
Abstract
Polyglutamine (polyQ) diseases are associated with a CAG/polyQ expansion mutation in unrelated proteins. Upon elongation of the glutamine tract, disease proteins aggregate within cells, mainly in the central nervous system (CNS) and this aggregation process is associated with neurotoxicity. However, it remains unclear to what extent and how this aggregation causes neuronal dysfunction in the CNS. Aiming at preventing neuronal dysfunction, it will be crucial to determine the links between aggregation and cellular dysfunction, understand the folding pathway of polyQ proteins and discover the relative neurotoxicity of polyQ protein species formed along the aggregation pathway. Here, we review what is known about conformations of polyQ peptides and proteins in their monomeric state from experimental and modelling data, how conformational changes of polyQ proteins relate to their oligomerisation and morphology of aggregates and which cellular function are impaired by oligomers, in vitro and in vivo. We also summarise the key modulatory cellular mechanisms and co-factors, which could affect the folding pathway and kinetics of polyQ aggregation. Although many studies have investigated the relationship between polyQ aggregation and toxicity, these have mainly focussed on investigating changes in the formation of the classical hallmark of polyQ diseases, i.e. microscopically visible inclusion bodies. However, recent studies in which oligomeric species have been considered start to shed light on the identity of neurotoxic oligomeric species. Initial evidence suggests that conformational changes induced by polyQ expansions and their surrounding sequence lead to the formation of particular oligomeric intermediates that may differentially affect neurotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polylglutamine (polyQ) diseases are a group of human disorders associated with a triplet CAG expansion mutation and can be classified together with fragile X syndrome, fragile X tremor/ataxia syndrome, Friedreich’s ataxia and the polyalanine diseases (e.g. oculopharyngeal muscular dystrophy) into the group of the trinucleotide repeat expansion diseases [118]. The trinucleotide expansions in the genes associated with these disorders occur in the gene coding regions and are translated into a mature protein. Currently, there are 10 known polyQ disorders (Table 1). Despite widespread expression of the disease-associated proteins outside the central nervous system (CNS), all polyQ disorders are slowly progressive, fatal neurodegenerative diseases of the CNS causing a large spectrum of neurological and psychiatric symptoms (Table 1) with no cure available. Patterns of affected brain regions and CNS cell types (mainly neurons) vary according to the disease (Table 2). As in other chronic CNS diseases such as Alzheimer’s- and Parkinson’s disease (AD, PD), neuropathology is relatively specific to certain brain regions and CNS cell types and varies for each polyQ disease (for a recent review see [10], see Table 2). With the exception of the androgen receptor (AR) that is X-linked, the genes associated with polyQ diseases are inherited in an autosomal dominant manner, in contrast to the majority of AD and PD sporadic cases. There is an inverse correlation between the length of the polyQ stretch and the age of onset of disease and its severity. The polyQ stretch is a strong determinant of disease and, e.g. in Huntington’s Disease (HD) determines ca. 50% of the variation in age of onset of HD [180]. Disease occurs above a CAG/polyQ threshold of approximately 35–40 (that is variable to some extent, see Table 1).
The proteins involved in polyQ diseases appear unrelated, are found at different intracellular locations and some functions of these proteins are known or are beginning to emerge (Table 2). Spinal bulbar muscular atrophy (SBMA) is caused by a polyQ expansion in the AR, a nuclear hormone receptor that is responsive to G-protein coupled receptor signalling. The AR binds DNA and regulates target gene expression by modulating chromatin complexes and their regulatory proteins and functions in many different physiological processes including male reproductive functions [105]. The transcriptional activity of the AR is inversely correlated with the length of its CAG repeat. The actions of ataxin-1 (the polyQ-expanded protein associated with spinocerebellar ataxia 1, SCA-1) also occur in the nucleus where it is involved in RNA processing and transcriptional repression. Ataxin-1 has also been suggested to modulate protein folding itself via interactions with the chaperone and ubiquitin–proteasome pathways [36, 50, 166]. Ataxin-2 has been implicated in RNA metabolism, the regulation of apoptosis and actin-polymerisation [115, 144, 181]. Ataxin-3 appears to mediate degradation of ubiquitinated proteins [43] via its activity as a polyubiquitin chain-editing enzyme [23]. The gene encoding ataxin-6 produces a subunit of a voltage-dependent calcium channel [53]. Ataxin-7 is a member of the transcription mediator complex STAGA [67] and hence is involved in the regulation of transcription. The human TATA-binding protein (TBP) contains 25–42 glutamines and upon expansion is a disease protein causing SCA17 [98]. Atrophin-1 that is associated with Dentatorubral-Pallidoluysian atrophy (DRPLA) acts as a transcriptional co-repressor [188] as suggested by a study in Drosophila melanogaster. A further role of atrophin-1 could be in assembling signalling complexes at the synapse by interacting with membrane-associated guanylate kinases [184]. Finally, huntingtin (htt) is a nucleocytoplasmic shuttling protein and directly involved in regulating transcription, but is also found in the cytoplasm, in axons and at the synapse. As a scaffolding protein, htt mediates many protein–protein interactions and plays a role in vesicle interactions, fast axonal transport and likely signalling in the post-synaptic density [42, 57, 61]. Htt further appears to regulate calcium signalling and bioenergetic homeostasis [90].
It is possible that the polyQ expansion leads to a loss of function of some of the properties of polyQ proteins (reviewed in [25]) but there is a significantly larger amount of evidence that a gain-of-function due to the polyQ mutations causes CNS pathology [41, 155]. In particular, the observed protein aggregation, due to CAG mutations, is likely to at least partly explain the disease-associated neurotoxicity. Therefore, misfolding and aggregation as a toxic gain-of-function in these diseases is the subject critically reviewed in this article. There is a strong correlation between proteins in each disease exceeding a critical polyQ threshold and, as a consequence, abnormal intracellular accumulation of expanded polyQ proteins occurs. The aggregation of polyQ proteins has been intensively studied for over 10 years, following seminal observations that neurons from the first mouse model of HD and brains of patients affected by HD and Machado-Joseph Disease (MJD) contained intraneuronal inclusion bodies (IBs), hallmarks of all polyQ disorders [37, 41, 99, 120]. These observations led to the speculation that aggregation of the polyQ proteins may be causally related, at least in part, to CNS pathology. This has led to the emergence of a large body of literature on the elucidation of structural mechanisms leading to aggregation and whether/how polyQ aggregates cause cytotoxicity. In recent years, it has been increasingly suggested that oligomeric forms of polyQ proteins are key players in the neuropathology of polyQ diseases. Hence, here we review relevant insights that have been made in understanding the folding and aggregation of polyQ proteins, particularly focussing on the formation of oligomeric polyQ species and how oligomerisation of polyQ proteins may be linked to neurotoxicity.
Folding pathways and aggregation of polyQ proteins
Unfortunately, structural understanding of polyQ proteins has proved difficult for a number of reasons, including the inevitable handling difficulties of an aggregation-prone protein and the difficulty in synthesising a polyQ peptide (for a review see [163]) and therefore there is only limited information regarding structures of polyQ proteins in their monomeric and aggregated forms. The field has, however, been assisted by the exploration of similarities in protein assemblies observed in other neurodegenerative diseases such as AD and PD and these similarities have sometimes led to hypothetical common mechanisms for aggregation and cellular toxicity of such amyloidogenic proteins [51]).
Conformation of polyQ peptides
With the sharp mind of a genius Max Perutz, proposed in 1994 that polyQ peptides could form “polar zippers”, which would form antiparallel β hairpins if formed intramolecularly and would induce aggregation of the peptide by forming intermolecular antiparallel β-sheets between main chain and side chain amides [124]. The proposition that polyQ proteins may aggregate followed the discovery in 1993 that the gene causing HD contained long stretches of CAG repeats and proved correct when IBs in neurons of the first mouse model were described 3 years afterwards [37]. Later, characteristic cross-β reflections from X-ray diffraction data of a polyQ peptide, D2Q15K, led to his proposal that a novel structure termed the “water-filled nanotube”, composed of a continuous β-strand running around a water-filled cylinder 31 Å in diameter, is formed [123]. However, this interpretation proved controversial and reanalysis of data collected by Perutz et al. revealed a more conventional cross-β sheet structure in which hydrogen bonds form between inter backbone amides and inter side chain amides [153]. An independent X-ray diffraction study of polyQ peptides of various lengths also suggested formation of β-sheets in slab-like structures [39]. Helical conformations have also been suggested from molecular mechanics calculations [88, 108], for example, a polyQ peptide was proposed to form a μ-helix with a cylindrical pore of 6.6 Å in diameter via hydrogen bonds with the side chain amides [108]. Other studies have indicated that polyQ peptides in aqueous solution adopt a random coil conformation [4, 31]. Fluorescence correlation spectroscopy using a G-Qn-C-K2 peptide suggested that the polyQ forms ensembles of interconvertable collapsed structures in aqueous solutions [34]. A study using a KK-C-Qn-W-KK peptide used the quenching of triplet state fluorescence of tryptophan by cysteine to conclude that polyQ is intrinsically stiff and adopts rod-like conformations and a rigid nature of a polyQ stretch was also suggested from single molecule force spectroscopy measurements [44]. Molecular simulation studies on homopolymeric constructs of polyQ at physiologically relevant temperatures indicated that they form ensembles of collapsed globules [169]. The apparent discrepancy as to whether polyQ forms rigid structures or fluctuating collapsed structures was addressed in a recent study using fluorescence resonance energy transfer (FRET) [174]. This study showed that in constructs with pairs of C- and N-terminal lysines, the collapse is not evident until the polyQ length exceeds 16 residues. Once this occurs, these collapsed conformations correlated with the propensity of the peptide to aggregate. The study also suggested that polyQ peptides containing C- and N-terminal lysines were likely to be affected by end-to-end electrostatic repulsions.
