
Abstract
Activation of the MAPK signaling pathway has been shown to be a unifying molecular feature in pilocytic astrocytoma (PA). Genetically, tandem duplications at chromosome 7q34 resulting in KIAA1549–BRAF fusion genes constitute the most common mechanism identified to date. To elucidate alternative mechanisms of aberrant MAPK activation in PA, we screened 125 primary tumors for RAF fusion genes and mutations in KRAS, NRAS, HRAS, PTPN11, BRAF and RAF1. Using microarray-based comparative genomic hybridization (aCGH), we identified in three cases an interstitial deletion of ~2.5 Mb as a novel recurrent mechanism forming BRAF gene fusions with FAM131B, a currently uncharacterized gene on chromosome 7q34. This deletion removes the BRAF N-terminal inhibitory domains, giving a constitutively active BRAF kinase. Functional characterization of the novel FAM131B–BRAF fusion demonstrated constitutive MEK phosphorylation potential and transforming activity in vitro. In addition, our study confirmed previously reported BRAF and RAF1 fusion variants in 72% (90/125) of PA. Mutations in BRAF (8/125), KRAS (2/125) and NF1 (4/125) and the rare RAF1 gene fusions (2/125) were mutually exclusive with BRAF rearrangements, with the exception of two cases in our series that concomitantly harbored more than one hit in the MAPK pathway. In summary, our findings further underline the fundamental role of RAF kinase fusion products as a tumor-specific marker and an ideally suited drug target for PA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pilocytic astrocytoma (PA) constitutes the second most common diagnosis in pediatric oncology after acute lymphoblastic leukemia, accounting for approximately 20% of pediatric brain tumors [16]. PA typically shows benign clinical behavior and, in contrast to World Health Organization (WHO) grade II astrocytoma, malignant progression is extraordinarily rare. Since radical resection remains the mainstay of therapy, the extent of surgical resection comprises the most important clinical determinant with regard to tumor recurrence and progression [29, 33]. However, complete resection may not be possible in up to 20% of cases, due to tumor localization in critical anatomic locations such as the brain stem or optic tract [8]. Another clinical observation in a large review of the SEER database was that infants with low-grade gliomas, particularly in their first year of life, have an inferior prognosis [30]. PA represents a heterogeneous morphologic spectrum ranging from pilocytic, bipolar cellular areas with Rosenthal fibers to less cellular protoplasmic areas with eosinophilic granular bodies and clear cells [25]. This varying morphological spectrum can make histopathological diagnosis extremely difficult [10]. Until recently, our knowledge about the molecular mechanisms contributing to PA development was restricted to a few consistent findings including the association of neurofibromatosis type 1 (NF1) with an increased incidence of low-grade gliomas, especially of the optic tract [21]. Widespread MAPK pathway activation in sporadic PA was first reported in 2005 [32], with activating mutation of KRAS being the first mechanism for aberrant MAPK activation to be identified in sporadic PA [12, 32]. The most prevalent point mutation reported to date, however, is a single base-pair substitution at codon 600 (V600E) of BRAF, leading to an amino acid exchange from valine to glutamate that is associated with constitutive kinase activation [27]. Alternatively, a 3-bp insertion in close proximity to the hot spot at position 600 confers the same phenotype [5, 15, 38]. Recent genome-wide DNA copy number studies enabled us and others to detect a tumor-specific copy number gain at 7q34 in the majority of PAs [5, 7, 9, 11, 13, 14, 18, 19, 27, 34]. Several of these studies show that this aberration results in a tandem duplication, leading to KIAA1549–BRAF fusion genes with constitutive kinase activity. Moreover, a small number of tumors were shown to harbor SRGAP3–RAF1 fusion genes, which arose through a tandem duplication at 3p25 [7, 15]. Notably, all fusion events reported to date included an in-frame RAF kinase domain, while lacking the N-terminal auto-inhibitory regions [7, 14, 34].
Since MAPK pathway downstream components (e.g., ERK) are phosphorylated in nearly all cases, growing evidence suggests that virtually all sporadic PAs show aberrant activation of MAPK signaling [32]. However, the exact molecular mechanism can so far only be deciphered for around 75% of cases. Thus, to define the spectrum of genetic alterations in PA more precisely, we focused on the identification and functional characterization of previously described and novel BRAF and RAF1 fusion genes.
Materials and methods
Tumor specimens—Heidelberg series
Pilocytic astrocytoma tissues in this series were collected at the Department of Neuropathology, Burdenko Neurosurgical Institute, Moscow, Russia or the Cnopf’sche Kinderklinik in Nürnberg, Germany. Informed consent was obtained for collection of specimens and scientific use. Diagnoses were made by histological assessment following the criteria of the WHO classification including a reference pathology evaluation [22]. Only histologically unambiguous cases of PA were enrolled in this study, whereas pilomyxoid astrocytomas and other pediatric low-grade gliomas were excluded. Clinical data are provided in Online Resource Table 1.
