
Abstract
Immunohistochemical detection of protein components of pathological inclusions is widely used for neuropathological diagnosis of neurodegenerative disorders. However, different antibodies and antigen unmasking methods may account for variability between research studies and thus may affect diagnostic accuracy. Using two different antibodies raised against either a segment (184–200 aa) or the full length of human recombinant brain-specific tubulin polymerization promoting protein TPPP/p25, we immunohistochemically screened neurodegenerative disorders, both with and without pathological α-synuclein structures. We tested three different epitope unmasking methods, we applied laser confocal microscopy to evaluate double immunolabelling, and we compared the amount of structures exhibiting TPPP/p25 and α-synuclein immunoreactivity. We demonstrate that there are a variety of staining patterns depending on the epitope retrieval method and antibody used. The antibody raised against aa 184–200 segment of TPPP/p25 is better in immunolabelling the majority of α-synuclein immunopositive neuronal and glial pathological profiles detectable in Parkinson’s disease, diffuse Lewy-body disease, and multiple system atrophy, in addition to immunostaining some extracellular huntingtin immunoreactive structures, lipofuscin, and neuromelanin particles. In contrast, the one raised against the full-length human recombinant TPPP/p25 is more suitable to immunodetect normal oligodendrocytes. Exposition of the segment aa 184–200 of TPPP/p25 in the aggregates of pathological inclusions renders this antibody a reliable marker of all types of α-synucleinopathies and suggests a role for TPPP/p25 in the aggregation process of some neurodegenerative conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intra- or extracellular accumulation of abnormal conformers of physiological proteins like β-amyloid derived from the amyloid precursor protein, the microtubule-associated protein tau, α-synuclein, prion protein, or proteins linked to trinucleotide repeat disorders like huntingtin characterizes neurodegenerative diseases [22]. Immunohistochemical detection of α-synuclein, phosphorylated tau, abnormal prion protein, and β-amyloid has become the histological technique of choice for the diagnosis [6]. However, as previous studies pointed out, variability across antibodies and antigen unmasking methods may account for the discrepancies between research studies and affect diagnostic accuracy (e.g. GFAP, ubiquitin, prion protein, α-synuclein) [5, 8, 12, 13]. This is of outmost importance when searching immunohistochemically for novel proteins, like TPPP/p25, related to pathological protein aggregates.
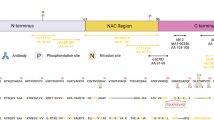
Tubulin polymerization promoting protein TPPP/p25 or p25α was originally co-purified with a tau kinase preparation from bovine brain [24]. In normal rat and human brain it was localized predominantly in oligodendrocytes [14, 18, 23, 25]. This protein must be distinguished from the extensively characterized p25 (hence introduction of the term TPPP/p25), which is a truncated form of p35 that deregulates cyclin-dependent kinase activity by causing prolonged activation and mislocalization of the kinase [20]. TPPP/p25 promotes the polymerization of tubulin into double-walled tubules and polymorphic aggregates [9]. It inhibits mitotic spindle assembly without affecting other cellular events like centrosome replication and segregation, nucleation of microtubules by the centrosomes, and nuclear growth [26]. In addition, it inhibits glycogen synthase kinase 3 that phosphorylates tau protein [16]. Purified recombinant human TPPP/p25 strongly stimulates the aggregation of α-synuclein in vitro [15].
Recently we demonstrated that TPPP/p25 is enriched in filamentous α-synuclein bearing Lewy-bodies of Parkinson’s (PD) and diffuse Lewy-body diseases (DLBD), as well as in glial cytoplasmic inclusions (Papp-Lantos bodies) of multiple system atrophy (MSA) [14]. Furthermore, it was reported in α-synuclein immunonegative neuronal inclusions in MSA [2, 10]. Previous studies on human diseased brain tissue agreed that TPPP/p25 is a marker of pathological α-synuclein immunoreactive structures [2, 14, 15], although the amount of immunolabelled neuronal inclusions of PD and DLBD was different [14, 15]. In our present study we evaluate antigen unmasking methods and antibodies raised against a small fragment and the full length of TPPP/p25. We extend the number of investigated neurodegenerative disorders to further specify the possible pathogenic pro-aggregatory role of TPPP/p25.
