
Abstract
Impairment of synaptic plasticity underlies memory dysfunction in Alzheimer’s disease (AD). Molecules involved in this plasticity such as PSD-95, a major postsynaptic scaffold protein at excitatory synapses, may play an important role in AD pathogenesis. We examined the distribution of PSD-95 in transgenic mice of amyloidopathy (5XFAD) and tauopathy (JNPL3) as well as in AD brains using double-labeling immunofluorescence and confocal microscopy. In wild type control mice, PSD-95 primarily labeled neuropil with distinct distribution in hippocampal apical dendrites. In 3-month-old 5XFAD mice, PSD-95 distribution was similar to that of wild type mice despite significant Aβ deposition. However, in 6-month-old 5XFAD mice, PSD-95 immunoreactivity in apical dendrites markedly decreased and prominent immunoreactivity was noted in neuronal soma in CA1 neurons. Similarly, PSD-95 immunoreactivity disappeared from apical dendrites and accumulated in neuronal soma in 14-month-old, but not in 3-month-old, JNPL3 mice. In AD brains, PSD-95 accumulated in Hirano bodies in hippocampal neurons. Our findings support the notion that either Aβ or tau can induce reduction of PSD-95 in excitatory synapses in hippocampus. Furthermore, this PSD-95 reduction is not an early event but occurs as the pathologies advance. Thus, the time-dependent PSD-95 reduction from synapses and accumulation in neuronal soma in transgenic mice and Hirano bodies in AD may mark postsynaptic degeneration that underlies long-term functional deficits.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It has been proposed that Alzheimer’s disease (AD) is a synaptic failure [33, 38]. Loss of synapses at a fine structural level as well as reduction in synaptic markers have been well documented in early and late stages of AD [8, 10, 25, 26, 37] and have been shown to correlate well with the extent of cognitive deficits [9, 38]. Whereas most of these studies have used presynaptic markers such as synaptophysin, postsynaptic changes have not been extensively investigated. As AD changes may evolve over years or decades prior to the onset of dementia, a question remains about the chronology of synaptic failure. It is believed that not all patients diagnosed with mild cognitive impairment (MCI) convert to AD [12] and, correspondingly, early accumulation of Aβ or tau in transgenic models does not, in and of itself, cause persistent memory deficits [28, 32]. Establishment of a reliable marker of synaptic structural damage could be of great help in guiding clinical diagnosis, therapy and management. As postsynaptic terminals are believed to be the site for memory formation and storage [36], postsynaptic structural proteins should be the most relevant markers in this regard. Furthermore, as transgenic models of Aβ overexpression do not produce significant tau pathology and vise versa, intrinsic biomarkers of synaptic degeneration are critically important for evaluation of disease progression.
In the present study, we focused on PSD-95, a major scaffold protein that determines the structural and functional integrity of excitatory synapses [4, 6, 11]. We hypothesized that neurotoxicity caused by Aβ and tau may ultimately damage postsynaptic terminals and cause reduction in PSD-95, leading to inevitable synaptic destruction and dysfunction. To establish the temporal relationship between postsynaptic damage and deposition of Aβ and tau, we examined transgenic models of 5XFAD mice (Tg6799 line) that co-overexpress human APP and PS1 harboring five familial AD mutations for amyloidopathy [28] and JNPL3 mice that express human tau encoding the frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) mutation for tauopathy [23]. To determine if PSD-95 destruction is an early or late event, we compared 3- with 6-month-old 5XFAD mice and 3- with 14-month-old JNPL3 mice, respectively. Our results indicate that both Aβ and tau accumulation can independently lead to PSD-95 reduction in excitatory synapses and that this postsynaptic damage is not an early event but one that becomes evident as Aβ and tau pathologies advance. In addition, we examined AD brains to identify PSD-95-related pathology. Changes in PSD-95 in AD brains have been reported; some noted a decrease in total protein level [31], whereas others described an increase in immunoreactivity [22]. Here, we demonstrate that PSD-95 accumulates in Hirano bodies, well-recognized dendritic inclusions frequently seen in hippocampal CA1 neurons in AD and other neurodegenerative diseases as well as, occasionally, in aging brains [16].
