
Abstract
TDP-43 was recently identified as the major disease protein in neuronal inclusions in frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U). TDP-43 becomes redistributed from the nucleus to the cytoplasm, ubiquitinated, hyperphosphorylated and cleaved to generate C-terminal fragments, thereby linking mismetabolism of TDP-43 to the pathogenesis of FTLD-U. The function of TDP-43 is unclear, however it has been shown that TDP-43 might act as transcription repressor and activator of exon skipping through interaction with proteins of the heterogeneous nuclear ribonucleoprotein (hnRNP) family as well as a scaffold for nuclear bodies through interactions with survival motor neuron protein.
To investigate whether these binding partners might be associated with TDP-43 pathology, we studied the expression and localization of proteins of the hnRNP family (hnRNP A1, A2/B1, C1/C2) and SMN protein in affected brain regions in patients with sporadic and familial FTLD-U and normal control brains by immunohistochemistry and biochemical analysis. In contrast to TDP-43, no changes in subcellular distribution, no labeling of pathologic inclusions and no biochemical alterations were detectable for the tested hnRNPs and SMN in FTLD-U brains compared to controls. These results argue against a role of these binding partners in the pathogenesis of FTLD-U and emphasize the specificity of TDP-43 as marker for FTLD-U pathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTDL-U) is the most common pathologic change underlying the clinical syndrome of frontotemporal dementia (FTD) [12, 16]. FTLD-U is characterized by ubiquitin-positive, tau- and α-synuclein-negative neuronal cytoplasmic and nuclear inclusions as well as dystrophic neurites. Based on the morphology and laminar distribution of inclusions, at least four subtypes of FTLD-U pathology can be delineated [13, 19, 25]. While mutations in the progranulin gene have been found to be pathogenic for familial FTLD-U [3, 9], the TAR DNA binding protein 43 (TDP-43) has recently been identified as the major disease protein in the characteristic inclusions in sporadic and familial FTLD-U as well as sporadic ALS [22] and was rapidly confirmed by others [1, 10].
TDP-43 is a ubiquitously expressed, highly conserved protein [2], that is physiologically primarily located to the nucleus. However, in FTLD-U, TDP-43 is redistributed to form predominantly cytoplasmic and neuritic inclusions associated with dramatic reduction of nuclear TDP-43 immunoreactivity [1, 10, 20–22]. Biochemically, pathologic TDP-43 is abnormally hyperphosphorylated, ubiquitinated and cleaved to generate C-terminal fragments [20–22]. Thus, the current evidence links TDP-43 to the pathogenesis of FTLD-U, although the pathologic mechanisms and significance of these alterations of TDP-43 remain unknown.
TDP-43 may act as scaffold for nuclear bodies through interaction with survival motor neuron protein (SMN) [27]. SMN is a ubiquitously expressed protein located in the cytoplasm and nucleus concentrated in specific nuclear structures called gems (gemini of coiled bodies) [17]. Loss of functional SMN protein leads to autosomal recessive spinal muscular atrophy (SMA) [15, 18]. In addition, TDP-43 may also act as transcription repressor and activator of exon skipping [4, 6]. TDP-43 has been shown to be capable of binding directly to several proteins of the heterogeneous nuclear ribonucleoprotein (hnRNP) family, in particular hnRNP A1, A2/B1, A3, and C1/C2, which seem to be necessary for the splicing inhibitory activity of TDP-43 [5].
The aim of the present study was to determine whether these identified binding partners of TDP-43 were also components of FTLD-U inclusions. Therefore, we analyzed several proteins of the hnRNP family (hnRNP A1, A2/B1, C1/C2) and SMN protein in affected brain regions in patients with sporadic and familial FTLD-U and normal control brains by immunohistochemistry and biochemical analysis. In contrast to TDP-43, no changes in subcellular distribution, no labeling of pathologic inclusions and no biochemical alterations were detectable for the tested hnRNPs and SMN in FTLD-U brains compared to controls, arguing for a specific role of TDP-43 in the pathogenic pathway leading to FTLD-U.