In summary, conformational studies of the polyQ stretch can be heavily influenced by differing experimental conditions in which measurements have been taken including various solvents, different sequences and temperature. The only studies that have been performed on true homopolymeric polyQ have been molecular simulations, with all experimental investigations using charged residues at the N- and/or C-termini, which can potentially influence intrinsic polyQ conformations and interactions [183]. It has been suggested that the random coil conformation is only visible when the polyQ peptide is fully soluble with beta-sheet and beta-hairpin structures only observed when the peptide is at least partially aggregated [104]. Indeed recent modelling data suggest that high β-content of monomeric polyQ is thermodynamically unfavourable with the formation of intermolecular interfaces promoting the formation of β-sheet structures [168]. This is consistent with recent biophysical data from a K2Q23K2 peptide, which indicate that large soluble oligomers have no regular secondary structure [89].
This recent biophysical study [89] has also contributed significantly to our understanding of the kinetic pathway of polyQ peptide aggregation. Previous analysis of kinetic data led to the proposal of a classic nucleation-elongation mechanism with a significant lag time, during which the peptide is monomeric. The monomer would then shift from random coil to a thermodynamically unfavourable conformation inducing aggregation of the peptide [17, 31, 32]. However, using laser light scattering and size exclusion chromatography, Lee et al. [89] showed that during the so-called lag time, the polyQ peptide is not solely monomeric, but also consists of large soluble aggregates before further aggregation into insoluble precipitates.
All of the above studies of polyQ aggregation have been performed with polyQ peptides, partly due to the very large size of the polyQ disease-causing proteins (Table 1), but also justified by the fact that the only similarity between the disease-causing polyQ proteins appears to be their polyQ stretch. In the various polyQ diseases, the location of the polyQ stretch varies within the corresponding disease-causing protein, and both the protein context surrounding the polyQ stretch and the cellular location and function of the polyQ proteins are different (see “Introduction” and Table 1). However, it is now clear that the context of the polyQ stretch, posttranslational modifications of the protein, cleavage of the full-length protein, intracellular location and many other cellular factors are important determinants in the aggregation of the protein, as outlined in “Factors affecting aggregation of polyQ proteins” (see below).
Effects of protein context on conformation of the polyQ stretch
A variety of conformations of polyQ stretches have been reported when the polyQ stretch is embedded within a protein. Indeed simply adding an oligoproline stretch C-terminal to a polyQ peptide affects its aggregation kinetics and the conformation [16]. In the presence of the oligoproline stretch, the alpha-helical content, as measured by circular dichroism (CD), of the polyQ peptide, is abolished, whereas the N-terminal fusion of oligoproline had no effect on conformation of the polyQ peptide [16]. A polyQ stretch with glutathione-S-transferase (GST) fused to the N-terminus of the stretch showed a random coil conformation [103]. Alpha-helical conformations of polyQ stretches fused to thioredoxin and in ataxin-3, i.e., in a disease-causing protein context, have also been observed with CD [113]. The alpha-helical content of the polyQ stretch in ataxin-3 is significantly reduced when the polyQ length is increased from 27 residues to 78 residues, with a corresponding increase in random coil, highlighting the importance of polyQ length in the determination of structure [15]. However, an expanded polyQ stretch in myoglobin formed an antiparallel beta-pleated sheet structure [162]. In contrast to the studies with polyQ peptides mentioned above [89, 168], fourier transform infrared spectroscopic analysis showed an increase in β-structure upon the appearance of small oligomers of htt Exon 1 (httex1) [126], consistent with CD data showing early oligomers of ataxin-3 are β-sheet-rich [56], indicating a shift to β structure early on in the aggregation process. As shown by molecular modelling data [9], it is likely that the polyQ stretch destabilises proteins to form partially unfolded intermediates, prone to aggregation potentially via inter-glutamine interactions. A significant step forward in understanding the conformation of polyQ stretches has been achieved by the recent determination of the first crystal structure of a polyQ-containing protein in its native context, i.e. httex1 fused to the maltose-binding protein (MBP), although it should be noted that the MBP fusion may have effects on conformation [80]. The structure of httex1 consists of an amino-terminal α-helix that precedes the polyQ stretch, which is then followed by a polyproline helix that was modelled, due to inconclusive electron densities. The polyQ region (consisting of 17Q) was crystallised in multiple conformations, including α-helix, random coil and extended loop. Httex1 with a polyQ length of 17Q does not aggregate and therefore it is still unclear as to which changes occur in the conformation of the polyQ stretch to induce aggregation of the protein: it is unknown whether the β-strand conformations, known to exist in fibrils formed by mutant httex1, also exist in the monomeric state of mutant htt. However, it is quite possible that different conformations do exist and that each conformation may form a different type of aggregate.
To date, there are limited data available on the kinetics of aggregation in an in vivo context. A recent FRET analysis of polyQ proteins containing a polyQ expansion within cells has been used to support a model of parallel β-sheet or a head-to-tail cylindrical β-sheet formation during aggregation, as signals were only detected when donor and acceptor fluorescent proteins were attached to the same side of the molecule [159]. As this experiment was performed with a polyQ stretch in atrophin-1, this model needs to be tested in other polyQ systems given the importance of flanking residues as discussed above. Another study using a cellular model addressed the proposal based on in vitro studies that polyQ aggregates consist of alternating beta-strand and beta-turns [165]. Cell models expressing httex1 engineered within the polyQ stretch to assume a proposed β-strand/β-turn structure formed similar aggregates to cells expressing the normal sequence of httex1 polyQ, whereas addition of proline residues to presumably prevent this β-strand/β-turn structure abrogated aggregation of the protein [125].
In summary, the conformations adopted by the polyQ stretch within a protein remain controversial and appear to be dependent on many conditions including protein context, temperature and polyQ length. Indeed recent crystallographic evidence suggests that in a protein context the polyQ stretch, at least in monomeric proteins, can adopt multiple conformations [80]. Although the conformation of the polyQ stretch in a protein context may be variable, it is generally accepted that in large insoluble aggregates, proteins containing a polyQ stretch can form a variety of amyloid conformations [114, 125], although these conformations may be different to the well-characterised β-amyloid-like fibrils formed in AD [35]. How and when the conformational transition to beta-sheet (if any) occurs is also a matter for debate and may indeed vary according to the polyQ-containing protein involved.
Morphological characterisation of the intermediates along the polyQ-aggregation pathway
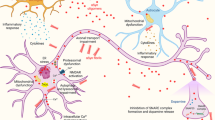
A variety of techniques have been used in order to investigate morphological, structural and kinetic features of the pathway of polyQ aggregation. It is clear that in vitro, peptides or proteins with a polyQ expansion form fibrillar aggregates with similar properties to those formed by amyloid β (Aβ) peptide. However, at least in vivo, investigations using the techniques mentioned above have shown that the pathway from soluble monomeric polyQ protein to the formation of fibrillar aggregates is a multistep process, outlined in Fig. 1 [164, 172]. As an additional step to the pathway in vivo, IBs, containing not only the polyQ protein, but a variety of other proteins and cellular components (like aggresomes), are formed. These IBs, far from being static end-stage structures, are also highly dynamic [26, 81, 157].

Model of aggregation of polyQ proteins based on several EM and AFM studies of oligomeric forms. a PolyQ proteins are likely to consist of several distinct monomeric conformations (as proposed by Muchowski and colleagues [172]), one or many of which may lead to the formation of the spherical, annular or amorphous oligomeric structures, as indicated. Some of these are “on-pathway” for the formation of fibrillar structures (which could also be formed directly from monomer), others of which are “off-pathway” for fibril formation, but may be “on-pathway” for inclusion body formation. Numbers in circles indicate where example AFM images of the structure are shown in b. b AFM analysis of httEx1Q53 aggregation reactions after cleavage from a GST-fusion protein. Scale bars 100 nm
Fibrillar polyQ protein aggregates have been well studied using EM and AFM (Fig. 1) and exhibit several defining features of amyloid, e.g. binding of thioflavin T, congo red birefringence, and they react with a “generic” anti-amyloid antibody [30, 147]. Fibrils are formed from short, sometimes curvilinear protofibrils [56, 126], tend to be approximately 10 nm in diameter and up to 2 μm in length with a beaded morphology [15, 46, 56], have a high β-content [126], and self-associate eventually forming large branched structures [35, 93] (also see Fig. 1).
A range of aggregation intermediates between monomeric and fibrillar polyQ have been identified. Elegant studies by the Muchowski lab using AFM to study the morphology of prefibrillar aggregates of httex1 and polyQ peptides, has identified a progression of aggregates from spherical structures with 5–65 nm diameter, rare annular structures composed of approximately 100 nm diameter (made of smaller annular and spherical structures), amorphous structures, protofibrils and fibrils [94, 172]. The intermediates formed were SDS-insoluble and could be altered by changes in concentration and incubation time, as well as by the addition of heat shock proteins (HSPs). The ability of HSPs (HSP40 and 70) to increase fibrillisation with a concomitant decrease in spherical structures led to the proposal that it is a particular conformation of monomer that leads to the formation of fibrils (which can be partitioned by HSPs), with spherical, annular and amorphous aggregates being “off-pathway” for fibril formation. The same study showed that the polyQ stretch is buried upon spherical oligomer and annular oligomer formation, whereas the stretch appears to be exposed in fibrils. Small oligomeric structures of approximately 5–20 nm diameter have also been detected by others using EM and AFM to monitor aggregation of httex1 constructs and ataxin-3 in vitro [46, 56, 68, 111, 126] (Fig. 1), and these oligomers have been shown to decorate fibrils formed by a GST-httex1 construct, indicating that these small oligomers are added to the ordered fibrillar structures to increase length or branching of the fibrils [68]. Annular structures of polyQ proteins, observed by AFM ([94, 172]; Fig. 1), similar to those reported to be formed by α-synuclein [87] have also been predicted by molecular modelling studies on polyQ peptides [101]. These annular oligomers have been suggested to be cytotoxic via the formation of ion-permeable pores in the membrane [87], further discussed below.