Tumor specimens—Cambridge series
These tissues were collected at the Karolinska Hospital, Stockholm and the Sahlgrenska University Hospital, Gothenberg, Sweden. This series included seven cases for which no prior MAPK alteration had been identified (cases PA1, PA3, PA15, PA18, PA27, PA60, PA65) [15], and from which only cDNA was available for the present study.
Tumor specimens—Montreal series
All samples were obtained with informed consent after approval of the institutional review board of the respective hospitals they were treated in and independently reviewed by senior pediatric neuropathologists, according to the WHO guidelines. All samples were taken at the time of the first surgery before further treatment, when needed. Tissues were obtained from the London/Ontario Tumor Bank, Ontario, Canada and from the Montreal Children’s Hospital, Montreal, Canada.
Nucleic acid extraction
Total RNA extraction was performed from snap-frozen tumor tissues using TRIZOL (Invitrogen, Carlsbad, CA, USA). RNA was analyzed on both NanoDrop (ND-1000, Thermo Scientific, Wilmington, DE, USA) and Agilent Bioanalyser (Agilent Technologies, Böblingen, Germany) for quality assessment. Only samples with an RNA integrity number (RIN) >5.0 and no evidence of ribosomal degradation were included in the study. Before use, RNA samples were treated with deoxyribonuclease at room temperature for 15 min. First strand cDNA was synthesized using oligo-dT primer or gene-specific primers. Genomic DNA was isolated using Qiagen DNA Blood and Tissue Midi Kit (Qiagen, Hilden, Germany).
Fusion detection in genomic DNA
For the mapping of breakpoints on the genomic level, long-distance inverse PCR (LDI-PCR) was performed as previously described [24]. DNA was digested with either SpeI or PciI according to the supplier’s specifications (New England Biolabs, Frankfurt, Germany). The ligation reaction was initiated by the addition of T4 DNA ligase to a concentration of 1 U and was carried out at 4°C overnight. The reaction was then terminated at 65°C for 10 min. The resulting DNA circles served as templates for LDI-PCR analyses using the Triple Master PCR System (VWR International, Darmstadt, Germany). PCR reactions were conducted according to the manufacturer’s recommendations. PCR amplimers were separated on 1% agarose gels. Non-germline DNA amplimers were gel-extracted and analyzed by direct sequencing. Primer sequences are available upon request.
Fusion detection in cDNA
To verify the cDNA transcripts derived from the fusion genes, we conducted RT-PCR using primers specific to exons of BRAF/RAF1 and the respective fusion partner gene in a conventional PCR. Primer sequences are available upon request.
FISH
Two-color interphase FISH was performed using custom-made probes: FITC (Fluorescein isothiocyanate)-labeled clone RP11-355D18 (Deutsches Resourcen-zentrum für Genomforschung) corresponding to KIAA1549 (green) and digoxigenin-labeled clone RP4-726N20 mapping to BRAF (red). Metaphase FISH was performed to verify the correct mapping of clones [20]. Pre-treatment of slides, hybridization, post-hybridization processing and signal detection were performed as reported elsewhere [28]. Samples showing sufficient FISH efficiency (>90% nuclei with signals) were evaluated by two independent investigators. Signals were scored in at least 100 non-overlapping, intact nuclei. Non-neoplastic specimens were used as a control. Chromosome gains at the 7q34 region were defined as >10% nuclei containing three or more signals for both probes. A KIAA1549–BRAF gene fusion was scored in cases showing fusion of one red signal and one green signal resulting in a yellow signal [18]. The analysis for SRGAP3–RAF1 was done in the same way, using probes RP11-334L22 and RP11-163D23 labeled with FITC and digoxigenin, respectively.
Array-based comparative genomic hybridization
Array-CGH was carried out as previously described [23, 35, 39]. Selection of genomic clones, isolation of BAC DNA, performance of degenerate oligonucleotide primed PCR, preparation of microarrays, labeling, hybridization and washing procedure, as well as data analysis, were performed as outlined elsewhere [23, 39].
Mutation analysis
We investigated tumor samples for activating mutations of MAPK pathway intermediates using either denaturing high-performance liquid chromatography (DHPLC [12]) followed by direct DNA sequencing or by PCR amplification and sequencing. Specific intronic primer pairs were used for PCR amplification of exons 11 and 15 of BRAF, exon 2 and 3 of KRAS and NRAS, and exon 13 of PTPN11. Mutation screening of RAF1 exon 2 to exon 7 was done in 19 samples, which showed no other MAPK alteration. Primer sequences are available upon request.
