
Abstract
The pathological distinctions between the various clinical and pathological manifestations of frontotemporal lobar degeneration (FTLD) remain unclear. Using monoclonal antibodies specific for 3- and 4-repeat isoforms of the microtubule associated protein, tau (3R- and 4R-tau), we have performed an immunohistochemical study of the tau pathology present in 14 cases of sporadic forms of FTLD, 12 cases with Pick bodies and two cases without and in 27 cases of familial FTLD associated with 12 different mutations in the tau gene (MAPT), five cases with Pick bodies and 22 cases without. In all 12 cases of sporadic FTLD where Pick bodies were present, these contained only 3R-tau isoforms. Clinically, ten of these cases had frontotemporal dementia and two had progressive apraxia. Only 3R-tau isoforms were present in Pick bodies in those patients with familial FTLD associated with L266V, Q336R, E342V, K369I or G389R MAPT mutations. Patients with familial FTLD associated with exon 10 N279K, N296H or +16 splice site mutations showed tau pathology characterised by neuronal neurofibrillary tangles (NFT) and glial cell tangles that contained only 4R-tau isoforms, as did the NFT in P301L MAPT mutation. With the R406W mutation, NFT contained both 3R- and 4R-tau isoforms. We also observed two patients with sporadic FTLD, but without Pick bodies, in whom the tau pathology comprised only of 4R-tau isoforms. We have therefore shown by immunohistochemistry that different specific tau isoform compositions underlie the various kinds of tau pathology present in sporadic and familial FTLD. The use of such tau isoform specific antibodies may refine pathological criteria underpinning FTLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Frontotemporal lobar degeneration (FTLD) describes a clinically and pathologically heterogeneous group of forms of dementia that have onset of illness usually between ages of 35 and 75 years and affect males and females equally [41, 56]. A previous family history of a similar disorder occurs in about half of patients [41, 53, 56] and in many such familial cases a mutation in the gene encoding the microtubule associated protein, tau (MAPT) on chromosome 17 seems causal (see Ref. [15, 32] for recent reviews). To date, about 35 causal MAPT mutations in around 150 families have been identified and the term frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) was adopted [12] to accommodate the clinical and genetic features of such cases. At autopsy, patients with FTLD generally show atrophy of the frontal and temporal lobes of the brain associated with a degeneration and loss of large pyramidal neurons from such regions irrespective of clinical subtype or family history [41, 56]. However, under this umbrella of pathological change, histopathological differences occur [41, 55, 62] and over the years there have been several attempts at classification based upon microscopic appearances.
In the first case reports, made over a century ago by Arnold Pick, the characterising features of intraneuronal argyrophilic inclusions (Pick bodies) and swollen or ballooned neurons (Pick cells) were described and the eponym Pick’s disease was coined to distinguish such cases from those of Alzheimer’s disease where senile plaques and neurofibrillary tangles were the key pathology. However, recognizing that clinically similar forms of FTLD occurred without such Pick- or Alzheimer-type changes being present led to the scheme by Constantinidis [7] in which three variants of FTLD were identified: one with Pick bodies and Pick cells (type A), one with only Pick cells (type B) and one with neither (type C). Later immunohistochemical studies [33] indicated either a Pick-type of histology based on the presence of tau-positive Pick bodies (equivalent to Constantinidis type A) or a microvacuolar-type histology in which no tau intraneuronal inclusions (Pick bodies) were seen (equivalent to Constantinidis types B and C). Recent surveys [22, 31, 37, 41, 55, 62] indicate that about half of cases of FTLD show a histopathology based on the accumulation of insoluble aggregates of tau protein within neurons and glial cells of the cerebral cortex and hippocampus. In most other cases, termed FTLD-U, tau negative but ubiquitin positive inclusions usually occur within cerebral cortex and hippocampus and when clinical motor neuron disease (MND) is also present the term FTLD-MND can be ascribed. Sometimes, neither tau nor ubiquitin inclusions are seen; such cases have been labelled as dementia lacking distinctive histology. These observations are broadly in line with the recommendations of a consensus conference held in 2001 in an attempt to establish internationally accepted clinical and neuropathological criteria for FTLD [34]. Nonetheless, at the time such criteria were put forward it was recognized that they were likely to be interim and subject to review in the light of expanding knowledge of this disorder—a point reiterated in some most recent surveys [37, 55]
Tau-based pathology in FTLD can occur in either sporadic or familial cases. In sporadic cases of FTLD, tau pathology usually takes the form of Pick bodies and Pick cells, though in such cases some tau positive glial cells and dystrophic neurites can often be seen [66], though if cases of corticobasal degeneration (CBD), progressive supranuclear palsy (PSP) or argyrophilic grain disease are included under the rubric of FTLD as has been the case in certain recent surveys [22, 31, 37] a neurofibrillary tangle-based tau pathology becomes more common.