Materials and methods
We included three neuropathologically characterized cases each of Alzheimer’s disease (AD), DLBD, PD, MSA, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD, only one case), Pick’s disease (PiD), motor neuron disease (MND, five cases), argyrophilic grain disease (AgD), frontotemporal lobar degeneration with ubiquitin-only immunoreactive neuronal changes (FTLD-U), Huntington’s disease (HD), and control brains (Co, five cases). Diagnoses of controls comprise extraneural carcinoma, HIV infection without affecting CNS, and alcoholic encephalopathy. Data of cases and examined anatomical regions are summarized in Table 1.
Production of TPPP/p25 antibodies
Two different antibodies were used to immunodetect TPPP/p25 in human brain tissue.
Antibody TPPP/p25-A
Immunogen (peptide of 184–200 aa, from sequence of hp25) was synthesized by standard Fmoc strategy. Extra Cys amino acid was added at the first N-terminal position for aiming conjugation. After conjugation of the unblocked peptide to KHL, rats were immunized by standard protocol, with a 4-week interval of boosting injections. After the third boosting, blood samples of the animals were withdrawn and tested against rechp25 by ELISA, and by western blotting on human cortex extract. This antibody was described already in detail [14].
Antibody TPPP/p25-B
This antibody is raised against full-length human recombinant TPPP/p25. His-tagged rechp25 was purified by IMAC chromatography from E. coli lysate and was used as immunogen. The purity of the protein was controlled on SDS-PAGE. Rats were immunized against the protein according to the same protocol as detailed above, but without conjugation. After the third boosting, blood samples of the animals were withdrawn and tested against rechp25 by ELISA, and by western blotting on human cortex extract. Specificity of this antibody was described earlier [19]. We also tested it in Western blots against human cortex extracts (data not shown). For immunohistochemistry we omitted the primary antibody or we incubated the peptide with the TPPP-B antibody before immunostaining and we did not observe any immunoreactivity as detailed below (data not shown).
Pilot study of pre-treatments
Using adjacent sections of pons from MSA and mesencephalon from PD cases, we tested both anti-TPPP/p25 antibodies applying the following epitope retrieval methods: (A) 10 min autoclaving (100°C) in 0.01 M citrate buffer (pH 6.0); (B) pre-treatment (A) followed by 1 min formic acid (88%) treatment; and (C) 0.03% proteinase K treatment for 10 min.
Immunohistochemistry
We immunostained adjacent sections of different anatomical regions (Table 1) using two different TPPP/p25 antibodies (A and B, both rat polyclonal, 1:200, over night incubation) and two different α-synuclein antibodies (mouse monoclonal, 1:10.000, clone 4D6, immunogen: human α-synuclein, Signet, Dedham, MA, USA; rabbit polyclonal, 1:200, immunogen C-terminus aa 111–132, Sigma, St. Louis, MO, USA) using the same epitope retrieval including 10 min 0.01 M citrate buffer (pH 6.0) followed by 1 min 88% formic acid. Additionally, in AD, PSP, CBD, PiD, AgD we applied anti-phospho-tau (mouse monoclonal, clone AT8, 1:200 Pierce Biotechnology, Rockford, IL, USA), in FTLD-U and MND anti-ubiquitin (rabbit polyclonal, 1:200, Dako, Glostrup, Denmark), and in HD anti-huntingtin antibody (mouse monoclonal, 1:100, Chemicon, Temecula, CA, USA). As a secondary system we used the Envision kit (Dako, Glostrup, Denmark). In addition, for double immunolabelling we used GFAP (mouse monoclonal, 1:50, Chemicon, Temecula, CA, USA) and S100 (rabbit polyclonal, 1:100, Dako, Glostrup, Denmark) antibodies.