Materials and methods
Tissue processing
5XFAD (APPSwFlLon, PSEN1 M146L and L286V) and JNPL3 (P301L) mice were purchased from Jackson Labs and Taconic, respectively. Three 3-month-old and three 6-month-old 5XFAD mice, and three 3-month-old and three 14-month-old JNPL3 mice, along with three wild type controls of 4–6 months of age, were used. All animal manipulations were performed in accordance with the guidelines of NYU Medical Center Animal Care and Use Committee. The mice were anesthetized with sodium pentobarbital (120 mg/kg, i.p.) and perfused transaortically with PBS, and the brains were processed as described previously [3]. Briefly, the right hemisphere was immersion fixed overnight in periodate-lysine-paraformaldehyde (PLP). After fixation, the brain was moved to a phosphate buffer solution containing 20% glycerol and 2% DMSO and stored at 4°C until sectioned. Serial coronal brain sections (40 μm) were cut on a freezing microtome and five series of sections at 0.2 mm intervals placed in ethylene glycol cryoprotectant and stored at −30°C until used for immunohistochemistry.
Autopsy brain tissue was obtained from six cases of neuropathologically confirmed cases of AD based on the CERAD criteria [27] and four control cases from individuals without AD (Table 1). Tissue was obtained from the Neuropathology Service at SUNY Downstate Medical Center and Kings County Hospital Center (Brooklyn, NY). The mean age of death was 80 years for the AD cases and 73.8 years for controls.
Immunohistochemistry
Mouse anti-PSD-95 monoclonal antibody was purchased from NeuroMab (clone K28/43, UC Davis, CA). Rabbit anti-Aβ polyclonal antibody was from Cell Signaling (Cat# 2454, Danvers, MA). Rabbit polyclonal antiserum against total tau was from Dako (code# A0024, Carpinteria, CA). The DNA marker, TO-PRO 3 was purchased from Invitrogen (Cat#T3605, Carlsbad, CA). For double-immunofluorescence, sections were incubated with a cocktail of the mouse anti-PSD-95 (1:400) and the rabbit anti-Aβ (1:1,000) or the rabbit anti-tau (1:1,000) in PBS buffer containing 0.01% Triton X-100 at 4°C overnight, following blocking non-specific binding with normal serum from the species that the secondary antibodies were raised in. As the mouse anti-PSD-95 antibody was used on mouse tissue, some sections were also pre-incubated with a mouse-over-mouse blocking solution (MOM kit, Vector Labs.). To control for specificity, the PSD-95 immunoreactivity was completely abolished following pre-absorption of the antibody with a full-length recombinant PSD-95 peptide (20 μg/ml). Additional controls included omissions of either primary antibody or secondary antibodies. Following the overnight incubation with primary antibodies, sections were subsequently labeled with secondary antibodies conjugated with Alexa Fluor 488 and Alexa Fluor 568 (Molecular Probe Inc., Eugene, OR), and counterstained with TO-PRO 3 (1:1,000). Sections were then mounted and coverslipped with the antifade medium Prolong Gold (Molecular Probe Inc., Eugene, OR).
For immunostaining of formalin-fixed, paraffin-embedded human brain tissue, 8-μm sections were cut from blocks of hippocampus with parahippocampal cortex and of frontal cortex. The sections were subject to deparaffinization, rehydration and antigen retrieval with microwave broiling for 5 min in citrate buffer pH 6.0. Sections were incubated with normal serum of the secondary antibody to block non-specific binding for 1 h and then incubated with primary antibodies overnight at 4°C. The double-immunofluorescence labeling in human brain sections was similar to that for mouse sections. In addition, a rabbit anti-PSD-95 antiserum (1: 500) (ab18258, Abcam) was paired with the mouse anti-actin IgG (1:500) (ab3280, Abcam) or the mouse anti-MAP2 IgG (1:400) (MAB3418, Chemicon). Following the secondary antibodies and TO-PRO 3 counterstain, the endogenous autofluorescence was blocked by Sudan black as previously described [34].
