. doi:10.1016/j.neuron.2011.09.010
Ronaghi M, Karamohamed S, Pettersson B, Uhlen M, Nyren P (1996) Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 242(1):84–89. doi:10.1006/abio.1996.0432
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362(6415):59–62. doi:10.1038/362059a0
Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, Hoon J, Simons JF, Marran D, Myers JW, Davidson JF, Branting A, Nobile JR, Puc BP, Light D, Clark TA, Huber M, Branciforte JT, Stoner IB, Cawley SE, Lyons M, Fu Y, Homer N, Sedova M, Miao X, Reed B, Sabina J, Feierstein E, Schorn M, Alanjary M, Dimalanta E, Dressman D, Kasinskas R, Sokolsky T, Fidanza JA, Namsaraev E, McKernan KJ, Williams A, Roth GT, Bustillo J (2011) An integrated semiconductor device enabling non-optical genome sequencing. Nature 475(7356):348–352. doi:10.1038/nature10242
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME (2011) Charcot–Marie–Tooth disease subtypes and genetic testing strategies. Ann Neurol 69(1):22–33. doi:10.1002/ana.22166
Sboner A, Mu XJ, Greenbaum D, Auerbach RK, Gerstein MB (2011) The real cost of sequencing: higher than you think! Genome Biol 12(8):125. doi:10.1186/gb-2011-12-8-125
Schwartz MK (1999) Genetic testing and the clinical laboratory improvement amendments of 1988: present and future. Clin Chem 45(5):739–745
Tennyson CN, Klamut HJ, Worton RG (1995) The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet 9(2):184–190. doi:10.1038/ng0295-184
Tewhey R, Warner JB, Nakano M, Libby B, Medkova M, David PH, Kotsopoulos SK, Samuels ML, Hutchison JB, Larson JW, Topol EJ, Weiner MP, Harismendy O, Olson J, Link DR, Frazer KA (2009) Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat Biotechnol 27(11):1025–1031. doi:10.1038/nbt.1583
Tosch V, Vasli N, Kretz C, Nicot AS, Gasnier C, Dondaine N, Oriot D, Barth M, Puissant H, Romero NB, Bonnemann CG, Heller B, Duval G, Biancalana V, Laporte J (2010) Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul Disord 20(6):375–381. doi:10.1016/j.nmd.2010.03.015
Trapnell C, Salzberg SL (2009) How to map billions of short reads onto genomes. Nat Biotechnol 27(5):455–457. doi:10.1038/nbt0509-455
Tucker EJ, Mimaki M, Compton AG, McKenzie M, Ryan MT, Thorburn DR (2011) Next-generation sequencing in molecular diagnosis: NUBPL mutations highlight the challenges of variant detection and interpretation. Hum Mutat 33(2):411–418. doi:10.1002/humu.21654
Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, Sidow A, Fire A, Johnson SM (2008) A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 18(7):1051–1063. doi:10.1101/gr.076463.108
Vasli N, Bohm J, Le Gras S, Muller J, Pizot C, Jost B, Echaniz-Laguna A, Laugel V, Tranchant C, Bernard R, Plewniak F, Vicaire S, Levy N, Chelly J, Mandel JL, Biancalana V, Laporte J (2012) Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol 124(2):273–283. doi:10.1007/s00401-012-0982-8
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. doi:10.1093/nar/gkq603
Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM (2008) The complete genome of an individual by massively parallel DNA sequencing. Nature 452(7189):872–876. doi:10.1038/nature06884
Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, Gruter AM, Hofker MH, Moerer P, Williamson R et al (1992) Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 2(1):26–30. doi:10.1038/ng0992-26
Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, Yan J, Azim AC, Cole N, Gascon G, Yagmour A, Ben-Hamida M, Pericak-Vance M, Hentati F, Siddique T (2001) The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 29(2):160–165. doi:10.1038/ng1001-160ng1001-160
Zimmern RL, Kroese M (2007) The evaluation of genetic tests. J Public Health (Oxf) 29(3):246–250. doi:10.1093/pubmed/fdm028
Acknowledgments
We thank Pr. Jean-Louis Mandel and Dr. Valérie Biancalana for discussions. This study was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the Centre National de la Recherche Scientifique (CNRS), University of Strasbourg (UdS), Collège de France, Agence Nationale de la Recherche (ANR-11-BSV1-026), Muscular Dystrophy Association (186985), and the Myotubular Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vasli, N., Laporte, J. Impacts of massively parallel sequencing for genetic diagnosis of neuromuscular disorders. Acta Neuropathol 125, 173–185 (2013). https://doi.org/10.1007/s00401-012-1072-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-012-1072-7