Double immunolabelling and laser confocal microscopy
Double immunofluorescent labelling was evaluated by a ZEISS LSM 510 confocal laser microscope. The fluorescent-labelled secondary antibody for TPPP/p25-A, and C was Alexa Fluor 633 goat anti-rabbit IgG (1:200, Molecular Probes Inc. Eugene, OR, USA), for ubiquitin and S100 was Alexa Fluor 488 goat anti-mouse IgG (1:200, Molecular Probes), and for AT8, GFAP, huntingtin, and alpha-synuclein Alexa Fluor 488 goat anti-mouse IgG (1:200, Molecular Probes). Primary antibodies were incubated over night. For TPPP/p25-A we applied rabbit anti-rat (1:200, Dako) as intermediary incubation step. We applied Sytox orange (1:8000, Molecular Probes) for 15 min to detect nuclear staining using Helium/Neon 546 nm laser. We used Argon 488 nm and Helium/Neon 633 nm lasers to elicit immunofluorescent staining. In order to control specific immunofluroescence we omitted the primary antibodies and used intermediary and/or secondary antibodies without detecting any fluorescence.
Quantitative evaluation
To evaluate the amount of TPPP/p25-A, B, and α-synuclein immunopositive pathological structures, we counted intra- and extracellular globular bodies in the same 1.0 × 1.0 cm framed temporal cortical area in adjacent sections in three DLBD cases using objective ×40. Similarly, we counted neuronal and glial cytoplasmic TPPP/p25-A, B, and α-synuclein immunoreactive profiles in the same 1.0 × 1.0 cm framed area in adjacent sections containing the pontine base in three MSA cases using objective ×40.
Results
Comparison of epitope unmasking methods
In our hands 10 min autoclaving (100°C) in 0.01 M citrate buffer (pH 6.0) followed by 1 min formic acid (88%) treatment (pre-treatment B) was the most efficient method for the immunodetection of pathological TPPP/p25 immunoreactive structures (Fig. 1a–c). Immunolabelling of oligodendrocytes was detectable with all three pre-treatments. Thus, to compare immunoreactivity for TPPP/p25-A and B we used pre-treatment B.

Testing of different epitope retrieval methods (a–c) and TPPP/p25 antibodies (d–r) in adjacent sections of pons of a MSA case (a–c, g–r) and mesencephalon of a PD case (d–f). Epitope retrieval methods include (a) 10 min autoclaving (100°C) in citrate buffer (pH 6.0), (b) the previous pre-treatment followed by 1 min formic acid (88%) treatment, and (c) 0.03% proteinase K treatment for 10 min. All photos represent antibody TPPP/p25-A and shows the same area with neurites and neuronal inclusions. Using the pre-treatment method shown in (b), TPPP/p25-A (e, h, k, n, q) gives better result when compared to immunoreactivity observed by using polyclonal anti-(-synuclein antibody (d, g, j, m, p), and TPPP/p25-B (f, i, l, o, r). Bar indicates 10 (m in a–c, 5 (m in d–i, m–o, and 30 (m in j–l and p–r
Comparison of antibodies
Results, including present and previous observations, are summarized in Tables 2 and 3.
In summary, TPPP/p25 immunoreactivity was observed in S100 immunopositive oligodendroglia-like cells and not in GFAP immunopositive astroglial cells (Fig. 2a, b) in all cases. TPPP/p25-A was less efficient in detecting oligodendroglial cells than antibody B. Lewy-bodies and Lewy-neurites in PD and DLBD cases (Fig. 1d–f), neuromelanin, and lipofuscin-like dots in all cases were immunostained predominantly with antibody TPPP/p25-A. It must be noted that anti-TPPP/p25-A immunolabelled practically all morphological types of pathological α-synuclein immunoreactive structures (Fig. 3). In contrast, TPPP/p25-B infrequently immunolabelled the rim of brainstem-type classical Lewy-bodies. This was never seen in cortical-type Lewy-bodies.