Image analysis for cell count and densitometry
Images were acquired using an Olympus FLUOVIEW FV1000 laser scanning confocal system, and processed and illustrated in Adobe Photoshop (version 5.5). Neuronal cell counts were performed in CA1 region as shown in the boxes in Figs. 1a and 2a. TO-PRO 3-labeled nuclei in 100 μm × 100 μm fields (upper panels in Figs. 1c, 2c) were counted in three consecutive fields in each section, and three separate sections from each animal were counted. Neuronal nuclei were recognized as uniformly large size (>5 μm) with complete nuclear membrane and at least one prominent nucleolus. The thickness (z-axis) of optical sections at the 40× objective was 0.64 μm, which effectively reduced overlapping.

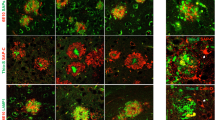
PSD-95 decreases in apical dendrites in 6-month-old, but not in 3-month-old, 5XFAD mice. a PSD-95 (green) and Aβ (red) immunoreactivity with TO-PRO 3 (blue) nuclear counterstain in hippocampus of wild type (WT), 3-month-old (3M) and 6-month-old (6M) 5XFAD mice. PSD-95 immunoreactivity highlights regions of apical dendrites in CA1, CA3 and DG (dentate gyrus). Boxes indicate the portion of CA1 region illustrated in b and c. Bar for all 500 μm. b High power images show PSD-95 and Aβ double labeling. PSD-95 immunoreactivity is prominent in apical dendrites and weak in soma in WT. Similar PSD-95 distribution is seen in 3M 5XFAD, despite the presence of Aβ plaques. In contrast, PSD-95 immunoreactivity is weak in apical dendrites and prominent in soma (arrow) in 6M 5XFAD. Bar for all 50 μm. c Images of 100 μm × 100 μm areas show TO-PRO 3 stained nuclei (upper panel) and PSD-95-labeled apical dendrites (lower panel) that were analyzed in d and e, respectively. Bar for all 50 μm. d Numbers of neuronal nuclei counted in CA1 pyramidal layer do not differ among the three groups: WT = 24.0 ± 0.4, 3M = 23.5 ± 0.5, 6M = 23.0 ± 0.6 (P > 0.05, ANOVA). e PSD-95 intensity measured in CA1 apical dendrites is reduced in 6M 5XFAD by 39.6 ± 1.3 and 41.8 ± 1.4% compared with WT and 3M 5XFAD mice, respectively (*P < 0.001, post hoc Tukey test)

PSD-95 decreases in apical dendrites in hippocampus in 14-month-old, but not in 3-month-old, JNPL3 mice. a PSD-95 (green) and tau (red) immunoreactivity with TO-PRO 3 (blue) nuclear counterstain in hippocampus of wild type (WT), 3-month-old (3M) and 14-month-old (14M) JNPL3 mice. Boxes indicate the portion of CA1 region illustrated in b and c. Bar for all 500 μm. b High power images show PSD-95 and tau double-labeling. In WT, PSD-95 immunoreactivity labels apical dendrites and no tau positive neurons are present. In 3M JNPL3, PSD-95 immunoreactivity also labels apical dendrites, with a number of tau positive neurons present. In 14M JNPL3, PSD-95 immunoreactivity disappears from apical dendrites and appears in a number of neuronal soma (arrow), some of which localize with tau (arrowhead). Bar for all 50 μm. c Images of 100 μm × 100 μm areas show TO-PRO 3 stained nuclei (upper panel) and PSD-95-labeled apical dendrites (lower panel) that were analyzed in d and e, respectively. Bar for all 50 μm. d Numbers of neuronal nuclei counted from CA1 pyramidal layer do not differ among the three groups: WT = 24.2 ± 0.7, 3M = 23.6 ± 0.3, 14M = 23.2 ± 0.2 (P > 0.05, ANOVA). e PSD-95 intensity measured from CA1 apical dendrites is significantly reduced in 14M by 57.7 ± 0.9 and 57.4 ± 5.1% compared to WT and 3M JNPL3 mice, respectively (*P < 0.001, post hoc Tukey test)
PSD-95 intensity was measured in 100× fields using the Image-Pro software (version 4.2). PSD-95 Immunofluorescence intensity was measured in 100 μm × 100 μm areas over CA1 apical dendrites (boxes in Figs. 1a, c, 2a, c). Three consecutive areas on each section and three separate sections from each animal were measured. Each reading was subsequently divided by reading over adjacent white matter to correct for background.