Visualisation of the morphology of polyQ oligomers formed in vivo is challenging, but has been achieved by tandem affinity purification of oligomers of httex1 with different affinity tags formed in cultured cells followed by detection by AFM and EM [111]. This revealed the formation of globular structures of 4–50 nm in diameter and the larger globules appeared to be formed from smaller structures. FCS has also been used to monitor oligomer formation in cultured cells, and shows a time-dependent increase in diffusion time and particle size [160]. This technique will be particularly useful in determining more precisely the effect of proposed polyQ-aggregation inhibitors, as demonstrated for Congo Red and QBP1 [160], which previously were usually characterised by their ability to prevent formation of SDS-insoluble material using a filter trap assay. Recently, the Seprion ligand has been used to isolate aggregated htt fragments from HD mouse models using an ELISA-based approach [143]. AFM and SDS-PAGE analysis of the aggregated protein showed that oligomeric, proto-fibrillar and fibrillar structures could be isolated and that these were similar in morphology and dimensions for two different HD mouse models and to species generated by recombinant httex1 protein. This provides useful evidence that in vitro similar aggregated structures can be produced to those found in vivo.
Factors affecting aggregation of polyQ proteins
Rather than discussing the many factors that affect polyQ aggregation in detail, which is beyond the scope of this review, we shortly summarise key factors and mechanisms that could have profound effects on the aggregation behaviour of polyQ proteins.
Postranslational modification and cleavage of polyQ proteins modulate polyQ aggregation
Recently, it has become clear that the polyQ expansion is not the only factor that determines the propensity of polyQ proteins to aggregate. Various posttranslational modifications may impact on the polyQ-aggregation process and its ensuing toxicity. For example, htt is both phosphorylated and SUMOylated at its first 17 N-terminal amino acids, which affect its aggregation properties [136, 156]. The most prevalent modification of httex1 is the acetylation and phosphorylation of threonine-3, and its phosphorylation alters httEx1 aggregation in cells and in vivo [2]. However, posttranslational modifications also determine whether htt and its cleavage fragments accumulate in the nucleus or not [136]. The nuclear space appears to be an aggregation-prone environment, perhaps due to the absence of autophagic activity (suggested to degrade misfolded polyQ species, reviewed in [138]) or more directly through interactions of polyQ proteins with DNA (see below). However, htt is also phosphorylated at S421 by Akt and SGK [71, 131] and at S1181 and 1201 by Cdk5 that significantly modulate its toxic properties. In neurons activation of the IGF-1/Akt pathway and phosphorylation of S421 reduces the number of IBs found in the nucleus [71]. Ataxin-3 phosphorylation at S256 by GSK3β has also been shown to modulate its aggregation [49]. Phosphorylation of Ataxin-1 at S776 by Akt appears critical to nuclear IB formation and S776A mutant protein does not form IBs in cultured cells [29, 47]. SUMOylation of the AR has also been shown to attenuate its aggregation [112]. It is currently unknown, how phosphorylation or indeed other modifications affect the folding/misfolding of polyQ proteins, as so far aggregation has either been recorded via IB counts within cells or measured biochemically by dot blot assays and hence effects on structural properties or oligomerisation remain to be determined.
Of course, posttranslational modifications also affect the degradation of polyQ proteins and hence their accumulation and aggregation indirectly, within a particular compartment. For example, acetylation of htt at K444 facilitates its trafficking to autophagosomes and increases its clearance via macroautophagy [77]. As many of these sites are located in the vicinity of caspase/other cleavage sites it is possible that phosphorylation also affects htt cleavage to a more aggregation-prone toxic fragment. Indeed Schilling and colleagues [150] found that mutation of S536, that is located in the proteolytic susceptibility domain of htt, to aspartic acid, inhibited calpain cleavage and reduced htt toxicity.
The cleavage of polyQ proteins such as enzymatic processing by caspases, calpains and unknown proteases is a common event during polyQ pathology, but likely also a normal physiological process for polyQ protein function [82]. Since the finding by Wellington and colleagues [179] that htt, ataxin-3, AR and atrophin-1 contain caspase cleavage sites, the in vitro and in vivo evidence for cleavage of polyQ proteins has steadily increased [173], but its role is uncertain. However, what has been clearly demonstrated is that cleavage of full-length polyQ proteins into smaller fragments modulates their intracellular localisation, aggregation and toxicity [62]. Such findings led Hayden and colleagues [63] to propose the “toxic fragment hypothesis” that has been confirmed by many cell- and animal studies. Therefore, it will be critical to explore how the cleavage and various posttranslational modifications of polyQ proteins directly influence their conformational and oligomeric states.
Effect of cellular factors and intracellular localisation on polyQ aggregation
It is well established that a change in intracellular localisation or cell type can have a significant effect on the aggregation state of polyQ proteins. For example, the AR forms large aggregates in the cytoplasm, but AR-transport into the nucleus prevented the formation of these aggregates [12]. On the other hand, nuclear targeting of an ataxin-3 fragment increased its aggregation [122]. Targeting httex1 to the endoplasmic reticulum, the nucleus or mitochondria was found to result in profound differences in the formation of SDS-insoluble material [137]. The effect of cellular localisation and cell type on polyQ aggregation is unsurprising given the demonstrated effects on polyQ aggregation of the cytoskeleton, e.g. [110], DNA, e.g. [119], lipids, e.g. [158], glucose, e.g. [133], metals, e.g. [52, 135] and osmolytes, e.g. [72], all of which are likely to vary between cellular compartments, in addition to cleavage enzymes and enzymes involved in posttranslational modification, as discussed above. Effects of macromolecular crowding in different cell compartments could also play an important role in affecting polyQ oligomerisation. Furthermore, key regulators of polyQ aggregation such as molecular chaperones (e.g. HSPs) also vary in their presence and level of expression in particular cell types and within specific compartments [130]. An effect on aggregation of htt in relation to the presence and state of prion protein has also been reported in yeast (Rnq1) and mammalian neuronal cell lines (PrPc) [91, 106]. Depletion of Rnq1 caused a reduction in polyQ aggregation in yeast and this effect was dependent on the prion conformation whereas in mammalian cells the depletion of PrPc increased and its overexpression decreased Httex1 aggregation.
It is currently unclear how the oligomerisation process of a polyQ protein is affected by the combination of such factors and future studies must show the relationships between these.
Flanking sequences and domains influence polyQ aggregation
As mentioned in “Effects of protein context on conformation of the polyQ stretch” sequences flanking the polyQ stretch significantly impact on aggregation and toxicity of polyQ proteins (for a review see [146]). A striking study in yeast showed that addition of particular sequences, e.g. a FLAG tag, flanking the polyQ region of htt could convert a non-toxic protein to a toxic species and modulate the morphology of aggregates formed [45]. The first 17 amino acids of htt directly preceding the polyQ stretch appear particularly influential, forming an amphipathic alpha-helical membrane-binding domain, disruption of which abolishes aggregation and increases toxicity, including the disruption of intracellular calcium homeostasis [7, 80, 136, 164]. In addition, the polyproline sequence following the polyQ stretch is highly influential in the aggregation and toxicity of htt [40]. Proline residues also follow the polyQ stretches in ataxin-1 and ataxin-7 and both proline and histidine residues within the polyQ stretch have been shown to mediate aggregation and toxicity of ataxin-1 [76, 151, 152]. The mutation of two residues significantly outside the polyQ stretch in the AR converted the wild-type receptor to an aggregation-prone toxic species, yet decreased the toxicity of the polyQ expanded, mutant receptor [55].
For at least some polyQ proteins (e.g. ataxin-1 and ataxin-3), regions outside the polyQ stretch are able to aggregate independently, e.g. [46], indicating that other regions can modulate and reinforce aggregation of the polyQ protein and indeed for ataxin-3, it has been reported that the formation of early ataxin-3 oligomers that can eventually lead to fibrillisation, is mediated by the Josephin domain [56]. The polyQ stretch may therefore act to destabilise surrounding domains, increasing their propensity to aggregate as has been implicated for htt [73].
What are the neurotoxic polyQ species?
The question of whether polyQ aggregation is itself neurotoxic is a highly debated issue. Many have argued that aggregation is toxic, whereas others have suggested that aggregation is simply a by-product of disease pathology and yet others imply that aggregates are protective, acting as a sink for otherwise toxic monomers. Although there have been a plethora of studies on aggregation inhibition and promotion, it is unfortunate that many previous studies testing the effects of compounds on polyQ aggregation did not investigate effects on toxicity.
Defining aggregate forms
One confounding factor in the debate on the nature of the neurotoxic polyQ species used to be the definition of the term “aggregate”. Traditionally many have used the term aggregate in the context of IBs, with measures of aggregation usually being simply a count of IBs and/or detection of SDS-insoluble material. However, now that methods to identify oligomeric species of polyQ proteins have been developed, along with the proposal of an aggregation pathway with intermediates on- and off-pathway to fibrillisation and insoluble species, future studies should address the relationship between the specific types of aggregates and neurotoxicity. Here, the use of the new, more sensitive technologies to identify the effects of aggregation inhibitors and promoters of oligomeric intermediates, previously used to show effects on toxicity, are required. For example, a recent study using agarose gel electrophoresis to detect oligomeric species in HD mouse and cell models, showed that oligomers could be detected before any measurable behavioural phenotype and the size of the aggregates correlated with disease progression [178]. FCS has also proven be a useful measure of alterations in aggregate size [34]. Now that different shapes and forms of polyQ species can be produced in vitro (Fig. 1) and characterised by AFM and biochemically, their neurotoxic potential can be tested. One difficulty, however, is to transfer a particular aggregate species of a sufficiently homogenous nature and quantity into cells. Recent work by Teplow and colleagues [116] in which specific oligomers of Aβ have been chemically stabilised via cross-linking and fractionated in pure form provides a promising step forward in this direction.