Double immunolabelling laser confocal microscopical images of S100 (a), GFAP (b), monoclonal anti-α-synuclein antibody (c, d), and anti-huntingtin (e) and anti-TPPP/p25-A (a–e). Sytox indicates the staining of nuclei. Bar indicates 10 μm in a–c; 5 μm. Note that TPPP/p25 co-localizes with S100 (a) and not with GFAP (b). There is a high degree overlap of TPPP/p25 and α-synuclein in glial cytoplasmic inclusions in MSA (c). In neuronal cytoplasmic inclusions, co-localization is present in contrast to nuclear pathological structures (arrowhead, d). In Huntington’s disease, TPPP/p25 is present only in some extracellular neuritic profiles and not in intranuclear huntingtin immunoreactive inclusions (e). Here, additional granular TPPP/p25-A immunoreactivity is seen in astroglia like cells

Immunostaining for TPPP/p25 in various pathological α-synuclein immunopositive neuronal structures, comprising (a) early dot-like accumulation of TPPP/p25 immunoreactivity (indicated by the white asterisk) beside neuromelanin granules, (b) classical Lewy-bodies, and (c) Lewy-neurites in a representative case of Parkinson’s disease. Similarly, in diffuse Lewy-body disease, different morphological variants of cortical Lewy-bodies (d, e) and α-synuclein immunopositive Lewy-neurites in the CA2/3 plexus (f) are strongly immunostained with anti-TPPP/p25. All photographs represent sections immunostained using anti-TPPP/p25-A antibody. Bars represent 10 μm in a–e and 25 μm in f
Both TPPP/p25 antibodies immunostained some granules in granulovacuolar degeneration, seen mostly in AD and PiD cases. In addition, glial and neuronal cytoplasmic inclusions and α-synuclein immunopositive threads in MSA cases were immunolabelled by both anti-TPPP/p25 antibodies (Figs. 1g–r, 2c, d). We observed immunoreactivity for neuronal nuclear inclusions in MSA cases very rarely with anti-TPPP/p25-A antibody, and these intranuclear inclusions were undetectable with anti-TPPP/p25-B antibody. Interestingly, huntingtin immunopositive extracellular globular immunodeposits, but not intranuclear inclusions, were occasionally co-immunostained with anti-TPPP/p25-A (Fig. 2e). This was not observed with two anti-α-synuclein and anti-TPPP/p25-B antibodies. In HD cases, in severely degenerated areas we noted granular TPPP/p25 immunoreactivity in astroglia-like cells not immunolabelled with anti-huntingtin (Fig. 2e).
Quantification of TPPP/p25 and α-synuclein immunoreactivity
The median value of intra- and extracellular globular bodies in DLBD immunolabelled with the monoclonal and polyclonal anti-α-synuclein antibodies was 127 and 128, and with anti-TPPP/p25-A antibody it was 103 (≈80% of α-synuclein immunopositive structures). The median value for glial cytoplasmic inclusions in MSA counted in sections immunostained with the monoclonal anti-α-synuclein antibody was 24, with polyclonal anti-α-synuclein antibody was 26, with anti-TPPP/p25-A was 20.6 (≈80%), and with TPPP/p25-B it was 14 (≈54%). The median value for neuronal cytoplasmic inclusions in MSA counted in sections immunostained with the monoclonal anti-α-synuclein antibody was 18, with the polyclonal anti-α-synuclein antibody was 23, and with the anti-TPPP/p25-A and B antibodies was 10 (≈50%) and none, respectively.
Discussion
In the present study we show that immunostaining for TPPP/p25 in various pathological structures in human brain depends on the antibody and pre-treatment used. This is similar for other antibodies, such as α-synuclein [5], and supports the need for harmonization of laboratory methodology in the evaluation of neurodegenerative disorders [1].