In some familial cases (i.e., those with FTDP-17) with missense mutations within coding regions of exons 1, 9, 11, 12 and 13 of MAPT there are swollen nerve cells and rounded neuronal inclusions within large and small pyramidal neurones of the cerebral cortex and pyramidal and granule cells of the hippocampus reminiscent of the Pick bodies seen in sporadic disease [4, 14, 23, 29, 30, 39, 42, 45, 47, 50, 52]. Such mutations affect all six isoforms of tau, generating mutated tau molecules that (variably) lose their ability to interact with microtubules [20, 21, 23, 39, 42, 50, 51], increasing their propensity to self-aggregate into fibrils [20, 21, 23, 42, 47, 51]. Other MAPT mutations cluster around, or lie within a predicted regulatory stem loop structure of a splice acceptor domain of MAPT pre-mRNA that determines the inclusion or exclusion of exon 10 by alternative splicing during gene transcription [2, 3, 5, 6, 9, 11, 17, 19, 20, 24, 25, 35, 40, 43, 46, 48, 52, 58–60], destabilizing the stem loop [24, 58] or strengthening [11, 18, 20] or destroying [11] splice enhancing, or splice silencing [11, 59] elements in the 5′ region of exon 10. Such cases show insoluble aggregated tau deposits as neurofibrillary tangle-like structures within large and smaller pyramidal cells of cortical layers III and V and prominently within glial cells in the deep white matter, globus pallidus and internal capsule [2, 17, 19, 35, 43, 46, 51, 58–60]. However, other exon 10 mutations do not affect the splicing of exon 10 [11, 24] but induce conformational changes in tau molecules containing exon 10 that interfere with microtubule function and lead to aggregation of the mutated tau into neurofibrillary tangles [18, 36, 59].
Although the brain tau isoform composition has been extensively analysed by western blotting both in cases of sporadic FTLD where Pick bodies [1, 8, 37, 53, 61, 65] or Pick-like bodies [37] are present and in many of the cases with FTDP-17 [2–6, 9, 14, 17, 19, 23, 25, 29, 30, 35, 36, 39, 42, 43, 45–47, 50, 52, 59, 60, 62, 65], certain ambiguities remain. For example, in cases of sporadic FTLD where Pick bodies are seen in histology, most western blotting studies have detected only tau isoforms with 3-repeat microtubule binding domains (3R-tau) [8, 37, 54], though other investigations have shown certain cases to show tau isoforms with both 3R-tau and 4-repeat microtubule binding domains (4R-tau) [1, 37, 62, 66] and in yet others only 4R-tau is seen [37, 62, 66]. Such disparities have led to controversies as to whether Pick bodies are composed of only 3R-tau, only 4R-tau or an admixture of 3R- and 4R-tau isoforms. Similarly, in some cases of FTDP-17 with Pick bodies (e.g., K257T MAPT mutation Ref. [50]) western blotting has likewise shown only 3R-tau to be present, whereas in other cases (e.g., L266V, G272V, G342V and G389R MAPT mutations) both 3R-tau and 4R-tau isoforms are seen [4, 14, 23, 31, 45]. One possible explanation for these apparent inconsistencies may lie in differing anatomical compartmentalizations of 3R- and 4R- tau isoforms between cases, a distinction that is lost upon the tissue homogenization required for western blotting. Specific immunohistological patterns associated with 3R- and 4R-tau isoforms cannot be distinguished using phospho-dependent and phospho-independent tau antibodies that recognize tau epitopes shared by all six tau isoforms.