Statistical analysis
For statistical analyses, nine data points for cell count or densitometry from each animal were collected and averaged. Three animals in each group (n = 3) were compared with other groups, and the intra- and inter-group variants were tested using one-way ANOVA followed by post hoc Tukey test with a minimal criterion of p < 0.05 (SigmaStat 3.5). Data were presented as mean ± SEM of each group.
Results
PSD-95 distribution in 5XFAD mice
To ask if and when Aβ accumulation affects postsynaptic terminals as measured by PSD-95 immunoreactivity, we compared PSD-95 distribution in 3- and 6-month-old 5XFAD mice versus wild type controls. Consistent with the original characterization of 5XFAD mice that showed Aβ plaques can be detected as early as 2 months of age in deep cortical layers and subiculum [28], Aβ deposits were seen in these regions and in hippocampus of the 3-month-old mice (Fig. 1a, b). In wild type mice, PSD-95 immunoreactivity was primarily observed in neuropil with dense labeling in dendrites and weak labeling in neuronal soma, as previously reported [18]. PSD-95 exhibited a distinct distribution in hippocampal apical dendrites that are known for their excitatory synapses (WT in Fig. 1a, b). In 3-month-old 5XFAD mice, PSD-95 immunoreactivity did not differ from that of the wild type control despite the presence of Aβ plaques (3M in Fig. 1a, b). However, in 6-month-old 5XFAD mice, PSD-95 immunoreactivity was significantly reduced, especially in hippocampal CA1 region, while it increased in neuronal soma relative to 3-month-old 5XFAD and wild type mice (6M in Fig. 1a–c). The modest increases in hippocampal somatic PSD-95 labeling appeared homogenous and likely related to the reduction in apical dendrites rather than to Aβ plaques. Quantitatively, PSD-95 intensity in hippocampal CA1 apical dendrites decreased by around 40% compared to wild type controls and 3-month-old 5XFAD mice, respectively (P < 0.001; Fig. 1e). As the PSD-95 reduction from apical dendrites would be a result of neuronal loss, we counted neuronal nuclei in CA1 pyramidal layer above the apical dendrites (boxes in Fig. 1a). As shown in the actual counted areas and the number of counts, no significant neuronal loss was detected (Fig. 1c, d). Thus, PSD-95 is quantitatively reduced from apical dendrites and qualitatively increased in neuronal soma in 6-month-old 5XFAD mice.
PSD-95 distribution in JNPL3 mice
To evaluate tau-related PSD-95 changes, we compared JNPL3 mice at 3 and 14 months of age with wild type controls. Tau immunoreactivity was minimally detectable in wild type controls, but markedly increased in 3- and 14-month-old JNPL3 mice (Fig. 2a). In hippocampal CA1 region, tau positive neurons were absent in wild type controls, but were frequently seen in 3- and 14-month-old JNPL3 mice (Fig. 2b). This somatodendritic tau immunoreactivity was described as ‘pre-tangles’ and can be detected as early as 2 months of age in the original study of the transgenic mice [23]. Similar to the 5XFAD mice, PSD-95 distribution in hippocampal apical dendrites was preserved in 3-month-old mice despite the presence of neuronal tau pathology, whereas PSD-95 immunoreactivity was extensively diminished and some neuronal soma exhibited increased PSD-95 immunoreactivity in 14-month-old JNPL3 mice (Fig. 2a, b). Quantitatively, PSD-95 intensity in hippocampal CA1 apical dendrites decreased by more than 55% in 14-month-old JNPL3 compared to wild type controls and 3-month-old JNPL3 mice (P < 0.001; Fig. 2e). Again, the marked reduction of PSD-95 might result from neuronal loss in this transgenic species. However, counting of the TO-PRO 3-labeled neuronal nuclei in the defined area indicated no detectable neuronal loss (Fig. 2c, d). Thus, PSD-95 decreased from apical dendrites and increased in soma of CA1 neurons in 14- but not 3-month-old JNPL3 mice.