We demonstrate that one antibody, raised against aa 184–200 segment of TPPP/p25 is better in immunolabelling the majority of α-synuclein immunopositive neuronal and glial pathological profiles detectable in PD, DLBD, and MSA, while another, raised against full-length human recombinant TPPP/p25 is more suitable to immunodetect normal oligodendrocytes, rendering the latter antibody more convenient to evaluate disease states affecting oligodendrocytes (e.g. brain tumours [21], myelination) (Table 3). Since pre-treatment with formic acid, which we selected in our study, is well known to enhance the immunodetection of aggregated proteins and those showing amyloid features [11], it is likely that TPPP/p25 is also in an altered, aggregated state. The process of pathological TPPP/p25 accumulation in oligodendrocytes comprises the expansion [15] of the regularly seen perinuclear cytoplasmic immunoreactivity and further its deposition in typical glial cytoplasmic inclusions (Papp-Lantos bodies) of MSA brains. Interestingly, α-synuclein may be observed in oligodendrocytes of normal brains [17], thus, the α-synuclein pro-aggregatory role of TPPP/p25 may lead to widespread accumulation in the form of cytoplasmic inclusions. Although neuronal expression of TPPP/p25 is considered as less physiological or at least lower than in oligodendroglia, an aggregation enhancing effect of TPPP/p25 may lead to the appearance of neuronal cytoplasmic inclusions in MSA and Lewy-body disorders.
It can be speculated that the segment between aa 184–200 of TPPP/p25 is more exposed in protein aggregates. The distinct immunolabelling patterns observed by anti-TPPP/P25-A and anti-TPPP/p25-B suggest different conformational structures for this protein within oligodendroglial or neuronal cytoplasm (soluble or attached to microtubular network), and within protein aggregates in the inclusions. Analogously, processing of α-synuclein in glial cytoplasmic inclusions and Lewy-bodies is different [3]. This is based on the observation that in MSA brains SDS-insoluble α-synuclein is not found in contrast to an elevated level of SDS-soluble and even TBS-soluble fraction of α-synuclein [3]. SDS-insoluble α-synuclein depicts less aggregated/membrane-associated forms of α-synuclein. Thus, our TPPP/p25-A antibody more likely associates both with the SDS-soluble and insoluble α-synuclein, contrasting our TPPP/p25-B antibody that detects predominantly epitopes of SDS or TBS soluble α-synuclein-associated forms of TPPP/p25. Nonetheless, this merits further immunohistochemical and biochemical studies using monoclonal antibodies against different epitopes of TPPP/p25. However, there is an additional issue that is also related to the distinct feature of the two antisera. The segment aa 184–200 of TPPP/p25 is localized at the C-terminal part of the protein that includes partly the predicted tubulin binding domain (J. Ovádi, unpublished data). Theoretically, this segment may be masked by microtubules or other interacting components under physiological conditions. During the pathological process that involves alterations in the microtubular network, this epitope might be exposed. An alternative explanation could be that the polyclonal antibody raised against the whole protein does not recognize the segment aa 184–200 of TPPP/p25. Interestingly, we found co-localization of TPPP/p25-A and huntingtin in some extracellular globular structures in HD cases (Fig. 2e). α-Synuclein immunoreactivity of huntingtin polyglutamine aggregates in HD was reported using another anti-α-synuclein antibody [4]. The anti-α-synuclein antibodies used in our study did not show similar immunoreactivity. This emphasizes the importance of optimized and comparable laboratory methodology. In addition, it suggests that TPPP/p25 may be either recruited to huntingtin aggregates together with α-synuclein, or theoretically may enhance extranuclear aggregation of huntingtin. While there is a lack of immunodetection of TPPP/p25 in normal astrocytes, the detection of granular TPPP/p25 immunoreactivity in reactive astrocytes may be indicative of a low level of expression in these cells related to the microtubule system that degenerates and accumulates, or less likely astrocytes might phagocytose breakdown products of other cells, e.g. oligodendroglia.
The detection of TPPP/p25 immunoreactivity in cells bearing lipofuscin and granulovacuolar degeneration may be due to the degeneration or breakdown of the microtubule system of the neuronal cytoskeleton, which may allow access of antibodies to epitopes. This suggests that neurons also possibly harbour low levels of TPPP/p25, although not detected with these immunohistochemical procedures. Since α-synuclein may be cross-linked to substantia nigra neuromelanin in PD [7] and subcellular proteomics revealed TPPP/p25 as a component of neuromelanin granules [27], it is not surprising to find TPPP/p25 immunoreactivity in neuromelanin granules of substantia nigra neurons.