In the present study, therefore, we have investigated in situ, using monoclonal antibodies specific for 3R- and 4R-tau isoform species, the isoform composition of the tau histopathological changes in 14 cases of sporadic FTLD and 27 cases of familial FTLD associated with 11 different mutations in MAPT, for which tau isoform patterns on western blotting have already been reported, in order to reconcile such inconsistencies and to provide further insight into disease classification by the use of tau antibodies not available when the last pathological criteria [34] were proposed.
Materials and methods
Brain tissues from 14 cases with sporadic FTLD (cases #1–14), in 12 of whom (cases #1–12) previous pathological investigations had shown Pick bodies to be present and five cases of familial FTLD (cases #30–34) with exon 10 +16 MAPT mutation were obtained from the Manchester Brain Bank. Seven of the sporadic FTLD cases (cases #1, 2, 4–7 and 13) had been included (as cases # 4–8, 10 and 12, respectively) in a previous study [66] investigating tau isoform composition in a series of 14 cases of FTLD. Eight of the sporadic FTLD cases (cases #1, 2, 4–7, 9 and 13) had been included (as cases #10, 11, 12, 13, 14, 15, 17 and 16, respectively) in a previous study of ours [62] also investigating tau isoform composition in FTLD. Tissues from the other 22 familial FTLD cases with MAPT mutations were kindly supplied in collaboration by colleagues from different centres across the world. Selected clinical and pathological details for all cases are given in Table 1. Full clinical and pathological descriptions for 25 of the 27 cases with MAPT mutations have been previously reported by the originating authors (see Table 1 for details of citation); the other two cases remain unreported to date.
Serial sections were cut at a thickness of 6 μm from formalin-fixed, wax-embedded blocks of frontal cortex (BA 8/9) from all 14 sporadic FTLD cases and 27 familial FTLD cases with MAPT mutations and from temporal cortex (BA 21/22) to include the hippocampus in the 14 sporadic FTLD cases alone and mounted onto APES-coated slides. One set of sections was immunostained for insoluble pathological tau proteins by a standard immunoperoxidase method using the phospho-dependent tau antibody AT8 (1:750) (Innogenetics, Belgium). AT8 antibody is raised against the phosphorylated Ser 202/Thr 205 epitope and immunoreacts with PHF-tau in AD [16]. It will detect all isoforms of tau in which this epitope is phosphorylated. Other sets of sections were stained with the 3R-tau specific monoclonal antibody RD3 [10] (1:3000; Upstate, Dundee, UK) and the 4R-tau specific monoclonal antibody ET3 (gift of P Davies, 1:100) as described [10]. Briefly, sections were deparaffinised in xylene and rehydrated in decreasing concentrations of alcohol. Endogenous peroxidase activity was blocked with 0.3% H2O2 in methanol for 10 min. Sections were pressure cooked for 10 min in 0.01 M citrate buffer pH6.0. Sections were incubated in 10% non-fat milk to block non-specific staining, then with the primary antibodies RD3 and ET3 for 1 h at room temperature. This was followed by several washes in PBS and treatment with biotinylated anti-mouse (Dako 1:200) for 30 min and ABC (Dako) for 30 min. Peroxidase activity was developed with diaminobenzidine/ H2O2 solution [10].
The specificity of ET3 has been demonstrated previously in Western blots of recombinant 3R- and 4R-tau [27]. It has also been characterised in immunohistochemical studies of argyrophilic grain disease [13] and other tauopathies [23].
Results
Sporadic FTLD
Semi-quantitative rating data for AT8, ET3 and RD3 immunostaining in the 14 sporadic FTLD cases is given in Table 2.
Cases with Pick bodies
Of the 14 cases with sporadic FTLD, 12 cases (cases #1–12) displayed Pick-type histology. Pick bodies were identified as defined by Kertesz et al. [28], as round or oval, compact intracytoplasmic neuronal inclusions, stained by Bielschowsky but not by Gallyas, tau-immunoreactive and located in dentate fascia, hippocampus and cerebral cortex. Clinically, nine cases (cases #1–7, 9 and 10) showed typical frontotemporal dementia, whereas case #8 had suffered from progressive aphasia and cases #11 and 12 from progressive apraxia. In 11 of these 12 cases (cases #1–4, 6–12), numerous Pick bodies were widespread within frontal and temporal cortex, chiefly in layers two and four and within dentate gyrus granule cells (Fig. 1a) and pyramidal cells of the hippocampus. However, in one elderly case (aged 83 years) (case #5) Pick bodies were widely present only in the granule cells of the dentate gyrus and pyramidal cells of the hippocampus, but less so within temporal neocortex. In this case occasional neurons showing diffuse cytoplasmic tau immunoreactivity (pretangles) and neurofibrillary tangle-like structures were seen in the frontal and temporal cortex (see also two cases reported by Mott et al. [37]. No other cases displayed such AD-type neurofibrillary changes. In cases #3, 4, 6 and 9, AT8-immunoreactive astrocytes were present within the cerebral cortex and white matter and a fine meshwork of dystrophic neurites (threads) was seen. These findings are typical of those seen using anti-tau antibodies such as AT8 which detect all hyperphosphorylated isoforms of tau and have been described on many occasions previously (e.g., Ref. [62, 66]).