PSD-95 association with AD pathology
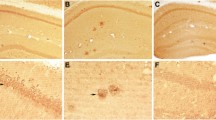
As both transgenic models of amyloidopathy and tauopathy showed reduction of PSD-95 in dendrites and accumulation in soma in hippocampal CA1 region, we sought to determine whether similar changes occur in AD. Similar to the wild type and 3-month-old transgenic mice, PSD-95 immunoreactivity exhibited a neuropil pattern in control human hippocampal CA1 regions (Fig. 3a). However, the intensity of PSD-95 immunoreactivity varied greatly among control and AD brains, which made its quantitation unreliable (see “Discussion”). Alternatively, we focused on qualitative changes that consistently occurred in association with AD pathology. In AD hippocampus, the PSD-95 antibody-labeled rod- or spindle-shaped structures that coexist but are disparate from tau positive neurofibrillary tangles (NFT) (Fig. 3b). These structures were further identified as Hirano bodies by their morphology and colocalization with MAP2 and actin [14, 17, 30] (Fig. 3c–f). The PSD-95 positive Hirano bodies were frequently seen in sections from AD cases, where Hirano bodies were readily identifiable. The sections from the control cases showed no identifiable Hirano bodies, so whether PSD-95 also present in Hirano bodies in normal aging brains was not determined. Interestingly, not all PSD-95 positive Hirano bodies were double-labeled with actin or MAP2, and vise versa. It is not clear whether PSD-95 accumulates in a subset of Hirano bodies or Hirano bodies lose some antigenicity over time. In summary, our data demonstrated that PSD-95 accumulated in Hirano bodies in AD brains.

PSD-95 immunoreactivity in Hirano bodies in AD brains. a PSD-95 (green) and tau (red) immunoreactivity with TO-PRO 3 counterstain (blue) in CA1 region of a control brain. PSD-95 immunoreactivity labels neuropil, while no tau positivity is detected. b CA1 region of an AD brain shows tau positive neurofibrillary tangles (NFT, arrowhead) and dystrophic neurons (DN) and PSD-95 positive spindle-like Hirano bodies (HB, arrows). c H&E stain shows an eosinophilic HB (arrow) in association with a neuron. d A HB (arrow) is labeled with PSD-95 antibody with immunoperoxidase-DAB reaction. e A HB (arrow) is double-labeled with antibodies to PSD-95 and MAP2, a somatodendritic marker. f A HB (arrow) is double-labeled with antibodies to PSD-95 and actin filament. Bar in b for a and b 50 μm. Bar in f for c–f 20 μm
Discussion
Using two lines of transgenic mouse models, we have shown that PSD-95, a major scaffold protein in excitatory synapses, is reduced in hippocampal apical dendrites, and accumulates in neuronal cell bodies, presumably in concert with synaptic damage and loss. Importantly, this magnitude of postsynaptic loss does not precede obvious Aβ and tau deposition but instead appears to occur as Aβ and tau pathologies advance. Thus, PSD-95 may serve as an intrinsic biomarker for progression of synaptic degeneration in both models.
PSD-95, a postsynaptic protein, has been shown to decline in 5XFAD mice, in association with progression of memory deficits [28]. Our finding of the age-related decrease in PSD-95 immunoreactivity is consistent with these observations. The prior study [28] also detected neuronal loss in deep cortical layers and subiculum, raising the possibility that the PSD-95 reduction may be a result of neuronal death. However, we detected no neuronal loss in the CA1 region of the transgenic mice we examined. Therefore, we believe that the loss of PSD-95 from CA1 apical dendrites most likely reflects postsynaptic degeneration. Postsynaptic damage has been demonstrated in neuronal cultures from transgenic and wild type mice, in which PSD-95 and dendritic spines appeared to be the primary targets for Aβ oligomers [1, 20]. Although, to our knowledge, PSD-95 has not been directly examined in JNPL3 mice, evidence for synaptic destruction in this model has been well documented in association with tau aggregation [5, 32]. Here, we have shown that, deposition of human Aβ in the 5XFAD mice or tau in the JNPL3 mice independently leads to deterioration of postsynaptic terminals as measured by PSD-95 immunoreactivity in dendrites.