Western blot analysis and MEK phosphorylation
Plat-E cells were transfected with the retroviral pMIG expression vector encoding N-terminally HA-tagged human BRAFWT, FAM131B-BRAF, BRAF V600E or BRAFinsT. At 48 h following transfection, cells were lysed and analyzed by Western blotting as described previously [1]. Polyclonal rabbit antibody raised against the C-terminus of BRAF (C-19; Santa Cruz Biotechnology, Heidelberg, Germany) and anti HA-tag 3F10 (Roche, Mannheim, Germany), as well as ERK and MEK (all from Cell Signaling Technologies, Frankfurt, Germany), were used in the dilution recommended by the manufacturer. Quantification of MEK phosphorylation in total cellular lysates expressing the indicated BRAF proteins was assessed by quantifying chemiluminescence signals using a Fuji LAS 4000 imager and Multi-Gauge software.
Transduction of NIH 3T3 cells
NIH 3T3 cells were infected with bi-cistronic pMIG retroviral vectors encoding BRAFWT, FAM131B-BRAF, BRAFV600E and the BRAFinsT protein, as well as the green fluorescent protein (GFP) as an infection marker. Cells were fed every 2 days and grown to confluence. Phase contrast and fluorescence micrographs were taken at days 13 and 20.
Statistical analysis
The median duration of follow-up was calculated according to Korn [17]. Estimation of survival time distribution was performed by the method of Kaplan and Meier. The prognostic value of clinical and molecular factors was assessed by their estimated hazard ratios including 95% confidence intervals. The proportional hazards regression model by Cox was applied to examine the association of single and multiple markers with the hazard of disease progression. The result of a test was judged as statistically significant when the corresponding two-sided p value was smaller than 5%. All statistical computations were performed with the statistical software environment R, version 2.11.0 [31].
Results
Identification and characterization of RAF fusion genes
We screened a total of 125 PA samples for all known KIAA1549–BRAF and SRGAP3–RAF1 fusion variants, as well as novel fusion partners of BRAF (Online Resource Table 1). Sequencing confirmed previously published breakpoints within KIAA1549 exon 16, BRAF exon 9 in 42 patient samples; KIAA1549 exon 15, BRAF exon 9 in 24; and KIAA1549 exon 16, BRAF exon 11 in 11 samples. In ten cases, we could only determine 7q34 duplication indicating KIAA1549–BRAF fusion, but localization of the exact breakpoint was not possible (Online Resource Table 1). Overall, we identified gene fusions targeting RAF kinases in 72% (90/125) of PA. Detailed analysis of genomic DNA (available only for a subset of the Heidelberg series) mapped 96% (52/54) of the breakpoints to the same breakpoint cluster region in intron 8 of the BRAF gene (Figs. 1a, b and 2a).

Distribution and clustering of (a) 52 breakpoints in the BRAF gene and (b) 51 breakpoints in KIAA1549

a Graphic illustration of the breakpoint region within the BRAF gene demonstrating that all breakpoints are located N-terminally of the BRAF kinase domain, leaving the catalytic domain intact. Schematic representation of bKIAA1549–BRAF, cFAM131B–BRAF and dSRGAP3–RAF1 fusion genes in pilocytic astrocytomas. e Schematic illustration of the FAM131B–BRAF fusion gene in 2A23 depicting the junction created by the fusion partners. Exon 1 and parts of exon 2 correspond to the 5′ UTR as indicated by the shorter bars
We determined the first non-intronic breakpoint variant in a single case, located between KIAA1549 intron 15 and BRAF exon 8. On the transcript level, we observed that exon 8 was skipped and the fusion was formed between KIAA1549 exon 15 and BRAF exon 9. In a single case, we identified a fusion gene in KIAA1549 intron 16 and BRAF intron 8, which additionally displayed an internal inversion of 1,623 bp in the remaining BRAF fragment of intron 8. In another case, we identified an insertion of a 550-bp fragment of the DENN/MADD domain containing 2A gene (DENND2A) within the breakpoint between KIAA1549 intron 15 and BRAF intron 8. At the genomic level, therefore, KIAA1549–BRAF rearrangements are more diverse and complex than previously identified.
A further interesting observation in terms of the mechanism behind these rearrangements was that a majority of cases showed inserts of ‘filler-DNA’ or mini-direct repeats (MDR) directly at the fusion sites. Both filler-DNA and MDRs are hallmarks for non-homologous end-joining (NHEJ) DNA repair processes [37].