Effects of altering aggregation on toxicity
A variety of compounds have been shown to alter the IB load in cells and/or mouse models. Compounds used to promote the formation of IBs have been shown to reduce cytotoxicity in polyQ cell models [20], and in addition, cells containing an htt IB exhibited improved survival compared to cells containing diffuse mutant htt fragments [6, 145]. Strikingly, IBs have been predominantly observed in spared interneurons in the striatum of HD patient brains, with few, if any IBs found within the vulnerable medium spiny striatal neurons (MSNs) [84]. A zebra fish model also revealed that apoptotic cells killed by expression of expanded polyQ htt fragments did not contain IBs [148].
In contrast, several different methods of reducing aggregation, including addition of peptides (e.g. QBP1), expression of intrabodies, molecular chaperones and their interacting proteins and addition of oligonucleotides and a variety of small molecules have been shown to be protective in cell and animal models of polyQ diseases. However, it is worth noting that all these methods require binding of the aggregation-inhibitor to the polyQ protein and therefore have the potential to alter monomeric and oligomeric species as well as affecting aggregation per se. Methods of increasing IB formation such as increasing ubiquitination [38] have also been shown to increase toxicity. On the other hand, some studies co-expressing molecular chaperones in cell models have indicated that toxicity is modified without changing the level of IB formation [189]. Again, one of the difficulties with all these studies is that, the method of monitoring aggregation is by IB count and/or SDS-insoluble material detection, without the effect on oligomeric species being monitored and therefore this could go some way in explaining the discrepancies, in addition to the variety of different models used. This issue is exemplified by a recent in vivo study of effects of aggregation, which shows that knockout of Hsp70 in the R6/2 HD mouse model significantly increased the average size and number of IBs, without affecting the total fibrillar aggregates measured biochemically, causing an increase in severity and behavioural phenotypes in this mouse model [171]. Therefore, Hsp70 may normally bind to monomeric and/or small oligomeric forms of polyQ protein that are off-pathway for fibril formation, but are toxic and lead to IB formation.
Methods which target specific aggregate structures provide great potential in investigating the most toxic species of aggregate. Towards this goal, a recent study has shown that an intrabody, which binds specifically to fibrils and increases fibrillisation of httex1, with no measurable effect on oligomers, increases toxicity of the polyQ protein [85]. A study using time-lapse live-cell fluorescence microscopy to measure aggregation of httex1 in PC12 cells was able to identify the rapid formation of “microaggregates” (<1 μm in diameter) visible by epifluorescent light microscopy, with a prolonged time required for formation of classical IBs. The detection of aggregation did not coincide with death, but detectable aggregation did correlate with an earlier cell death and the larger the final aggregate formed, the longer the cell survived [60]. Microaggregates, i.e. soluble high molecular weight complexes with a size of 700–7,000 kDa (i.e. approximately 20–200 molecules) have also been observed in a mouse model expressing polyQ-expanded ataxin-3, which are found in the soluble fraction of brain lysates, but are SDS-resistant [182]. The appearance of microaggregates in cell and mouse model systems described above, favours the “multi-nucleation” hypothesis of polyQ aggregation. Increasing the formation of microaggregates by downregulating the ubiquitin ligase, CHIP, accelerated disease progression in these mice, also indicating that the microaggregates are toxic [182]. Another study supporting the toxicity of soluble microaggregates used a yeast model expressing polyQ-expanded AR, in which a 67Q stretch formed SDS-insoluble IBs, whereas a 102Q stretch formed some irregularly shaped soluble microaggregates, but the 102Q stretch was the more toxic form [149]. In another yeast model, httex1 IBs were found to be non-toxic, whilst amorphous aggregates tended to be toxic, although this was not always the case, suggesting that morphology alone does not necessarily determine cytotoxicity [45]. It is possible that the increase in surface area of microaggregates compared to large foci like IBs increases their toxicity, for example, by an increased ability to sequester other proteins. However, two studies show that downregulation of chaperonins causes an increase in IB formation and soluble aggregates of polyQ proteins, which is associated with an increase in toxicity but that upregulation of a chaperonin can cause the formation of multiple smaller foci, which are linked with a decrease in toxicity [83, 161].
The toxicity of polyQ proteins at the monomeric level is supported by the increased toxicity after microinjection of a monomeric thioredoxin-polyQ fusion protein with a β-sheet conformation compared to the toxicity induced by an α-helical conformation of the same protein. However, the injection of soluble oligomeric and fibrillar forms of this protein were also toxic to some extent [113]. In addition, insertion of proline residues to disrupt the monomeric conformation of the thioredoxin-polyQ protein also significantly reduced toxicity in a cell model [127]. It has also been suggested that the promotion of oligomeric species “off pathway” from fibril production can be cytoprotective. For example, the promotion of “benign” oligomers by the cooperation of chaperones (Hsp70/40) and a chaperonin (TriC) has been reported to prevent toxicity of htt [13].
In summary, it is currently unclear what the exact neurotoxic polyQ species are, but from the studies discussed above, it seems that both monomeric- and oligomeric polyQ states and assemblies confer increased cytotoxicity, compared to fibrillar structures and IBs.
Why might oligomeric polyQ species be cytotoxic?
As outlined above, there is mixed evidence regarding the toxicity of aggregation, and virtually all species of polyQ protein from monomeric polyQ, through small oligomeric and larger microscopically visible aggregate structures have been implicated in toxicity in some way. However, below we focus particularly on how oligomer formation of polyQ proteins may be toxic, although some proposed mechanisms are likely to be inseparable from toxicity caused by larger aggregates and/or IB formation.
Gain versus loss of function
As summarised in the introduction, polyQ proteins have defined roles in a normal cellular context and hence oligomerisation of polyQ proteins may lead to a loss of function (see “Introduction” for references on gain vs. loss of function during polyQ disease). Aggregation of polyQ-containing proteins into oligomers can prevent them from performing their normal cellular function either by sequestering the proteins away from their normal cellular location (see below) or by preventing interactions with their protein network. Indeed htt protein has been shown to have a substantial protein network and has been implicated in a wide variety of cellular functions. Furthermore, mutant htt has been shown to promote fibrillogenesis of wild-type htt [24]. However, individuals that are hemizygous for htt do not show any symptoms of HD [5], indicating that a loss of function of htt is not the central cause of this disease. However, it is likely that loss of function of polyQ proteins goes some way to exacerbate the disease phenotype. A toxic gain of function is likely given that the polyQ expansion appears to be the only aspect that links the various polyQ proteins to brain pathology and that polyQ peptides are intrinsically cytotoxic [74, 102], even in organisms such as Caenorhabditis elegans, which have no functional homologs of the polyQ disease proteins [48].
Oligomerisation itself could cause a change in cellular localisation, which may lead to further aggregation due to abnormal interactions with cellular material. For example, a polyQ expansion in htt is known to impair its nuclear export and it can be envisaged that oligomerisation of the protein could prevent shuttling between the nucleus and cytoplasm due to an increase in size and/or a change in conformation of the protein preventing the interaction with the nuclear pore protein complex [33]. Changes in nucleocytoplasmic shuttling of ataxin-3 have also been implicated in MJD [97]. The physical accumulation of protein oligomers in unusual cellular compartments can clearly cause a whole host of problems for neurons, including synaptic dysfunction and axonal transport deficits [92, 107], that are not further discussed here.
Membrane disruption, insertion and translocation
It has been suggested for other aggregation-prone proteins, including α-synuclein, that annular oligomers can form pores in the membranes of cells, implicating membrane permeabilisation as an inducer method of toxicity for protein aggregation. Htt has been shown to associate with membranes [158]. Recently htt with a wild-type polyQ stretch has been shown to interact in vitro with lipid vesicles and lipid rafts in a HD mouse model with mutant htt interacting with lipids not recognised by the wild-type htt and having a greater affinity with lipid rafts [78]. Therefore, mutant htt could disturb membrane-dependent signalling pathways and membrane trafficking. It has been previously shown that polyQ peptides can form channels in lipid bilayers [109] and recently it was shown that an expanded polyQ stretch can cause penetration of htt into lipid bilayers [79]. The effect of membrane insertion could be due to oligomerisation, and could affect membrane protein function and/or lipid composition of the membrane, causing the membrane to be dysfunctional and possibly forming pores, as predicted for α-synuclein. Such pores have, however, never been observed in vivo. Membrane dysfunction can be linked to many of the defects observed in polyQ disease models, including defects at the synapse and with mitochondria [65]. Indeed htt has been reported to associate with mitochondria [117]. No data are currently available for polyQ proteins other than htt in relation to membrane interactions.
In addition, fibrils formed by polyQ peptides applied extracellularly have recently been demonstrated to have the ability to cross the membrane and enter the cytoplasm [134]. This is of particular interest, as recent investigations with various other amyloid-forming proteins including tau and α-synuclein have suggested a degree of “infectivity” of these proteins due to a templated conformational change (for a review see [54]). Therefore, even if the fibrillar form of a polyQ protein is not toxic, its ability if released, e.g. due to neuronal death, to seed aggregation in another cell, could induce toxicity and perhaps propagating this “aggregation signal” throughout the CNS.