The significance of such postsynaptic damages can be postulated in terms of memory deficit, at least, in 5XFAD mice. It has been shown that although Aβ burden was significantly increased in 1.5- 3-month-old mice, the loss of spatial memory only occurred at 4–5 months but not at 2 months of age [28]. Further electrophysiological and behavioral studies demonstrated impairment in long-term memory consolidation in 6-month-old 5XFAD mice, but not in mice younger than 4 months of age [19, 29]. In addition, there appeared to be no further cognitive deterioration from 6 to 15 months of age, compared with age-matched controls [29]. As these behavioral studies have pinpointed the deterioration between 3 and 6 months, the biomarker indicating structural changes is still lacking. Here, we show that the altered pattern of PSD-95 immunoreactivity may underline the memory deficits.
Changes of PSD-95 in AD have been reported but some discrepancies exist. A study measuring total protein against recombinant truncated protein standards found a significant decrease in AD temporal cortex but not in less affected occipital cortex [31]. A decrease in PSD-95 protein has also been reported in synaptosomes prepared from cerebral cortex [15]. In contrast, studies using immunohistochemistry showed an increase in the postsynaptic marker PSD-95 with a decrease in the presynaptic marker synaptophysin in the cerebral cortex and hippocampus in AD brains [21, 22]. The discrepancy among these studies remains to be clarified. Studies have shown that, although PSD-95 can remain stable for 72 h postmortem, the variability among individuals at a given time point is large and that ApoE genotype may influence the individual variability of PSD-95 [24, 35]. From the present study, factors such as disease duration and severity may also influence PSD-95; however, unlike transgenic mice, these factors are difficult to control in human cases. The high individual variability among our AD and control cases prevented meaningful quantitation of PSD-95 immunostain. Instead, we focused on qualitative changes that consistently occurred in association with AD pathology. We found that PSD-95 immunoreactivity was detectable in Hirano bodies in hippocampal CA1 region. Hirano bodies are well known for their location in and proximal to pyramidal neurons and contain dendritic elements such as actin and MAP2 [2, 13, 14, 30]. The presence of postsynaptic proteins such as PSD-95, together with AMPA receptors and atypical PKCs [2, 7, 34], further suggests that Hirano bodies may represent aberrant formation of postsynaptic structures in response to distal synaptic destruction.
Overall, our findings indicate that PSD-95 loss from apical dendrites and its accumulation in neurons and Hirano bodies, as measured by immunoreactivity, may serve as a marker for severe postsynaptic damage that correlates with behavioral deficit. It is conceivable that early deficits prior to the loss of PSD-95 may primarily involve transmitter/receptor alterations that can be corrected by enhancing transmitter/receptor efficacy; while after significant loss of PSD-95 growth factors may be required to restore neuronal connections. In the final stage of significant cell loss, stem cell transplant may become necessary. It will be interesting to know if the changes of PSD-95 can be prevented or even restored with various therapeutic remedies.
Abbreviations
- PSD-95:
-
Postsynaptic density 95
- AD:
-
Alzheimer’s disease
- 5XFAD:
-
Five familial AD mutations
- JNPL3:
-
Founder line of P301L mice that express human tau encoding the frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17)
- CA1:
-
Cornu Ammonis area 1 of hippocampus
References
Almeida CG, Tampellini D, Takahashi RH et al (2005) Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20:187–198
Aronica E, Dickson DW, Kress Y, Morrison JH, Zukin RS (1998) Non-plaque dystrophic dendrites in Alzheimer hippocampus: a new pathological structure revealed by glutamate receptor immunocytochemistry. Neuroscience 82:979–991
Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM (2007) Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 27:9115–9129
Blanpied TA, Kerr JM, Ehlers MD (2008) Structural plasticity with preserved topology in the postsynaptic protein network. Proc Natl Acad Sci USA 105:12587–12592
Boekhoorn K, Terwel D, Biemans B et al (2006) Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci 26:3514–3523
Chen X, Winters C, Azzam R et al (2008) Organization of the core structure of the postsynaptic density. Proc Natl Acad Sci USA 105:4453–4458