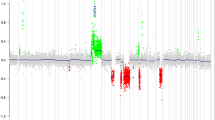
Most importantly, however, LDI-PCR analysis revealed a novel BRAF fusion partner other than KIAA1549. The novel fusion genes are composed of the 5′ part of the FAM131B gene and the 3′ part of the BRAF gene (Fig. 2b, c). Sequence analysis mapped the breakpoint of the first case (2A23) to the exon 2 of the FAM131B gene and exon 9 of BRAF (Online Resource Fig. 1a). The full-length FAM131B–BRAF transcript for this first case (2A23) consists of 1,223 bp. The full sequence is provided in Online Resource Table 2. Moreover, in a case which we received for consultation (BT-005), we detected a second FAM131B–BRAF fusion gene. Further investigation using LDI-PCR located the breakpoint to intron 1 of FAM131B and intron 9 of BRAF, forming an even smaller fusion gene of 1,152 bp. This led us to additionally screen cDNA from further tumors (Cambridge series) for FAM131B–BRAF fusion. We identified a third case (PA60, 21-year-old male with a cerebellar tumor) with two splice variants of a similar fusion gene, with a junction between FAM131B exon 2 or exon 3 and BRAF exon 9, respectively (Online Resource, Fig. 1b–d). These cases were histologically indistinguishable from cases with KIAA1549–BRAF fusion or other alterations. The genomic order of BRAF and FAM131B suggested interstitial deletion rather than tandem duplication as the most likely mechanism to form fusion genes. This hypothesis was confirmed by aCGH analysis of samples 2A23 and BT-005, demonstrating a deleted region encompassing ~2.5 Mb (Fig. 3a, b). We conducted further functional characterization with the fusion transcript from 2A23, since this was also the more prevalent variant in PA60. In total, we identified three novel FAM131B–BRAF fusion variants in three cases, suggesting that further fusion variants may also occur.

Array-CGH trace of sample BT-005 (a) and 2A23 (b) showing a deletion at 7q34 leading to a fusion between FAM131B and BRAF
Investigating tumors without any BRAF alteration for SRGAP3–RAF1 fusions, we identified two novel fusion variants, bringing the total identified to date to four. The first shows breakpoints in intron 11 of SRGAP3 and intron 6 of RAF1, including a one base pair deletion and a cryptic fragment of 12 bp of intron 11. The second novel fusion gene is formed between intron 10 of SRGAP3 and intron 8 of RAF1. Although the mode of fusion and the exact breakpoints vary within the respective introns of BRAF and RAF1, all of the fusion genes lose the RAF N-terminal auto-inhibitory regions while retaining an intact C-terminal kinase domain (Fig. 2a).
Altogether, we identified BRAF and RAF1 fusion genes in 82% (62/76) of cerebellar tumors, whereas in non-cerebellar tumors, fusions were significantly less frequent at 57% (28/49; p < 0.005, two-tailed Fisher’s exact test; Online Resource Table 1). The two SRGAP3–RAF1 fusion genes were also found in cerebellar tumors. This is in line with previous reports on RAF1 fusion genes, which were also detected in cerebellar tumors. Notably, PFS was not associated with the presence of fusion genes (p = 0.47, log-rank test).
Confirmation of fusion genes by FISH
In tumors for which formalin-fixed and paraffin-embedded (FFPE) tissues were available (61/125), breakpoints identified by PCR-based methods (n = 42) were validated by two-color FISH with probes corresponding to the BRAF and KIAA1549 loci or SRGAP3 and RAF1, respectively (Online Resource Table 1). Strikingly, carrying out analyses in a blinded manner, the fusion was confirmed in 39/42 cases (one of the negative cases being the FAM131B–BRAF fusion) indicating a high sensitivity of 92.9% for this FISH assay. Only one false positive case was identified, giving a specificity of 94.4%. These figures clearly indicate the potential clinical utility of this screening method as a diagnostic tool.
Mutational analysis of MAPK pathway components
Screening the samples for activating mutations of MAPK pathway intermediates, we found six BRAF V600E mutations, two cases with a BRAF ins598T insertion and two tumors harboring a KRAS mutation (Online Resource Table 1). No mutations were seen in HRAS, NRAS or PTPN11. Most mutations were detected in a mutually exclusive fashion to RAF gene fusions, suggesting that BRAF and KRAS mutations as well as loss of NF1 (clinically diagnosed) may constitute alternative mechanisms of MAPK activation in this tumor. However, we found that mutations and fusion genes can occasionally occur together, with two cases in our series harboring concomitant BRAF mutation and BRAF fusion. One of the cases harbored a BRAF V600E mutation together with a KIAA1549–BRAF fusion. This 7-year-old patient after complete resection of her cerebellar tumor has been tumor free for 43 months without requiring adjuvant therapy. The other patient (3 years old at diagnosis) in addition to the same two hits was also diagnosed with NF1. Her diencephalic tumor could not be completely resected and she experienced tumor progression after an observation time of 36 months. Our study thus demonstrates the co-existence of mutations and fusions, indicating that more than one alteration of MAPK pathway components can arise in the same tumor.