Materials and methods
Immunohistochemistry
Tissue blocks from frontal and temporal neocortex and hippocampus from FTLD-U (n = 10) and control brains (n = 3) were fixed with either 70% ethanol in 150 mmol/l NaCl or phosphate-buffered 3.65% formaldehyde and embedded in paraffin. FTLD-U cases consisted of eight sporadic FTLD-U cases further classified as subtype 1 (n = 3), subtype 2 (n = 3) and subtype 3 (n = 2) according to Sampathu et al. [25], one case with a progranulin mutation (R493X), and one case with a VCP mutation (R155H). Immunohistochemistry was carried out as described previously using the avidin–biotin complex detection system (Vector Laboratories, Burlingame, CA, USA) and 3,3 diaminobenzidine as chromogen. Antigen retrieval was done by boiling the sections in 10 mmol/l citrate buffer (pH 6.0) in a microwave oven. Antibodies used in this study included affinity-purified rabbit polyclonal anti-TDP-43 (ProteinTech Group, Chicago, IL, USA), mouse monoclonal antibody (mAb) anti-hnRNP A1 clone 9H10 [7] and clone 4B10 [24], mAb anti-hnRNP A2/B1 clone DP3B3 [14], mAb anti-hnRNP C1/C2 clone 4F4 [8], and mAb anti-SMN clone 62E7 [30] and clone 2B1 [29]. Double-labeling immunofluorescence was performed as previously described using Alexa Fluor 488 and 594 conjugated secondary antibodies (Molecular Probes, Eugene, OR, USA).
Sequential biochemical fractionation and immunoblot analysis
Frozen tissues from frontal gray matter from FTLD-U (n = 10, including one case with progranulin gene mutation and one case with a VCP gene mutation) and control cases (n = 3) were used for the sequential extraction of proteins with buffers of increasing stringency, as described [22, 25]. Briefly, gray matter was extracted at 5 ml/g (v/w) with low-salt buffer (10 mmol/l Tris, pH 7.5, 5 mmol/l EDTA, 1 mmol/l dithiothreitol, 10% sucrose, and a cocktail of protease inhibitors), high salt-Triton X buffer (low salt buffer + 1% Triton X-100 + 0.5 mol/l NaCl), myelin flotation buffer (Triton X buffer containing 30% sucrose), and Sarkosyl buffer (low salt buffer + 1% N-lauroyl-sarcosine + 0.5 mol/l NaCl). The detergent-insoluble materials were extracted in 0.25 ml/g of urea buffer (7 mol/l urea, 2 mol/l thiourea, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 30 mmol/l Tris, pH 8.5).
For Western blot analysis, fractions from different samples were resolved by Tris-glycine 5 to 20% gradient sodium dodecyl sulfatepolyacrylamide gel electrophoresis, transferred to nitrocellulose and probed with primary and secondary antibodies (horseradish peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG (Jackson ImmunoReasearch, West Grove, PA, USA). Blots were developed with Renaissance Enhanced Luminol Reagents (NEN Life Science Product, Inc., Boston, MA, USA), and digital images were acquired using a Fujifilm Intelligent Darkbox II (Fuji Systems USA, Stamford, CT, USA).
Results
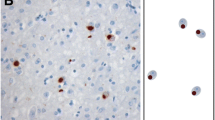
We performed immunohistochemistry (IHC) and double-label immunofluorescence studies on brain sections from hippocampus and neocortical brain regions from controls and FTLD-U cases, including different FTLD-U subtypes according to Sampathu et al. [25] as well as single familial cases with progranulin and VCP gene mutations. In accordance with previous studies, anti-TDP-43 IHC labeled numerous cytoplasmic inclusions in the dentate granule cells (Fig. 1a) as well as cytoplasmic and neuritic inclusions in affected cortical brain regions (Fig. 1f–h) in FTLD-U cases. While cells without inclusions showed a strong nuclear TDP-43 staining (arrows in Fig. 1a), there was a dramatic reduction in the labeling intensity of nuclear TDP-43 in inclusion-bearing cells (arrowheads in Fig. 1a). Standard IHC on adjacent tissue sections with antibodies against hnRNPs revealed a robust, predominantly nuclear staining for hnRNP A1 (Fig. 1b), A2/B1 (Fig. 1c), and C1/C2 (Fig. 1d) in FTLD-U brains and controls (data not shown), but no labeling of cytoplasmic inclusions in FTLD-U. Double-label immunofluorescence experiments confirmed that TDP-43 positive cytoplasmic and neuritic inclusions in FTLD-U were not immunoreactive for hnRNP A1 (Fig. 1f), A2/B1 (Fig. 1g) and C1/C2 (Fig. 1h).

Immunohistochemistry of TDP-43, hnRNPs and SMN in FTLD-U. a Sections of the dentate gyrus of FTLD-U case immunolabeled with antibody against TDP-43. In addition to physiological nuclear TDP-43 staining (arrow in a), cytoplasmic TDP-43 positive inclusions are detectable (arrowhead in a). Note the absence of nuclear TDP-43 staining in inclusion bearing cells. Adjacent sections were immunostained with antibodies against hnRNP A1 (b), hnRNP A2/B1 (c), hnRNP C1/C2 (d) and SMN protein (e) showing predominantly nuclear staining in all neurons for hnRNPs and nuclear and cytoplasmic staining for SMN, but no labeling of pathologic inclusions. Double fluorescence labeling confirming absence of hnRNP A1 (f, red), hnRNP A2/B1 (g, red) and C1/C2 (h, red) in TDP-43 positive cytoplasmic and neuritic inclusions (f–h, green). Scale bar in a corresponds to 20 μm (a–h)
Anti-SMN IHC demonstrated nuclear and cytoplasmic labeling of neurons in FTLD-U (Fig. 1e) and controls (data not shown), but also absence of immunoreactivity in pathologic TDP-43 positive inclusions in sporadic and familial FTLD-U.