In summary, we demonstrate that TPPP/p25 is consistently associated with pathological α-synuclein profiles. We identify aa 184–200 as a more exposed segment of TPPP/p25 in pathological protein aggregates related predominantly to altered α-synuclein or less frequently huntingtin, or in some circumstances associated with early stages of tangle formation composed of hyperphosphorylated tau [14]. This suggests a possible role of TPPP/p25 in protein aggregation during some neurodegenerative processes.
References
Alafuzoff I, Pikkarainen M, Al-Sarraj S, Arzberger T, Bell J, Bodi I, Bogdanovic N, Budka H, Bugiani O, Ferrer I, Gelpi E, Giaccone G, Graeber MB, Hauw JJ, Kamphorst W, King A, Kopp N, Korkolopoulou P, Kovacs GG, Meyronet D, Parchi P, Patsouris E, Preusser M, Ravid R, Roggendorf W, Seilhean D, Streichenberger N, Thal DR, Kretzschmar H (2006) Interlaboratory comparison of assessments of Alzheimer disease-related lesions: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol 65:740–757
Baker KG, Huang Y, McCann H, Gai WP, Jensen PH, Halliday GM (2006) P25alpha immunoreactive but alpha-synuclein immunonegative neuronal inclusions in multiple system atrophy. Acta Neuropathol (Berl) 111:193–195
Campbell BC, McLean CA, Culvenor JG, Gai WP, Blumbergs PC, Jakala P, Beyreuther K, Masters CL, Li QX (2001) The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J Neurochem 76:87–96
Charles V, Mezey E, Reddy PH, Dehejia A, Young TA, Polymeropoulos MH, Brownstein MJ, Tagle DA (2000) Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci Lett 289:29–32
Croisier E, MRes DE, Deprez M, Goldring K, Dexter DT, Pearce RK, Graeber MB, Roncaroli F (2006) Comparative study of commercially available anti-alpha-synuclein antibodies. Neuropathol Appl Neurobiol 32:351–356
Dickson DW (2005) Required techniques and useful molecular markers in the neuropathologic diagnosis of neurodegenerative diseases. Acta Neuropathol (Berl) 109:14–24
Fasano M, Giraudo S, Coha S, Bergamasco B, Lopiano L (2003) Residual substantia nigra neuromelanin in Parkinson’s disease is cross-linked to alpha-synuclein. Neurochem Int 42:603–606
Halliday GM, Cullen KM, Kril JJ, Harding AJ, Harasty J (1996) Glial fibrillary acidic protein (GFAP) immunohistochemistry in human cortex: a quantitative study using different antisera. Neurosci Lett 209:29–32
Hlavanda E, Kovacs J, Olah J, Orosz F, Medzihradszky KF, Ovadi J (2002) Brain-specific p25 protein binds to tubulin and microtubules and induces aberrant microtubule assemblies at substoichiometric concentrations. Biochemistry 41:8657–8664
Jellinger KA (2006) P25alpha immunoreactivity in multiple system atrophy and Parkinson disease. Acta Neuropathol (Berl) 112:112
Kitamoto T, Ogomori K, Tateishi J, Prusiner SB (1987) Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest 57:230–236
Kovacs GG, Head MW, Hegyi I, Bunn TJ, Flicker H, Hainfellner JA, McCardle L, Laszlo L, Jarius C, Ironside JW, Budka H (2002) Immunohistochemistry for the prion protein: comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol 12:1–11
Kovacs GG, Flicker H, Budka H (2003) Immunostaining for ubiquitin: efficient pretreatment. Neuropathol Appl Neurobiol 29:174–177
Kovacs GG, Laszlo L, Kovacs J, Jensen PH, Lindersson E, Botond G, Molnar T, Perczel A, Hudecz F, Mezo G, Erdei A, Tirian L, Lehotzky A, Gelpi E, Budka H, Ovadi J (2004) Natively unfolded tubulin polymerization promoting protein TPPP/p25 is a common marker of alpha-synucleinopathies. Neurobiol Dis 17:155–162