Impairment of the protein turnover machineries
Although the autophagic/lysosomal system appears to deal with the degradation of aggregation-prone molecules [187], the proteasome may be able to deal with existing polyQ aggregates, if expression of the protein in neurons is switched off [187] and there is evidence that polyQ stretches that are targeted to the UPS are at least partially degraded in cell systems [69]. These findings argue against the fact that polyQ oligomers could be trapped irreversibly in the proteasome. Although previous studies of proteasomal degradation of polyQ peptides in vitro indicate that the eukaryotic proteasome is unable to cleave within polyQ stretches [167], a recent study using mass spectrometry suggests that in the presence of a proteasome activator, PA28gamma (K188E), a fluorescein labelled polyQ peptide was able to be cleaved after each glutamine residue [128]. Despite this, oligomerisation of polyQ proteins is at least likely to increase the time required for breakdown of the protein, leading to slower degradation of the polyQ protein and also of other proteins reliant on proteasomal degradation, possibly leading to the accumulation of proteins, affecting signalling cascades (as has been suggested for anaphase-promoting complex substrates [19]) and synaptic function [176]. An increase in pure polyQ stretches being released by the proteasome would place further demand on peptidases that are able to cleave polyQ stretches, in particular puromycin-sensitive aminopeptidase [18] and could cause a further increase in aggregation and a further decline in proteasomal and peptidase activity [132]. Proteasome dysfunction has been shown to occur before the accumulation of IBs in cells [14] and therefore oligomer formation may be the cause of the decrease in efficiency of the proteasome. Fluorescence live-cell imaging showed that the proteasome can be irreversibly sequestered within aggregates formed by overexpressed mutant httex1 or simple polyQ expansions [69] and hence as aggregates grow larger, physical “trapping” of the proteasome may also play a role in decreased efficiency of proteasomal function.
Sequestration of other cellular proteins and transcriptional impairment
The demonstration that proteins other than the polyQ disease protein itself can be found in IBs opens up the possibility that aggregation of the polyQ protein sequesters other polyQ proteins, disabling their function in some way. For example, and as discussed above, the aggregation of polyQ proteins can alter their conformation and therefore binding of other polyQ stretch-containing proteins by oligomers could cause the protein to switch to an inactive conformational state. An important class of polyQ-containing proteins are transcription factors, including CBP, TBP and Sp1. The glutamine-rich regions in these proteins are critical for the control of interactions between activator and suppressor proteins and general mediators that affect RNA polymerase II activity. Indeed several of the polyQ disease proteins have been implicated to play a role in transcription with transcriptional dysregulation being a common feature of polyQ diseases. Incorporation of these transcription factors into oligomers could severely alter the function of these factors, and direct interactions between transcription factors and polyQ proteins have been widely reported [28]. However, it is not only transcription factors, but also other cellular proteins that are sequestered and/or bind reversibly to polyQ aggregates such as molecular chaperones [185] and proteins of the UPS and autophagic-lysosomal system [75]. Proteins containing not only pure polyQ stretches but also Q/N-rich stretches have been reported to interact with expanded polyQ proteins, directly affecting their functions. For example, mutant htt efficiently seeds the fibrillisation of the RNA-binding protein T-cell intracellular antigen-1 (TIA-1), which is sequestered into IBs in an HD mouse model and polyQ tracts are potent inducers of amyloid formation by prion proteins, Sup35 and Rnq1, in yeast [59]. Given that the total number of Q/N-rich domains has been estimated to be 200 in the human proteome, this suggests that effects of changes in conformation and aggregation states of these proteins must somehow contribute to the pathology of polyQ diseases [66]. PolyQ aggregates have been shown to contain other proteins involved in neurodegeneration such as alpha-synuclein (involved in PD) [27], which causes a translocation of alpha-synuclein away from the synapse. Htt has also been associated with intracellular tangles and inclusions found in AD and Pick’s Disease, especially associated with cytoskeletal filaments [154], providing evidence that an abnormal interaction with neurofilaments could contribute to HD pathology.
An even more general mechanism of polyQ-induced toxicity has been proposed, based on alterations in protein homeostasis (for a review see [8]). The expression of a polyQ stretch in several diverse temperature-sensitive mutations in C. elegans caused the temperature-sensitive mutation to exhibit their phenotypes at the permissive conditions, showing that the function of structurally and functionally unrelated proteins is affected by the expression of the aggregation-prone polyQ protein and indicating that aggregation of the polyQ protein may prevent the cell from dealing with unstable proteins that it would otherwise cope with by (re)-folding and clearance mechanisms [58]. This finding would provide a mechanism for the characteristic slow progression of polyQ diseases, i.e. the cell initially manages the aggregation of polyQ protein, but the accumulation of other misfolded and damaged proteins eventually overwhelms the cell’s capabilities (for a review see [65]).
Most studies have addressed the issue of sequestration of normal cellular proteins by co-immunostaining of IBs in vitro or in vivo, rather than investigating the sequestration of cellular proteins to polyQ oligomers. The relevance of sequestration of cellular proteins for polyQ disease is difficult to assess as most of the published studies did not quantify the degree of abnormal sequestration into polyQ aggregates.
ROS production
Another proposed mechanism of toxicity due to expanded polyQ proteins is the production of reactive oxygen species (ROS). Htt and ataxin-3 have both been shown to bind redox-active metals and the effect of metals in increasing aggregation of full-length polyQ proteins has also been reported [52, 135]. Our observations show that copper increases the fibrillisation of recombinant httex1 in vitro as measured by AFM and increases httex1 IB formation within cells [64]. It is therefore possible that oligomerisation of polyQ proteins creates an environment in which redox-active metals can react with molecular oxygen to form ROS, as suggested for other amyloidogenic proteins [3]. Alteration of ROS-producing systems (e.g. by mitochondria) by polyQ oligomers is another way by which oligomerisation could modulate the cellular redox-homeostasis. Both wildtype and mutant htt and htt fragments have indeed been localised to the outer mitochondrial membrane and appear to induce several mitochondrial dysfunctions (reviewed in [129]). Additionally, polyQ-expanded httex1 appears to disrupt the mitochondrial fusion–fission machinery [175] and to recruit a number of glycolytic enzymes such as GAPDH, enolase and fructose bis-phosphate into aggregates [177]. It is currently unclear whether and how polyQ oligomerisation disrupts the cellular redox-homeostasis.
PolyQ aggregation and the evolution of brain- and peripheral polyQ pathology
As mentioned above polyQ diseases are primarily disorders of the CNS, despite the polyQ-containing proteins being expressed in the CNS and the periphery at similar levels. PolyQ aggregation in the CNS during polyQ pathology is widespread and although immunohistochemical analysis of IBs in DRPLA, SCA3, SCA-7 and HD postmortem brain shows an overlap with the neuropathology that occurs in these disorders, it appears that polyQ aggregation is more widespread [11, 41, 70]. The brain-region-specific- and intracellular localisation of aggregated polyQ protein varies with the degree of polyQ pathology and disease progression. For example in HD, DiFiglia and colleagues [41] showed that N-terminal htt forms neuronal intranuclear inclusion bodies (NIIs) that are highly heterogenous in composition containing a mixture of several forms of filaments/fibrils and granules/oligomers, something that is also observed in brains of MJD patients [186]. Interestingly, it has been recently shown that oligomers of cortical neurons observed by EM analysis in the human HD brain were similar in lateral dimension to oligomers observed by AFM using recombinant httex1 ([94], see Fig. 1). Sapp et al. [140] reported that NIIs are more frequent in the cortex than in the striatum and that in earlier stages of disease accumulation of N-terminal mutant htt also occurs in the cytoplasm. Furthermore, NIIs are found in the allocortex in addition to the neostriatum and neocortex and NII frequency in the neocortex is highest in juvenile patients [96]. The frequency of the normally large ubiquitinated nuclear and cytoplasmic aggregates of mutant htt is 1–7% of cortical, striatal and thalamic neurons and neurophil of patients with adult onset of symptoms and up to 21% of cortical neurons in individuals with juvenile onset ([170] and refs therein) and therefore much lower compared to some HD rodent models (see below). However, systematic correlative biochemical studies on polyQ aggregation that also consider polyQ precursors such as oligomeric structures in the HD brain and indeed in other polyQ diseases are missing. Therefore, it is currently unclear how polyQ oligomerisation relates to the complex CNS pathology in HD and polyQ diseases and as noted in a recent review on HD neuropathology, increased effort should go into analysing human diseased brains, rather than deducing too many aspects of polyQ pathology from animal models [170].
Numerous reports identified significant abnormalities in the peripheral tissues of HD patients including altered glucose homeostasis and (sub)- cellular alterations in fibroblasts, lymphocytes and erythrocytes, but the evidence of polyQ aggregation in such tissues is rare or non-existent (reviewed in [141]). This is in contrast to some of the mouse models of polyQ disorders that have been extensively analysed in relation to polyQ aggregation. The most studied HD rodent model is the R6/2 line expressing httex1 with a CAG expansion of 145 [99]. In the R6/2 line, IBs are first detected in the cortex and striatum in week 3–5 after birth, before onset of symptoms co-incident with brain weight loss [142]. Later a massive production of IBs occurs in many CNS regions, and currently novel approaches for more sensitive biochemical aggregate detection are being used to understand the role of polyQ oligomerisation and pathology in R6/2 and other mouse models [86, 143, 178]. Such studies must now be extended to human CNS and peripheral tissues of polyQ pathologies to learn more about the role of oligomerisation during the evolution of pathology. For a report on peripheral events and the wide spectrum of the clinically heterogenous group of SCAs the reader is referred to the paper by Manto [100].