Biochemically, FTLD-U brains show a disease-specific signature of pathologically altered TDP-43 in detergent-insoluble, urea-soluble proteins extracts. To examine the biochemical properties of hnRNPs and SMN in FTLD-U and controls compared to TDP-43, we performed Western blot analysis on protein samples sequentially extracted from cortical gray matter of FTLD-U and control brains (Fig. 2). Full-length TDP-43 was detected in all soluble and insoluble protein fraction in FTLD-U and controls, while additional proteins bands of ∼25 and 45 kD, as well as a high molecular smear were detected in urea samples from FTLD-U brains but not controls, as described [22]. hnRNPs and SMN proteins were predominantly found in the sarcosyl and urea fractions of controls and FTLD-U brains. There was some variation in the intensity of labeled bands among cases. However, no changes in solubility or banding pattern were detectable for hnRNPs and SMN protein between FTLD-U and control brains.

Biochemical analysis of TDP-43, hnRNPs and SMN in FTLD-U and controls. Proteins were sequentially extracted from frontal cortex of FTLD-U and controls with buffers of increasing strength and subjected to SDS-PAGE. Western blots were probed with antibodies against TDP-43, hnRNP A1, A2/B1, C1/C2 and SMN, as indicated to the left. In addition to physiological TDP-43 running as ∼43 kD band in soluble and insoluble brain fraction in control and FTLD-U, rabbit anti-TDP-43 showed pathological ∼ 25-kD (*), ∼45-kD (**) bands and high molecular smear (***) in the urea fraction of FTLD-U cases. No changes in solubility and/or changes in banding pattern were detectable for tested hnRNPs and SMN protein in FTLD-U brains compared to controls. Lane 1 low salt, 2 high salt with triton X-100; 3 sarkosyl, 4 urea fraction
Discussion
TDP-43 is the major disease protein in FTLD-U that forms the signature intraneuronal cytoplasmic and intranuclear inclusions predominantly in the frontal and temporal cortex. Pathologic TDP-43 in FTLD-U becomes hyperphosphorylated, ubiquitinated and cleaved to generate C-terminal fragments [1, 22]. Thus, current evidence links abnormal metabolism of TDP-43 to the pathogenesis of FTLD-U, although the pathologic mechanisms and the significance of these alterations are unknown.
Very little is known about the normal function of TDP-43. It is a highly conserved nuclear protein, as closely related proteins have been identified in Drosophila and Caenorhabditis [2]. TDP-43 contains two RNA-recognition motifs (RRMs) and a glycine-rich domain, both of which are functional modules commonly found in RNA-binding proteins, such as hnRNPs [11]. At least the first RRM of TDP-43 is required for RNA binding, thereby acting as transcription repressor and activator of exon skipping [4]. The C-terminal region of TDP-43 has been shown to be capable of interacting with several proteins of the hnRNP family, in particular hnRNP A1, A2/B1, C1/C2 and A3. This interaction leading to formation of a hnRNP-rich complex seems to be essential for the splicing inhibitory activity of TDP-43 [5]. In addition to the role as splicing factor, TDP-43 might act as a bridge between the various nuclear bodies, possibly through interaction with SMN protein [28], an ubiquitously expressed cytoplasmic and nuclear protein. SMN interacts with a number of proteins to form a stable complex which functions in the assembly and metabolism of small nuclear ribonucleoproteins (snRNP) [23, 26]. Mutations in the SMN gene leading to reduced levels of functional SMN are associated with autosomal recessive spinal muscular atrophy, a disease characterized by the degeneration of motor neurons in the anterior horn of the spinal cord [15, 18].