Crary JF, Shao CY, Mirra SS, Hernandez AI, Sacktor TC (2006) Atypical protein kinase C in neurodegenerative disease I: PKMzeta aggregates with limbic neurofibrillary tangles and AMPA receptors in Alzheimer disease. J Neuropathol Exp Neurol 65:319–326
Davies CA, Mann DM, Sumpter PQ, Yates PO (1987) A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci 78:151–164
DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457–464
Dickson DW, Crystal HA, Bevona C et al (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 16:285–298 (discussion 98–304)
El-Husseini Ael D, Schnell E, Dakoji S et al (2002) Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108:849–863
Fischer P, Jungwirth S, Zehetmayer S et al (2007) Conversion from subtypes of mild cognitive impairment to Alzheimer dementia. Neurology 68:288–291
Galloway PG, Perry G, Gambetti P (1987) Hirano body filaments contain actin and actin-associated proteins. J Neuropathol Exp Neurol 46:185–199
Goldman JE (1983) The association of actin with Hirano bodies. J Neuropathol Exp Neurol 42:146–152
Gylys KH, Fein JA, Yang F et al (2004) Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol 165:1809–1817
Hirano A (1994) Hirano bodies and related neuronal inclusions. Neuropathol Appl Neurobiol 20:3–11
Hirano A, Dembitzer HM, Kurland LT, Zimmerman HM (1968) The fine structure of some intraganglionic alterations. Neurofibrillary tangles, granulovacuolar bodies and “rod-like” structures as seen in Guam amyotrophic lateral sclerosis and parkinsonism-dementia complex. J Neuropathol Exp Neurol 27:167–182
Hunt CA, Schenker LJ, Kennedy MB (1996) PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J Neurosci 16:1380–1388
Kimura R, Ohno M (2009) Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis 33:229–235
Lacor PN, Buniel MC, Chang L et al (2004) Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci 24:10191–10200
Leuba G, Savioz A, Vernay A et al (2008) Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimers Dis 15:139–151
Leuba G, Walzer C, Vernay A et al (2008) Postsynaptic density protein PSD-95 expression in Alzheimer’s disease and okadaic acid induced neuritic retraction. Neurobiol Dis 30:408–419
Lewis J, McGowan E, Rockwood J et al (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25:402–405
Love S, Siew LK, Dawbarn D et al (2006) Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging 27:797–803
Masliah E, Mallory M, Alford M et al (2001) Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 56:127–129
Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA (1991) Cortical and subcortical patterns of synaptophysin like immunoreactivity in Alzheimer’s disease. Am J Pathol 138:235–246
Mirra SS, Heyman A, McKeel D et al (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Oakley H, Cole SL, Logan S et al (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140
Ohno M (2009) Failures to reconsolidate memory in a mouse model of Alzheimer’s disease. Neurobiol Learn Mem 92:455–459
Peterson C, Kress Y, Vallee R, Goldman JE (1988) High molecular weight microtubule-associated proteins bind to actin lattices (Hirano bodies). Acta Neuropathol (Berl) 77:168–174
Proctor DT, Coulson EJ, Dodd PR (2010) Reduction in post-synaptic scaffolding PSD-95 and SAP-102 protein levels in the Alzheimer inferior temporal cortex is correlated with disease pathology. J Alzheimers Dis 21:795–811
Ramsden M, Kotilinek L, Forster C et al (2005) Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25:10637–10647
Selkoe DJ (1994) Alzheimer’s disease: a central role for amyloid. J Neuropathol Exp Neurol 53:438–447
Shao CY, Crary JF, Rao C, Sacktor TC, Mirra SS (2006) Atypical protein kinase C in neurodegenerative disease II: PKCiota/lambda in tauopathies and alpha-synucleinopathies. J Neuropathol Exp Neurol 65:327–335
Siew LK, Love S, Dawbarn D, Wilcock GK, Allen SJ (2004) Measurement of pre- and post-synaptic proteins in cerebral cortex: effects of post-mortem delay. J Neurosci Methods 139:153–159
Sjostrom PJ, Rancz EA, Roth A, Hausser M (2008) Dendritic excitability and synaptic plasticity. Physiol Rev 88:769–840
Sze CI, Troncoso JC, Kawas C et al (1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56:933–944
Terry RD, Masliah E, Salmon DP et al (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580
Acknowledgments
This work was supported by NIH Grants R01 AG020197 and R01 AG032611, and the Alzheimer’s Association to E.M.S., and by NIH Grants RO1 MH53576 and MH57068 to T.C.S.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shao, C.Y., Mirra, S.S., Sait, H.B.R. et al. Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Aβ and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol 122, 285–292 (2011). https://doi.org/10.1007/s00401-011-0843-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0843-x