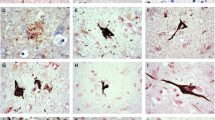
Tau pathology in patients with sporadic FTLD. In patient #4 (a–c) Pick bodies within granule cells of the dentate gyrus of the hippocampus are immunoreactive with AT8 (a) and RD3 (b) antibodies, but not with ET3 (c) antibody. In patient #13 without Pick bodies (d–f) neurones with amorphous tau deposits or neurofibrillary tangle-like structures are immunoreactive with AT8 (d) and ET3 (f) antibodies, but not with RD3 antibody (e). Immunoperoxidase-haematoxylin, AT8 antibody (a, d), RD3 antibody (b, e), ET3 antibody (c, f). All 250 times microscope magnification
In all 12 cases, the Pick bodies were strongly stained with RD3 antibody (Fig. 1b), usually to approximately the same extent as with AT8, but none were stained with ET3 (Fig. 1c). In case #5 a minority of the Pick bodies within temporal neocortex were also stained with RD3, as were a few of the NFT-like structures in the frontal cortex, though neither were stained with ET3. In case #6 rare glial cells were diffusely stained with ET3.
Cases without Pick bodies
In the other two sporadic FTLD cases (cases #13 and 14) typical Pick bodies were absent from hippocampal dentate gyrus granule cells and pyramidal cells. In these, AT8 staining revealed numerous cells in both hippocampal regions containing fine, granular deposits of tau protein, which sometimes had NFT-like appearance. In both cases a few of the pyramidal cells, but none of the dentate gyrus granule cells, were stained with ET3, but not RD3, antibody. In frontal and temporal cortex fine amorphous deposits of tau protein which sometimes adopted a rounded, ring or crescent shape, other times a more NFT-like structure, were seen with AT8 staining (Fig. 1d). The amorphous tau deposits were sometimes similarly stained with RD3 and may be associated with ribosomes (Nissl bodies) (see also Papasozomenos Ref. [44]), whereas the rounded, ring or crescent shaped inclusions were only stained with ET3 (Fig. 1f) and not RD3 (Fig. 1e). In neither case were glial cell inclusions, or astrocytic plaques, of the type seen in CBD, present. These cases presented clinically with FTD and showed no features to distinguish them from the other 12 sporadic FTLD cases.
Familial FTLD
Semi-quantitative rating data for AT8, ET3 and RD3 immunostaining in the 27 familial FTLD cases is given in Table 3.
MAPT mutations in exons 9, 12 and 13
In cases #15 (exon 9, L266V), #36 (exon 12, Q336R), #37 (exon 12, E342V) and #38 (exon 12, K369I), many neurons containing diffuse deposits of tau protein were seen in frontal cortex in AT8 immunostained sections. In some cells, in each mutation, the insoluble tau was aggregated into rounded structures resembling the Pick bodies seen in the sporadic FTLD cases (Fig. 2a). These Pick-like bodies were likewise stained with RD3 antibody (Fig. 2b) but not (or only very rarely so) with ET3 antibody (Fig. 2c). However, in L266V and E342V MAPT mutations numerous diffusely stained astrocytes were seen with AT8 antibody (Fig. 2a). These were also stained with ET3 (Fig. 2c), but not RD3 (Fig. 2b), antibody. In case #40 (exon 13, G389R), a few or a moderate number of pyramidal neurons with amorphous tau deposits were seen in AT8 stained sections, these sometimes having a rounded appearance resembling Pick bodies. Many thread-like structures were also seen within deeper layers of the frontal cortex. Occasionally, the Pick-like bodies were stained with RD3 antibody, but not with ET3.