To investigate whether these identified interactors with TDP-43 might be associated with TDP-43 pathology in FTLD-U and might therefore help to elucidate the underlying mechanisms leading to neurodegeneration in FTLD-U and ALS, we performed immunohistochemical and biochemical studies on FTLD-U and control brains. We demonstrated that hnRNPs A1, A2/B1 and C1/C2 as well as SMN protein were not detectable in the TDP-43 positive inclusions in FTLD-U brains. While the nuclear staining of TDP-43 in inclusion bearing cells was dramatically reduced, in accordance with previous reports [1, 10, 21, 22], no obvious changes of staining intensity and subcellular distribution was seen for hnRNPs and SMN protein in cells with cytoplasmic and nuclear TDP-43 inclusions. In addition, no biochemical alterations of hnRNPs with respect to posttranslational modification and change in solubility were present in FTLD-U, in contrast to the pathological alterations of TDP-43 in insoluble protein fractions extracted from sporadic and familial FTLD-U brains [22].
In conclusion, we found no evidence that previously identified proteins interacting with TDP-43, including proteins from the hnRNP family and SMN protein, are involved in TDP-43 aggregation and FTLD-U pathogenesis. These results emphasize the specificity of TDP-43 as marker protein for FTLD-U pathology, however, further studies, including identification of additional binding partners of TDP-43, will need to address mechanistic aspects of TDP-43 aggregation and their role in the pathogenesis of neurodegeneration.
References
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda M (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis Biochem Biophys Res Commun 351:602–611
Ayala YM, Pantano S, D’Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE (2005) Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function J Mol Biol 348:575–588
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17 Nature 442:916–919
Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276:36337–36343
Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280:37572–37584
Buratti E, Brindisi A, Pagani F, Baralle FE (2004) Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8 and causes skipping of exon 9: a functional link with disease penetrance. Am J Hum Genet 74:1322–1325
Burd CG, Dreyfuss G (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science 265:615–621
Choi YD, Dreyfuss G (1984) Monoclonal antibody characterization of the C proteins of heterogeneous nuclear ribonucleoprotein complexes in vertebrate cells. J Cell Biol 99:1997–1204
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924
Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, Mann DM (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol (Berl) Epub
Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG (1993) hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem 62:289–321
Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, Chatterjee A, Hurtig HI, Karlawish JH, Rosen HJ, Van Deerlin V, Lee VM, Miller BL, Trojanowski JQ, Grossman M (2006) Frontotemporal dementia: clinicopathological correlations. Ann Neurol 59:952–962
Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu PH, Watts GD, Markesbery WR, Smith CD, Kimonis VE (2006) Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol 65:571–581
Kamma H, Horiguchi H, Wan L, Matsui M, Fujiwara M, Fujimoto M, Yazawa T, Dreyfuss G (1999) Molecular characterization of the hnRNP A2/B1 proteins: tissue-specific expression and novel isoforms. Exp Cell Res 246:399–411
Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16:265–269
Lipton AM, White CL 3rd, Bigio EH (2004) Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol (Berl) 108:379–385
Liu Q, Fischer U, Wang F, Dreyfuss G (1997) The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 90:1013–1021
Lorson CL, Hahnen E, Androphy EJ, Wirth B (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 96:6307–6311
Mackenzie IR, Baborie A, Pickering-Brown S, Plessis DD, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM (2006) Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol (Berl) 112:539–549
Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowsk JQ, Lee VM (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66:177–183
Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS (2007) TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 66:152–157
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Pellizzoni L, Yong J, Dreyfuss G (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science 298:1775–1779
Pinol-Roma S, Choi YD, Matunis MJ, Dreyfuss G (1988) Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 2:215–227
Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM (2006) Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 169:1343–1352
Wan L, Battle DJ, Yong J, Gubitz AK, Kolb SJ, Wang J, Dreyfuss G (2005) The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol Cell Biol 25:5543–5551
Wang HY, Wang IF, Bose J, Shen CK (2004) Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83:130–139
Wang IF, Reddy NM, Shen CK (2002) Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci USA 99:13583–13588
Wang J, Dreyfuss G (2001) A cell system with targeted disruption of the SMN gene: functional conservation of the SMN protein and dependence of Gemin2 on SMN. J Biol Chem 276:9599–9605
Wang J, Dreyfuss G (2001) Characterization of functional domains of the SMN protein in vivo. J Biol Chem 276:45387–45393
Acknowledgment
This work was funded by the National Institutes of Health (AG10124, AG17586) and the German Federal Ministry of Education and Research (01GI0505). VM-YL is the John H. Ware III Chair of Alzheimer’s Research and JQT is the William Maul Measey-Truman G. Schnabel, Jr., M.D. Professor of Geriatric Medicine and Gerontology. The authors would like to thank Christopher G. Dengler for help with immunohistochemistry and the families of patients who made this research possible.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Neumann, M., Igaz, L.M., Kwong, L.K. et al. Absence of heterogeneous nuclear ribonucleoproteins and survival motor neuron protein in TDP-43 positive inclusions in frontotemporal lobar degeneration. Acta Neuropathol 113, 543–548 (2007). https://doi.org/10.1007/s00401-007-0221-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-007-0221-x