Notably, we observed no mutations in RAF1 in a selected cohort of tumors without detectable BRAF alterations (n = 19). This is likely due to differences in the charged residues, meaning RAF1 has a lower basal kinase activity than BRAF and, therefore, two synergistic mutations would be needed to induce elevated kinase activity [6]. In contrast to the high frequency of BRAF and RAF1 fusions in cerebellar tumors, activating mutations of either BRAF or KRAS were predominantly found in non-cerebellar tumors (10/49; 20%), whereas analysis of cerebellar tumors revealed mutations in only 4% (3/76) of cases (p = 0.005, two-tailed Fisher’s exact test; Online Resource Table 1). As with BRAF and RAF1 fusion events, PFS was not significantly associated with the presence of an activating mutation in either BRAF or KRAS (p = 0.11, log-rank test).
Functional characterization of the novel FAM131B–BRAF fusion protein
To elucidate the activating potential of the novel FAM131B–BRAF fusion gene, we compared its protein product with the BRAF V600E and BRAF insT mutants by Western blot analysis. Using an antibody raised against the HA-tag of our constructs, we were able to analyze and compare the expression levels of the BRAF proteins. As expected, BRAFWT, BRAFV600E and BRAFinsT are detected as 95 kDa proteins. In contrast, due to the absent N-terminal regions, FAM131B–BRAF migrates with the theoretically predicted mass of 47 kDa in SDS-PAGE. As shown in Fig. 4a, all of the altered BRAF variants show higher levels of MEK and ERK phosphorylation, indicating activation of downstream effectors of the MAPK pathway due to these BRAF alterations. Interestingly, as shown in Fig. 4b, the cellular MEK phosphorylation elicited by the FAM131B–BRAF fusion protein was significantly lower than that of BRAFV600E (p = 0.006; ANOVA single factor analysis) and at the same time significantly elevated compared to BRAFWT (p = 0.0004).

FAM131B-BRAF migrates as a 47 kDa protein and represents a potent activator of the MEK/ERK pathway. Plat-E cells were transfected with expression vectors encoding N-terminally HA-tagged human BRAF wild type (BRAF wt; WT), FAM131B–BRAF (FAM), BRAF V600E or BRAF insT. a At 48 h following transfection, cells were lysed and analyzed by Western blotting using the indicated antibodies. The correct expression of FAM131B–BRAF was additionally confirmed by probing the membrane using an antibody raised against the C-terminus of BRAF, which also recognizes endogenous BRAF as indicated by the faint band present in the lysate generated from empty vector transfected cells. b Quantification of MEK phosphorylation in total cellular lysates expressing the indicated BRAF proteins. The signal elicited by the internal reference (BRAFwt) was set in each analysis to 1. Data represent the arithmetic mean from four independent transfections, and error bars indicate standard deviation from the mean
FAM131B–BRAF induces the transformation of NIH 3T3 cells
To functionally assess the transforming potential of the novel fusion gene FAM131B–BRAF, NIH 3T3 cells were retrovirally transduced with either empty pMIG vector or pMIG vectors containing either the BRAF WT, FAM131B–BRAF, BRAF V600E or BRAF insT cDNAs. The bi-cistronic pMIG vector system was chosen as it also encodes a GFP marker allowing the tracking of infected cells [1]. As shown in Fig. 5, GFP-positive cells expressing FAM131B–BRAF or the two oncogenic full-length BRAF mutants display a refractile morphology and loss of contact inhibition, clearly demonstrating transforming activity.

NIH 3T3 cells were infected with retroviral vectors encoding the indicated BRAF proteins and GFP as an infection marker. The medium was replenished every 2 days, and cells were grown to confluency and photographed at days 13 and 20. Note that GFP-positive cells expressing BRAFwt display a normal morphology and are well integrated into the monolayer. In contrast, cells expressing FAM131B-BRAF, BRAFV600E and BRAFinsT display a refractile morphology, criss-cross growth and have overridden contact inhibition
Analysis of clinical data
Median follow-up for the series was 54 months, and the 5-year PFS rate was 71%. Unlike for all tested molecular variables, univariable proportional hazards regression analysis revealed a significant association of progression-free survival (PFS) and radical tumor resection (HR = 0.11, 95% CI 0.04–0.32, p < 0.001), with PFS being better after gross total resection as assessed by post-operative MRI (5-year PFS was 91% vs. 47% after total or subtotal resection, respectively). An increased risk of progression was also observed for patients aged 1 year or younger (HR = 4.46, 95% CI 1.53–13.0, p = 0.006, Table 1a, Online Resource Fig. 2), with a 5-year PFS in this group of 33%, compared with 72% in the older group.