Lindersson E, Lundvig D, Petersen C, Madsen P, Nyengaard JR, Hojrup P, Moos T, Otzen D, Gai WP, Blumbergs PC, Jensen PH (2005) p25alpha Stimulates alpha-synuclein aggregation and is co-localized with aggregated alpha-synuclein in alpha-synucleinopathies. J Biol Chem 280:5703–5715
Martin CP, Vazquez J, Avila J, Moreno FJ (2002) P24, a glycogen synthase kinase 3 (GSK 3) inhibitor. Biochim Biophys Acta 1586:113–122
Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K (2002) Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp Neurol 176:98–104
Nishie M, Mori F, Houzen H, Yamaguchi J, Jensen PH, Wakabayashi K (2006) Oligodendrocytes within astrocytes (“emperipolesis”) in the cerebral white matter in hepatic and hypoglycemic encephalopathy. Neuropathology 26:62–65
Olah J, Tokesi N, Vincze O, Horvath I, Lehotzky A, Erdei A, Szajli E, Medzihradszky KF, Orosz F, Kovacs GG, Ovadi J (2006) Interaction of TPPP/p25 protein with glyceraldehyde-3-phosphate dehydrogenase and their co-localization in Lewy bodies. FEBS Lett 580:5807–5814
Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402:615–622
Preusser M, Lehotzky A, Budka H, Ovádi J, Kovács GG (2006) TPPP/p25 in brain tumours: expression in non-neoplastic oligodendrocytes but not oligodendroglioma cells. Acta Neuropathol (Berl) (in press)
Prusiner SB (2001) Shattuck lecture–neurodegenerative diseases and prions. N Engl J Med 344:1516–1526
Skjoerringe T, Lundvig DM, Jensen PH, Moos T (2006) P25alpha/Tubulin polymerization promoting protein expression by myelinating oligodendrocytes of the developing rat brain. J Neurochem 99:333–342
Takahashi M, Tomizawa K, Ishiguro K, Sato K, Omori A, Sato S, Shiratsuchi A, Uchida T, Imahori K (1991) A novel brain-specific 25 kDa protein (p25) is phosphorylated by a Ser/Thr-Pro kinase (TPK II) from tau protein kinase fractions. FEBS Lett 289:37–43
Takahashi M, Tomizawa K, Fujita SC, Sato K, Uchida T, Imahori K (1993) A brain-specific protein p25 is localized and associated with oligodendrocytes, neuropil, and fiber-like structures of the CA3 hippocampal region in the rat brain. J Neurochem 60:228–235
Tirian L, Hlavanda E, Olah J, Horvath I, Orosz F, Szabo B, Kovacs J, Szabad J, Ovadi J (2003) TPPP/p25 promotes tubulin assemblies and blocks mitotic spindle formation. Proc Natl Acad Sci USA 100:13976–13981
Tribl F, Marcus K, Meyer HE, Bringmann G, Gerlach M, Riederer P (2006) Subcellular proteomics reveals neuromelanin granules to be a lysosome-related organelle. J Neural Transm 113:741–749
Acknowledgment
We are grateful to Gerda Ricken and Helga Flicker for their technical assistance. This work was supported in part by EU Grant FP6, BNEII No LSHM-CT-2004-503039, by the Austrian-Hungarian Intergovernmental Cooperation (A14/04), by FP6-2003-LIFESCIHEALTH-I: Bio-Sim, by NKFP-MediChem2 1/A/005/2004, and by OTKA T-046071 to JO.GGK receives Bolyai fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kovács, G.G., Gelpi, E., Lehotzky, A. et al. The brain-specific protein TPPP/p25 in pathological protein deposits of neurodegenerative diseases. Acta Neuropathol 113, 153–161 (2007). https://doi.org/10.1007/s00401-006-0167-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-006-0167-4