Non-CNS polyQ IBs in the R6/2 HD mouse model appear to occur later, but indeed in many peripheral tissues such as skeletal muscle, heart, liver, adrenal glands, pancreas, kidney, and this correlates with a progressive decrease in size of these organs [142]. Such abnormalities could be related to the polyQ-aggregation process, but other cellular dysfunctions (e.g. altered transcription and endo/exocytosis, see above) unrelated to toxic polyQ oligomerisation may also be important. IBs in peripheral tissue have also been reported in SBMA [95] and in an SBMA mouse model [1]. The factors that determine polyQ cell-specific aggregation are unclear and likely include a multitude of parameters (see “Effect of cellular factors and intracellular localisation on polyQ aggregation”). One obvious important reason for aggregation pathology in the CNS is that the post-mitotic nature of neurons (or muscle in the periphery) prevents a reduction of aggregate load by cell division, whereas in cycling cells the aggregated material would be distributed between daughter cells. Cycling cells also have the opportunity to redistribute aggregates from the nucleus into the cytoplasm during cell division when the nuclear membrane disintegrates hence potentially reducing toxicity via autophagic/lysosomal degradation that only occurs in the cytoplasm (see “Effect of cellular factors and intracellular localisation on polyQ aggregation”). However, this reasoning would not explain neuronal subtype-specific toxicity and the sometimes observed polyQ aggregation and altered cellular function in glial cells (e.g. astrocytes or microglia) that are increasingly believed to contribute to polyQ pathology via non-cell autonomous mechanisms [21, 22, 121, 139].
Conclusion
The current evidence indicates that a conformational change induced by the polyQ expansion, also determined by the protein sequence surrounding the polyQ stretch, significantly impacts on aggregation and toxicity. It is likely that several different conformations of monomeric polyQ protein exist and lead to particular oligomeric intermediates. It is becoming clear that although IBs are clear pathological hallmarks of the polyQ diseases, earlier intermediates are the likely cause of neuronal dysfunction. If this were the case then accumulation into larger aggregates and IBs may well be a cell-protective mechanism, at least in the short term. However, it is difficult to imagine how the presence of such a large intracellular structure, would not at some point lead to neurotoxicity, whether it be due to its large physical size disrupting cellular transport and other processes, its ability to sequester other essential cellular proteins and impair the protein turnover machinery or a combination of these factors.
As structurally defined polyQ peptides and proteins can now be produced in vitro and the polyQ-aggregation behaviour manipulated in vivo, correlative studies can be performed. This will determine the relative neurotoxic impact of different polyQ species. However, in vivo studies investigating the causal relationships between polyQ aggregation and toxicity remain technically challenging and the effects of oligomers on toxicity remain unclear. As no reports in the literature have been found of polyQ oligomers being present in normal individuals, it would seem prudent to focus efforts on preventing the early stages of aggregation. Thus, it is important to invest time in obtaining more detail on the structures of monomeric and oligomeric polyQ protein species produced in vivo and to make further attempts to model these species in order to evaluate their respective toxicities. Such studies will assist in the design of effective modifiers of early aggregation, such as intrabodies, in the hope of producing treatments to prevent and/or delay the devastating symptoms of the polyQ diseases.
References
Adachi H, Kume A, Li M et al (2001) Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet 10:1039–1048
Aiken CT, Steffan JS, Guerrero CM et al (2009) Phosphorylation of threonine-3: implications for huntingtin aggregation and neurotoxicity. J Biol Chem 284:29427–29436
Allsop D, Mayes J, Moore S, Masad A, Tabner BJ (2008) Metal-dependent generation of reactive oxygen species from amyloid proteins implicated in neurodegenerative disease. Biochem Soc Trans 36:1293–1298
Altschuler EL, Hud NV, Mazrimas JA, Rupp B (1997) Random coil conformation for extended polyglutamine stretches in aqueous soluble monomeric peptides. J Pept Res 50:73–75
Ambrose CM, Duyao MP, Barnes G et al (1994) Structure and expression of the Huntington’s disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat Cell Mol Genet 20:27–38
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810
Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet 16:2600–2615
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319:916–919
Barton S, Jacak R, Khare SD, Ding F, Dokholyan NV (2007) The length dependence of the PolyQ-mediated protein aggregation. J Biol Chem 282:25487–25492
Bauer PO, Nukina N (2009) The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem 110:1737–1765
Becher MW, Kotzuk JA, Sharp AH et al (1998) Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis 4:387–397
Becker M, Martin E, Schneikert J, Krug HF, Cato ACB (2000) Cytoplasmic localization and the choice of ligand determine aggregate formation by androgen receptor with amplified polyglutamine stretch. J Cell Biol 149:255–262
Behrends C, Langer CA, Boteva R et al (2006) Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell 23:887–897
Bennett EJ, Bence NF, Jayakumar R, Kopito RR (2005) Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell 17:351–365
Bevivino AE, Loll PJ (2001) An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel beta-fibrils. Proc Natl Acad Sci USA 98:11955–11960
Bhattacharyya A, Thakur AK, Chellgren VM et al (2006) Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol 355:524–535
Bhattacharyya AM, Thakur AK, Wetzel R (2005) Polyglutamine aggregation nucleation: thermodynamics of a highly unfavorable protein folding reaction. Proc Natl Acad Sci USA 102:15400–15405
Bhutani N, Venkatraman P, Goldberg AL (2007) Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO J 26:1385–1396
Bocharova NA, Sokolov SS, Knorre DA, Skulachev VP, Severin FF (2008) Unexpected link between anaphase promoting complex and the toxicity of expanded polyglutamines expressed in yeast. Cell Cycle 7:3943–3946
Bodner RA, Outeiro TF, Altmann S et al (2006) Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. Proc Natl Acad Sci USA 103:4246–4251
Bradford J, Shin J-Y, Roberts M, Wang C-E, Li X-J, Li S (2009) Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci USA 106:22480–22485
Bradford J, Shin J-Y, Roberts M et al (2010) Mutant huntingtin in glial cells exacerbates neurological symptoms of huntington disease mice. J Biol Chem 285:10653–10661
Burnett B, Li F, Pittman RN (2003) The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet 12:3195–3205
Busch A, Engemann S, Lurz R, Okazawa H, Lehrach H, Wanker EE (2003) Mutant huntingtin promotes the fibrillogenesis of wild-type huntingtin. J Biol Chem 278:41452–41461
Cattaneo E, Zuccato C, Tartari M (2005) Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci 6:919–930
Chai Y, Shao J, Miller VM, Williams A, Paulson HL (2002) Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci USA 99:9310–9315
Charles V, Mezey E, Reddy PH et al (2000) Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci Lett 289:29–32
Chen-Plotkin AS, Sadri-Vakili G, Yohrling GJ et al (2006) Decreased association of the transcription factor Sp1 with genes downregulated in Huntington’s disease. Neurobiol Dis 22:233–241
Chen H-K, Fernandez-Funez P, Acevedo SF et al (2003) Interaction of Akt-phosphorylated Ataxin-1 with 14–3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113:457–468
Chen S, Berthelier V, Hamilton JB, O’Nuallai B, Wetzel R (2002) Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 41:7391–7399
Chen S, Berthelier V, Yang W, Wetzel R (2001) Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol 311:173–182
Chen S, Ferrone FA, Wetzel R (2002) Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci USA 99:11884–11889
Cornett J, Cao F, Wang C-E et al (2005) Polyglutamine expansion of huntingtin impairs its nuclear export. Nat Genet 37:198–204
Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV (2006) Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc Natl Acad Sci USA 103:16764–16769
Dahlgren PR, Karymov MA, Bankston J et al (2005) Atomic force microscopy analysis of the huntington protein nanofibril formation. Nanomedicine 1:52–57
Davidson JD, Riley B, Burright EN, Duvick LA, Zoghbi HY, Orr HT (2000) Identification and characterization of an ataxin-1-interacting protein: A1Up, a ubiquitin-like nuclear protein. Hum Mol Genet 9:2305–2312
Davies S, Turmaine M, Cozens B et al (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90:537–548
de Pril R, Fischer DF, Roos RAC, van Leeuwen FW (2007) Ubiquitin-conjugating enzyme E2–25 K increases aggregate formation and cell death in polyglutamine diseases. Mol Cell Neurosci 34:10–19
Deepak S, Leonid MS, Hideyo I, Ronald W, Daniel AK (2005) Polyglutamine homopolymers having 8–45 residues form slablike beta-crystallite assemblies. Proteins 61:398–411
Dehay B, Weber C, Trottier Y, Bertolotti A (2007) Mapping of the epitope of monoclonal antibody 2B4 to the proline-rich region of human huntingtin, a region critical for aggregation and toxicity. Biotechnol J 2:559–564
DiFiglia M, Sapp E, Chase K et al (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993
DiFiglia M, Sapp E, Chase K et al (1995) Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14:1075–1081
Doss-Pepe EW, Stenroos ES, Johnson WG, Madura K (2003) Ataxin-3 interactions with Rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol Cell Biol 23:6469–6483
Dougan L, Li J, Badilla CL, Berne BJ, Fernandez JM (2009) Single homopolypeptide chains collapse into mechanically rigid conformations. Proc Natl Acad Sci USA 106:12605–12610
Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S (2006) Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc Natl Acad Sci USA 103:11045–11050
Ellisdon AM, Thomas B, Bottomley SP (2006) The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem 281:16888–16896