Confirming previous results, multivariable Cox proportional hazards regression analysis showed that radical surgery was the most important clinical prognostic factor for PA (HR = 0.06, 95% CI 0.02–0.22, p < 0.001). Young age at diagnosis (≤1 year) was also independently associated with a worse prognosis (HR = 4.03, 95% CI 1.24–13.1, p = 0.02, Table 1b, Online Resource Fig. 2). Since we had only a single patient in our cohort who died during follow-up, we did not perform statistical analysis for overall survival.
Discussion
The tumorigenesis of pilocytic astrocytoma has been shown to center around aberrant activation of the MAPK pathway, which regulates a wide range of substrates from transcription factors to additional protein kinases that control cell proliferation, growth, differentiation and apoptosis [4, 36]. In particular, activating (BRAF, KRAS) or inactivating (NF1) point mutations and fusion genes involving members of this pathway have been shown to be the most common genetic lesions. Recent studies have identified BRAF and RAF1 fusion genes arising from tandem duplications in up to 80% of PAs [7, 14, 27, 34]. A total of five different fusion variants for KIAA1549–BRAF and two for SRGAP3–RAF1 have been identified to date [7, 14, 34].
The study presented here extends the spectrum of MAPK alterations in PAs, and reveals further evidence that RAF kinase fusion genes are a key oncogenic mechanism in constitutively activating MAPK signaling in this entity. Applying LDI-PCR as a robust and reliable way to detect novel fusion partners in genomic DNA, we identified a novel BRAF fusion partner, FAM131B, which is currently uncharacterized and has not previously been implicated in tumorigenesis. Moreover, aCGH data demonstrated that this novel fusion gene results from an interstitial deletion at 7q34, rather than a tandem duplication as seen with prior fusions. Despite several recent studies reporting RAF fusion genes, deletion has not previously been implicated as a mechanism for promoting these fusions. This finding provides support for the involvement of multiple mechanisms in the formation of fusion genes and proto-oncogene activation, and also suggests a likelihood for the presence of further fusion variants targeting RAF genes. The demonstration of alternative splice variants resulting in the formation of two distinct fusion gene isoforms is also believed to be a novel finding.
As with all previously reported RAF fusion genes, such as KIAA1549–BRAF and SRGAP3–RAF1 in PA, AKAP9–BRAF in thyroid cancer, FCHSD1–BRAF in melanocytic nevi and, more recently, SLC45A3–BRAF and ESRP1–RAF1 in prostate cancer and AGTRAP–BRAF in gastric cancer, the novel FAM131B–BRAF fusion products share a lack of the RAF auto-inhibitory domain [2, 3, 26]. We have shown here that removal of the N-terminal regulatory regions in FAM131B–BRAF results in constitutive kinase activity and that the fusion gene is able to transform NIH 3T3 cells. Strikingly, the novel fusion genes contain only a small number of exons of FAM131B, which comprise mostly the 5’ UTR. This strongly suggests that the role of the RAF fusion partners is restricted to providing an efficient promoter and a splice donor site, thereby facilitating constitutive activation of the respective RAF gene.
Analysis of junction sequences at the fusion sites provided a mechanistic insight into the formation of tandem duplications at 7q34 and 3p25, since filler-DNA and MDRs were found to be present in a majority of cases, indicating the participation of NHEJ in the promotion of fusion events. NHEJ is initiated in response to DNA double-strand break repair and is referred to as “non-homologous”, because the break ends can be directly ligated without the need for extensive sequence homology. Therefore, it is predicted to cause translocations with microhomology at the repair junction. Genetic translocations consistent with NHEJ have also been discussed in several hematological and other malignancies with known translocations (discussed in [37]).
The vast majority of MAPK aberrations in our series were fusions of RAF family oncogenes, removing the auto-inhibitory domain and rendering the kinase constitutively active. However, we also diagnosed neurofibromatosis type 1 and/or detected BRAF/KRAS mutations in 10% of patients with PA. Interestingly, fusions of RAF1 and BRAF in our series were highly prevalent in cerebellar tumors (62/76, 82%), whereas point mutations were found in only 4% (3/76) of tumors in this location. In contrast, fusion genes were much less frequent in non-cerebellar PAs (28/49, 57%), while mutations were detected at a higher frequency (10/49; 20%). These findings further substantiate the hypothesis of localization-specific genetic events activating MAPK signaling in PA [9]. The significance of these findings in terms of tumor origin and potential additional alterations warrants further investigation.
The clinical behavior of tumors in our cohort, in terms of PFS, strongly correlates with the degree of surgical resection, in line with previous reports [30, 33]. Patients with gross total resection showed an excellent PFS. Also, clinically interesting was the finding that patients aged 1 year or below showed a significantly inferior prognosis, which remained an independent marker in multivariate analysis. It would be of interest to investigate the interrelation of clinical behavior and the presence of specific genetic events in a larger cohort of these very young infants.
Taken together, our findings further stress the role of RAF kinase fusions as a central oncogenic mechanism in the development of PA and strengthen their potential role as both a tumor-specific marker for molecular diagnosis and an ideally suited molecular target for future therapeutic strategies. Since almost all genetic events detected in PA to date converge on MAPK pathway activation, established multikinase inhibitors such as sorafenib, which is already in the market, currently in phase I clinical trials in children and effective in vitro against BRAF fusion genes [26], may be good first candidates to treat these patients. However, drugs directly targeting the constitutively active RAF kinase domain may show even greater efficacy, compared with unselective MAPK inhibitors, and would certainly constitute a promising next step in drug development.
References
Brummer T, Martin P, Herzog S, Misawa Y, Daly RJ, Reth M (2006) Functional analysis of the regulatory requirements of B-Raf and the B-Raf(V600E) oncoprotein. Oncogene 25:6262–6276
Ciampi R, Knauf JA, Rabes HM, Fagin JA, Nikiforov YE (2005) BRAF kinase activation via chromosomal rearrangement in radiation-induced and sporadic thyroid cancer. Cell Cycle 4:547–548
Dessars B, De Raeve LE, Housni HE, Debouck CJ, Sidon PJ, Morandini R, Roseeuw D, Ghanem GE, Vassart G, Heimann P (2007) Chromosomal translocations as a mechanism of BRAF activation in two cases of large congenital melanocytic nevi. J Invest Dermatol 127:1468–1470
Dibb NJ, Dilworth SM, Mol CD (2004) Switching on kinases: oncogenic activation of BRAF and the PDGFR family. Nat Rev Cancer 4:718–727
Eisenhardt AE, Olbrich H, Röring M, Janzarik W, Van Anh TN, Cin H, Remke M, Witt H, Korshunov A, Pfister SM, Omran H, Brummer T (2010) Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int J Cancer [Epub ahead of print]
Emuss V, Garnett M, Mason C, Marais R (2005) Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res 65:9719–9726
Forshew T, Tatevossian R, Lawson A, Ma J, Neale G, Ogunkolade B, Jones T, Aarum J, Dalton J, Bailey S, Chaplin T, Carter R, Gajjar A, Broniscer A, Young B, Ellison D, Sheer D (2009) Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 218:172–181
Gajjar A, Sanford RA, Heideman R, Jenkins JJ, Walter A, Li Y, Langston JW, Muhlbauer M, Boyett JM, Kun LE (1997) Low-grade astrocytoma: a decade of experience at St. Jude Children’s Research Hospital. J Clin Oncol 15:2792–2799
Horbinski C, Hamilton R, Nikiforov Y, Pollack I (2010) Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol 119:641–649
Ichimura K, Ohgaki H, Kleihues P, Collins VP (2004) Molecular pathogenesis of astrocytic tumours. J Neurooncol 70:137–160
Jacob K, Albrecht S, Sollier C, Faury D, Sader E, Montpetit A, Serre D, Hauser P, Garami M, Bognar L, Hanzely Z, Montes JL, Atkinson J, Farmer JP, Bouffet E, Hawkins C, Tabori U, Jabado N (2009) Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br J Cancer 101:722–733
Janzarik W, Kratz C, Loges N, Olbrich H, Klein C, Schaefer T, Scheurlen W, Roggendorf W, Weiller C, Niemeyer C, Korinthenberg R, Pfister S, Omran H (2007) Further evidence for a somatic KRAS mutation in a low-grade astrocytoma. Neuropediatrics 38:1–3
Jones D, Ichimura K, Liu L, Pearson D, Plant K, Collins V (2006) Genomic analysis of pilocytic astrocytomas at 0.97 Mb resolution shows an increasing tendency toward chromosomal copy number change with age. J Neuropathol Exp Neurol 65:1049–1058
Jones DTW, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673–8677
Jones DTW, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP (2009) Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 28:2119–2123
Kaatsch P (2010) Epidemiology of childhood cancer. Cancer Treat Rev 36:277–285
Korn EL (1986) Censoring distributions as a measure of follow-up in survival analysis. Stat Med 5:255–260
Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, Pfister S, von Deimling A, Hartmann C (2009) Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 118:401–405
Lawson A, Tatevossian R, Phipps K, Picker S, Michalski A, Sheer D, Jacques T, Forshew T (2010) RAF gene fusions are specific to pilocytic astrocytoma in a broad paediatric brain tumour cohort. Acta Neuropathol 120:271–273
Lichter P, Cremer T, Borden J, Manuelidis L, Ward D (1988) Delineation of individual human chromosomes in metaphase and interphase cells by in situ suppression hybridization using recombinant DNA libraries. Hum Genet 80:224–234
Listernick R, Ferner R, Liu G, Gutmann D (2007) Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol 61:189–198