Emamian ES, Kaytor MD, Duvick LA et al (2003) Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38:375–387
Faber PW, Alter JR, MacDonald ME, Hart AC (1999) Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci USA 96:179–184
Fei E, Jia N, Zhang T et al (2007) Phosphorylation of ataxin-3 by glycogen synthase kinase 3 beta at serine 256 regulates the aggregation of ataxin-3. Biochem Biophys Res Com 357:487–492
Fernandez-Funez P, Nino-Rosales ML, de Gouyon B et al (2000) Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 408:101–106
Forman MS, Trojanowski JQ, Lee VMY (2004) Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 10:1055–1063
Fox JH, Kama JA, Lieberman G et al (2007) Mechanisms of copper ion mediated huntington’s disease progression. PLoS One 2:e334
Frontali M (2001) Spinocerebellar ataxia type 6: channelopathy or glutamine repeat disorder? Brain Res Bull 56:227–231
Frost B, Diamond MI (2009) The expanding realm of prion phenomena in neurodegenerative disease. Prion 3:74–77
Funderburk SF, Shatkina L, Mink S, Weis Q, Weg-Remers S, Cato ACB (2009) Specific N-terminal mutations in the human androgen receptor induce cytotoxicity. Neurobiol Aging 30:1851–1864
Gales L, Cortes L, Almeida C et al (2005) Towards a structural understanding of the fibrillization pathway in Machado-Joseph’s disease: trapping early oligomers of non-expanded ataxin-3. J Mol Biol 353:642–654
Gauthier LR, Charrin BC, Borrell-Pagès M et al (2004) Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138
Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI (2006) Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311:1471–1474
Goehler H, Droge A, Lurz R, Schnoegl S, Chernoff YO, Wanker EE (2010) Pathogenic polyglutamine tracts are potent inducers of spontaneous Sup35 and Rnq1 amyloidogenesis. PLoS One 5:e9642
Gong B, Lim MCY, Wanderer J, Wyttenbach A, Morton AJ (2008) Time-lapse analysis of aggregate formation in an inducible PC12 cell model of Huntington’s disease reveals time-dependent aggregate formation that transiently delays cell death. Brain Res Bull 75:146–157
Gunawardena S, Her L-S, Brusch RG et al (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic PolyQ proteins in drosophila. Neuron 40:25–40
Hackam AS, Singaraja R, Wellington CL et al (1998) The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol 141:1097–1105
Hackam AS, Wellington CL, Hayden MR (1998) The fatal attraction of polyglutamine-containing proteins. Clin Genet 53:233–242
Hands S, Mason R, Sajjad MU, Giorgini F, Wyttenbach A (2010) Metallothioneins and copper metabolism are candidate therapeutic targets in Huntington’s disease. Biochem Soc Trans 38:552–558
Hands S, Sinadinos C, Wyttenbach A (2008) Polyglutamine gene function and dysfunction in the ageing brain. Biochim Biophys Acta 1779:507–521
Harrison P, Gerstein M (2003) A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol 4:R40
Helmlinger D, Hardy S, Abou-Sleymane G et al (2006) Glutamine-expanded Ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol 4:e67
Hollenbach B, Scherzinger E, Schweiger K, Lurz R, Lehrach H, Wanker EE (1999) Aggregation of truncated GST–HD exon 1 fusion proteins containing normal range and expanded glutamine repeats. Philos Trans R Soc B Biol Sci 354:991–994
Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI (2004) Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J 23:4307–4318
Holmberg M, Duyckaerts C, Durr A et al (1998) Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet 7:913–918
Humbert S, Bryson EA, Cordelières FP et al (2002) The IGF-1/Akt pathway is neuroprotective in Huntington’s Disease and involves huntingtin phosphorylation by Akt. Dev Cell 2:831–837
Ignatova Z, Gierasch LM (2006) Inhibition of protein aggregation in vitro and in vivo by a natural osmoprotectant. Proc Natl Acad Sci USA 103:13357–13361
Ignatova Z, Thakur AK, Wetzel R, Gierasch LM (2007) In-cell aggregation of a polyglutamine-containing chimera is a multistep process initiated by the flanking sequence. J Biol Chem 282:36736–36743
Ikeda H, Yamaguchi M, Sugai S, Aze Y, Narumiya S, Kakizuka A (1996) Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat Genet 13:196–202
Iwata A, Christianson JC, Bucci M et al (2005) Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci USA 102:13135–13140
Jayaraman M, Kodali R, Wetzel R (2009) The impact of ataxin-1-like histidine insertions on polyglutamine aggregation. Protein Eng Des Sel 22:469–478
Jeong H, Then F, Melia TJ et al (2009) Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137:60–72
Kegel KB, Sapp E, Alexander J et al (2009) Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J Neurochem 110:1585–1597
Kegel KB, Schewkunow V, Sapp E et al (2009) Polyglutamine expansion in huntingtin increases its insertion into lipid bilayers. Biochem Biophys Res Comm 387:472–475
Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I (2009) Secondary structure of huntingtin amino-terminal region. Structure 17:1205–1212
Kim S, Nollen EAA, Kitagawa K, Bindokas VP, Morimoto RI (2002) Polyglutamine protein aggregates are dynamic. Nat Cell Biol 4:826–831
Kim YJ, Yi Y, Sapp E et al (2001) Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci USA 98:12784–12789
Kitamura A, Kubota H, Pack C-G et al (2006) Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol 8:1163–1169
Kuemmerle S, Gutekunst CA, Klein AM et al (1999) Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol 46:842–849
Kvam E, Nannenga BL, Wang MS, Jia Z, Sierks MR, Messer A (2009) Conformational targeting of fibrillar polyglutamine proteins in live cells escalates aggregation and cytotoxicity. PLoS One 4:e5727
Landles C, Sathasivam K, Weiss A et al (2010) Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington Disease. J Biol Chem 285:8808–8823
Lashuel HA, Petre BM, Wall J et al (2002) Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 322:1089–1102
Lathrop RH, Casale M, Tobias DJ, Marsh JL, Thompson L (1998) Modeling protein homopolymeric repeats: possible polyglutamine structural motifs for Huntington’s disease. Proc Int Conf Intell Syst Mol Biol 6:105–114
Lee CC, Walters RH, Murphy RM (2007) Reconsidering the mechanism of polyglutamine peptide aggregation. Biochemistry 46:12810–12820
Lee J-M, Ivanova EV, Seong IS et al (2007) Unbiased gene expression analysis implicates the huntingtin polyglutamine tract in extra-mitochondrial energy metabolism. PLoS Genet 3:e135
Lee K-J, Panzera A, Rogawski D, Greene LE, Eisenberg E (2007) Cellular prion protein (PrPC) protects neuronal cells from the effect of huntingtin aggregation. J Cell Sci 120:2663–2671
Lee W-CM, Yoshihara M, Littleton JT (2004) Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc Natl Acad Sci USA 101:3224–3229
Legleiter J, Lotz GP, Miller J et al (2009) Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J Biol Chem 284:21647–21658
Legleiter J, Mitchell E, Lotz GP et al (2010) Mutant Huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J Biol Chem 285:14777–14790
Li M, Miwa S, Kobayashi Y et al (1998) Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol 44:249–254
Maat-Shieman ML, Dorsman JC, Smoor MA et al (1999) Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J Neuropathol Exp Neurol 58:129–137
Macedo-Ribeiro S, Cortes L, Maciel P, Carvalho A (2009) Nucleocytoplasmic shuttling activity of Ataxin-3. PLoS One 4:e5834
Maltecca F, Filla A, Castaldo I et al (2003) Intergenerational instability and marked anticipation in SCA-17. Neurology 61:1441–1443
Mangiarini L, Sathasivam K, Seller M et al (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87:493–506
Manto M-U (2005) The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum 4:2–6
Marchut AJ, Hall CK (2006) Spontaneous formation of annular structures observed in molecular dynamics simulations of polyglutamine peptides. Comput Biol Chem 30:215–218
Marsh JL, Walker H, Theisen H et al (2000) Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet 9:13–25
Masino L, Kelly G, Leonard K, Trottier Y, Pastore A (2002) Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett 513:267–272
Masino L, Pastore A (2002) Glutamine repeats: structural hypotheses and neurodegeneration. Biochem Soc Trans 30:548–551
McEwan I (2004) Molecular mechanisms of androgen receptor-mediated gene regulation: structure-function analysis of the AF-1 domain. Endocr Relat Cancer 11:281–293
Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY (2002) Huntingtin toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 157:997–1004
Milnerwood AJ, Raymond LA (2007) Corticostriatal synaptic function in mouse models of Huntington’s disease: early effects of huntingtin repeat length and protein load. J Physiol 585:817–831
Monoi H (1995) New tubular single-stranded helix of poly-l-amino acids suggested by molecular mechanics calculations: I. Homopolypeptides in isolated environments. Biophys J 69:1130–1141
Monoi H, Futaki S, Kugimiya S-I, Minakata H, Yoshihara K (2000) Poly-l-glutamine forms cation channels: relevance to the pathogenesis of the polyglutamine diseases. Biophys J 78:2892–2899
Muchowski PJ, Ning K, D’Souza-Schorey C, Fields S (2002) Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proc Natl Acad Sci USA 99:727–732
Mukai H, Isagawa T, Goyama E et al (2005) Formation of morphologically similar globular aggregates from diverse aggregation-prone proteins in mammalian cells. Proc Natl Acad Sci USA 102:10887–10892
Mukherjee S, Thomas M, Dadgar N, Lieberman AP, Iniquez-Lluhi JA (2009) Small ubiquitin-like modifier (SUMO) modification of the androgen receptor attenuates polyglutamine-mediated aggregation. J Biol Chem 284:21296–21306
Nagai Y, Inui T, Popiel HA et al (2007) A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol 14:332–340
Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci USA 106:9679–9684
Nonhoff U, Ralser M, Welzel F et al (2007) Ataxin-2 interacts with the DEAD/H-Box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell 18:1385–1396
Ono K, Condron MM, Teplow DB (2009) Structure neurotoxicity relationships of amyloid-beta protein oligomers. Proc Natl Acad Sci USA 106:14745–14750
Orr AL, Li S, Wang C-E et al (2008) N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci 28:2783–2792
Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Ann Rev Neurosci 30:575–621