Louis D, Ohgaki H, Wiestler O, Cavenee W, Burger P, Jouvet A, Scheithauer B, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Mendrzyk F, Radlwimmer B, Joos S, Kokocinski F, Benner A, Stange DE, Neben K, Fiegler H, Carter NP, Reifenberger G, Korshunov A, Lichter P (2005) Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J Clin Oncol 23:8853–8862
Meyer C, Schneider B, Reichel M, Angermueller S, Strehl S, Schnittger S, Schoch C, Jansen MWJC, van Dongen JJ, Pieters R, Haas OA, Dingermann T, Klingebiel T, Marschalek R (2005) Diagnostic tool for the identification of MLL rearrangements including unknown partner genes. Proc Natl Acad Sci USA 102:449–454
Ohgaki H, Kleihues P (2005) Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 64:479–489
Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, Han B, Cao Q, Cao X, Suleman K, Kumar-Sinha C, Dhanasekaran SM, Chen Y-b, Esgueva R, Banerjee S, LaFargue CJ, Siddiqui J, Demichelis F, Moeller P, Bismar TA, Kuefer R, Fullen DR, Johnson TM, Greenson JK, Giordano TJ, Tan P, Tomlins SA, Varambally S, Rubin MA, Maher CA, Chinnaiyan AM (2010) Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 16:793–798
Pfister S, Janzarik W, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, Radlwimmer B, Kulozik A, Pietsch T, Herold-Mende C, Gnekow A, Reifenberger G, Korshunov A, Scheurlen W, Omran H, Lichter P (2008) BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118(5):1739–1749
Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, Wittmann A, Devens F, Gerber NU, Joos S, Kulozik A, Reifenberger G, Rutkowski S, Wiestler OD, Radlwimmer B, Scheurlen W, Lichter P, Korshunov A (2009) Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27:1627–1636
Pfister S, Witt O (2009) Pediatric gliomas. Recent Results Cancer Res 171:67–81
Qaddoumi I, Sultan I, Gajjar A (2009) Outcome and prognostic features in pediatric gliomas. Cancer 115:5761–5770
R Development Core Team (2010) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Sharma M, Zehnbauer B, Watson M, Gutmann D (2005) RAS pathway activation and an oncogenic RAS mutation in sporadic pilocytic astrocytoma. Neurology 65:1335–1336
Sievert A, Fisher M (2009) Pediatric low-grade gliomas. J Child Neurol 24:1397–1408
Sievert A, Jackson E, Gai X, Hakonarson H, Judkins A, Resnick A, Sutton L, Storm P, Shaikh T, Biegel J (2009) Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol 19:449–458
Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Dohner H, Cremer T, Lichter P (1997) Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 20:399–407
Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Project CG, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867
Weinstock DM, Elliott B, Jasin M (2006) A model of oncogenic rearrangements: differences between chromosomal translocation mechanisms and simple double-strand break repair. Blood 107:777–780
Yu J, Deshmukh H, Gutmann RJ, Emnett RJ, Rodriguez FJ, Watson MA, Nagarajan R, Gutmann DH (2009) Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology 73:1526–1531
Zielinski B, Gratias S, Toedt G, Mendrzyk F, Stange DE, Radlwimmer B, Lohmann DR, Lichter P (2005) Detection of chromosomal imbalances in retinoblastoma by matrix-based comparative genomic hybridization. Genes Chromosomes Cancer 43:294–301
Acknowledgments
Frauke Devens, Andrea Wittmann, Anna Schöttler, Stephanie Riester, Julia Hofmann and Danita M. Pearson are gratefully acknowledged for excellent technical assistance. This project was partially funded by the Sibylle Assmus Award 2009 to SP. TB was supported by the Deutsche Forschungsgemeinschaft via the Emmy-Noether Program and the Collaborative Research Centre 850 and RM by the grant 107819 from the Deutsche Krebshilfe.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2011_817_MOESM1_ESM.eps
Supplementary material 1 (Online Resource Fig. 1 Identification of FAM131B-BRAF fusion genes. Sequence trace of the breakpoints in (a) first case 2A23, (b) and (c) in the second case (PA60) of both FAM131B-BRAF splice variants and (d) in BT-005. (e) Amplification of the breakpoint of both splice variant fusion genes in sample PA60)
401_2011_817_MOESM2_ESM.eps
Supplementary material 2 (Online Resource Fig. 2 Kaplan–Meier survival estimates for progression-free survival (a) comparing patients after gross total tumor resection versus subtotal resection, and (b) patients 1 year of age or younger versus patients above 1 year of age)
Rights and permissions
About this article
Cite this article
Cin, H., Meyer, C., Herr, R. et al. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol 121, 763–774 (2011). https://doi.org/10.1007/s00401-011-0817-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0817-z