Parekh-Olmedo H, Wang J, Gusella J, Kmiec E (2004) Modified single-stranded oligonucleotides inhibit aggregate formation and toxicity induced by expanded polyglutamine. J Mol Neurosci 24:257–267
Paulson HL, Perez MK, Trottier Y et al (1997) Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19:333–344
Pavese N, Gerhard A, Tai YF et al (2006) Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology 66:1638–1643
Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN (1998) Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol 143:1457–1470
Perutz MF, Finch JT, Berriman J, Lesk A (2002) Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci USA 99:5591–5595
Perutz MF, Johnson T, Suzuki M, Finch JT (1994) Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA 91:5355–5358
Poirier MA, Jiang H, Ross CA (2005) A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet structure. Hum Mol Genet 14:765–774
Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem 277:41032–41037
Popiel HA, Nagai Y, Onodera O et al (2004) Disruption of the toxic conformation of the expanded polyglutamine stretch leads to suppression of aggregate formation and cytotoxicity. Biochem Biophys Res Comm 317:1200–1206
Pratt G, Rechsteiner M (2008) Proteasomes cleave at multiple sites within polyglutamine tracts. J Biol Chem 283:12919–12925
Quintanilla RA, Johnson GVW (2009) Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res Bull 80:242–247
Quraishe S, Asuni A, Boelens WC, O’Connor V, Wyttenbach A (2008) Expression of the small heat shock protein family in the mouse CNS: differential anatomical and biochemical compartmentalization. Neuroscience 153:483–491
Rangone H, Poizat G, Troncoso J et al (2004) The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur J Neurosci 19:273–279
Raspe M, Gillis J, Krol H et al (2009) Mimicking proteasomal release of polyglutamine peptides initiates aggregation and toxicity. J Cell Sci 122:3262–3271
Ravikumar B, Stewart A, Kita H, Kato K, Duden R, Rubinsztein DC (2003) Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy. Hum Mol Genet 12:985–994
Ren P-H, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 11:219–225
Ricchelli F, Fusi P, Tortora P et al (2007) Destabilization of non-pathological variants of ataxin-3 by metal ions results in aggregation/fibrillogenesis. Int J Biochem Cell Biol 39:966–977
Rockabrand E, Slepko N, Pantalone A et al (2007) The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet 16:61–77
Rousseau E, Dehay B, Ben-Haïem L, Trottier Y, Morange M, Bertolotti A (2004) Targeting expression of expanded polyglutamine proteins to the endoplasmic reticulum or mitochondria prevents their aggregation. Proc Natl Acad Sci USA 101:9648–9653
Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786
Sapp E, Kegel KB, Aronin N et al (2001) Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol 60:161–172
Sapp E, Penney J, Young A, Aronin N, Vonsattel J-P, DiFiglia M (1999) Axonal transport of N-terminal huntingtin suggests early pathology of corticostriatal projections in Huntington disease. J Neuropathol Exp Neurol 58:165–173
Sassone J, Colciago C, Cislaghi G, Silani V, Ciammola A (2009) Huntington’s disease: the current state of research with peripheral tissues. Exp Neurol 219:385–397
Sathasivam K, Hobbs C, Turmaine M et al (1999) Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet 8:813–822
Sathasivam K, Lane A, Legleiter J et al (2010) Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet 19:65–78
Satterfield TF, Jackson SM, Pallanck LJ (2002) A drosophila homolog of the polyglutamine disease gene sca2 is a dosage-sensitive regulator of actin filament formation. Genetics 162:1687–1702
Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55–66
Saunders HM, Bottomley SP (2009) Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng Des Sel 22:447–451
Scherzinger E, Lurz R, Turmaine M et al (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90:549–558
Schiffer NW, Broadley SA, Hirschberger T et al (2007) Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J Biol Chem 282:9195–9203
Schiffer NW, Ceraline J, Hartl FU, Broadley SA (2008) N-terminal polyglutamine-containing fragments inhibit androgen receptor transactivation function. Biol Chem 389:1455–1466
Schilling B, Gafni J, Torcassi C et al (2006) Huntingtin phosphorylation sites mapped by mass spectrometry. J Biol Chem 281:23686–23697
Sen S, Dash D, Pasha S, Brahmachari SK (2003) Role of histidine interruption in mitigating the pathological effects of long polyglutamine stretches in SCA1: a molecular approach. Protein Sci 12:953–962
Sharma D, Sharma S, Pasha S, Brahmachari SK (1999) Peptide models for inherited neurodegenerative disorders: conformation and aggregation properties of long polyglutamine peptides with and without interruptions. FEBS Lett 456:181–185
Sikorski P, Atkins E (2005) New model for crystalline polyglutamine assemblies and their connection with amyloid fibrils. Biomacromolecule 6:425–432
Singhrao SK, Thomas P, Wood JD et al (1998) Huntingtin protein colocalizes with lesions of neurodegenerative diseases: an investigation in Huntington’s, Alzheimer’s, and Pick’s Diseases. Exp Neurol 150:213–222
Squitieri F, Gellera C, Cannella M et al (2003) Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain 126:946–955
Steffan JS, Agrawal N, Pallos J et al (2004) SUMO modification of huntingtin and Huntington’s Disease pathology. Science 304:100–104
Stenoien DL, Mielke M, Mancini MA (2002) Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components. Nat Cell Biol 4:806–810
Suopanki J, Götz C, Lutsch G et al (2006) Interaction of huntingtin fragments with brain membranes; clues to early dysfunction in Huntington’s disease. J Neurochem 96:870–884
Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O (2008) Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet 17:345–356
Takahashi Y, Okamoto Y, Popiel HA et al (2007) Detection of polyglutamine protein oligomers in cells by fluorescence correlation spectroscopy. J Biol Chem 282:24039–24048
Tam S, Geller R, Spiess C, Frydman J (2006) The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol 8:1155–1162
Tanaka M, Morishima I, Akagi T, Hashikawa T, Nukina N (2001) Intra- and intermolecular beta-pleated sheet formation in glutamine-repeat inserted myoglobin as a model for polyglutamine diseases. J Biol Chem 276:45470–45475
Temussi PA, Masino L, Pastore A (2003) From Alzheimer to Huntington: why is a structural understanding so difficult? EMBO J 22:355–361
Thakur AK, Jayaraman M, Mishra R et al (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol 16:380–389
Thakur AK, Wetzel R (2002) Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci USA 99:17014–17019
Tsai C-C, Kao H-Y, Mitzutani A et al (2004) Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci USA 101:4047–4052
Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 14:95–104
Vitalis A, Lyle N, Pappu RV (2009) Thermodynamics of β-Sheet formation in polyglutamine. Biophys J 97:303–311
Vitalis A, Wang X, Pappu RV (2008) Atomistic simulations of the effects of polyglutamine chain length and solvent quality on conformational equilibria and spontaneous homodimerization. J Mol Biol 384:279–297
Vonsattel J (2008) Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol 115:55–69
Wacker JL, Huang S-Y, Steele AD et al (2009) Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s Disease. J Neurosci 29:9104–9114
Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ (2004) Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol 11:1215–1222
Walsh R, Storey E, Stefani D, Kelly L, Turnbull V (2005) The roles of proteolysis and nuclear localisation in the toxicity of the polyglutamine diseases. A review. Neurotox Res 7:43–57
Walters RH, Murphy RM (2009) Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. J Mol Biol 393:978–992
Wang H, Lim PJ, Karbowski M, Monteiro MJ (2009) Effects of overexpression of Huntingtin proteins on mitochondrial integrity. Hum Mol Genet 18:737–752
Wang J, Wang C-E, Orr A, Tydlacka S, Li S-H, Li X-J (2008) Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J Cell Biol 180:1177–1189
Wang Y, Meriin AB, Costello CE, Sherman MY (2007) Characterization of proteins associated with polyglutamine aggregates: a novel approach towards isolation of aggregates from protein conformation disorders. Prion 1:128–135
Weiss A, Klein C, Woodman B et al (2008) Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington’s disease. J Neurochem 104:846–858
Wellington CL, Ellerby LM, Hackam AS et al (1998) Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem 273:9158–9167
Wexler NS (2004) Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci USA 101:3498–3503
Wiedemeyer R, Westermann F, Wittke I, Nowock J, Schwab M (2003) Ataxin-2 promotes apoptosis of human neuroblastoma cells. Oncogene 22:401–411
Williams AJ, Knutson TM, Colomer Gould VF, Paulson HL (2009) In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol Dis 33:342–353
Williamson TE, Vitalis A, Crick SL, Pappu RV (2010) Modulation of polyglutamine conformations and dimer formation by the N-terminus of huntingtin. J Mol Biol 396:1230–1295
Wood JD, Yuan J, Margolis RL et al (1998) Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol Cell Neurosci 11:149–160
Wyttenbach A, Swartz J, Kita H et al (2001) Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington’s disease. Hum Mol Genet 10:1829–1845
Yamada M, Sato T, Shimohata T et al (2001) Interaction between neuronal intranuclear inclusions and promyelocytic leukemia protein nuclear and coiled bodies in CAG repeat diseases. Am J Pathol 159:1785–1795
Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s Disease. Cell 101:57–66
Zhang S, Xu L, Lee J, Xu T (2002) Drosophila atrophin homolog functions as a transcriptional corepressor in multiple developmental processes. Cell 108:45–56
Zhou H, Li S-H, Li X-J (2001) Chaperone suppression of cellular toxicity of huntingtin is independent of polyglutamine aggregation. J Biol Chem 276:48417–48424
Acknowledgments
AW and SH thank the Medical Research Council for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hands, S.L., Wyttenbach, A. Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta Neuropathol 120, 419–437 (2010). https://doi.org/10.1007/s00401-010-0703-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0703-0