Tau pathology in patients with familial FTLD. In patient #15 with MAPT L266V mutation (a–c), Pick bodies within pyramidal cells of the frontal cortex are immunoreactive with AT8 (a) and RD3 (b) antibodies, but not with ET3 (c) antibody. However, there are also numerous astrocytes within grey matter of frontal cortex that are immunoreactive with AT8 antibody (a), some of which are also reactive with ET3 antibody (c), but not with RD3 (b) antibody. In patient #40 with MAPT R406W mutation (d–f) neurones show amorphous tau deposits or neurofibrillary tangle-like structures that are immunoreactive with AT8 (d), RD3 (e) and ET3 (f) antibodies. In this patient numerous neuropil threads are seen but these are reactive only with AT8 antibody (d). Immunoperoxidase-haematoxylin, AT8 antibody (a, d), RD3 antibody (b, e), ET3 antibody (c, f). All 250 times microscope magnification
In cases #40 and 41 (exon 13, R406W) many nerve cells containing diffuse cytoplasmic tau deposits or well-formed NFT were seen in AT8 immunostained sections (Fig. 2d). These NFT were strongly stained with RD3 (Fig. 2e), but less so with ET3 (Fig. 2f).
MAPT mutations in exon 10
A similar tau pathology was seen in the 8 cases with N279K MAPT mutation in exon 10 (patients #16–23), albeit these cases were from two different kindreds and of different ethnic background (Japanese, cases #16 and 17 and North American/Caucasians, cases # 18–23). This was characterised in AT8 immunostained sections by the presence of pyramidal neurons in frontal cortex containing either amorphous cytoplasmic tau deposits or deposits resembling NFT (Fig. 3a), along with variable numbers of ballooned neurons and widespread glial cell tangles within grey matter and, especially within, white matter (Fig. 3d). These neuronal and glial cell tau deposits were strongly stained with ET3 (Fig. 3c, f) antibody, but not, or only rarely so in occasional cases, with RD3 antibody (Fig. 3b, e). The patient with MAPT N296H mutation (case #24) and those with MAPT exon 10 +16 splice site mutation (cases #30–35) showed similar changes to those with MAPT N279K mutation.

Tau pathology in patients with familial FTLD. In patient #23 with MAPT N279K mutation (a–f), pyramidal cells of the frontal cortex are immunoreactive with AT8 (a) and ET3 (c) antibodies, but not with RD3 (b) antibody. Similarly, numerous glial cells within white matter of the frontal cortex contain tangles that are immunoreactive with AT8 antibody (d) and most of which are also reactive with ET3 antibody (f), but not with RD3 (e) antibody. In patient #25 with MAPT P301L mutation (g–i) neurones show amorphous tau deposits or neurofibrillary tangle-like structures that are immunoreactive with AT8 (g) and ET3 (i) antibodies, but not with RD3 antibody (h). In this patient numerous neuropil threads reactive only with AT8 antibody (g) are seen. Immunoperoxidase-haematoxylin, AT8 antibody (a, d, g), RD3 antibody (b, e, h), ET3 antibody (c, f, i). All 250 times microscope magnification
In the case with MAPT S305S mutation (case #29), a moderate number of small curvilinear, ring or crescent-shaped bodies were seen in neurons on AT8 staining. These were also stained with ET3 but not RD3 antibody. In cases with P301L mutation (cases #25–28), a moderate number of, or many, diffuse cytoplasmic and NFT-like tau aggregates were seen in pyramidal neurons on AT8 immunostaining, along with many neuropil threads (Fig. 3g). Many of the AT8-immunoreactive neurons were also stained with ET3 antibody, adopting a NFT-like appearance in some instances, but in others a perinuclear ring or crescent shaped structure was seen (Fig. 3i). In cases #25 and 26 none of the AT8-positive structures in nerve cells were stained with RD3 antibody (Fig. 3h), though in cases #27 and 28, respectively, rare or occasional nerve cells were RD3 reactive.
Discussion
In the present report, we have investigated by immunohistochemistry the tau isoform composition of the tau pathology present in 14 cases of sporadic FTLD and 27 cases of familial FTLD association with mutations in MAPT employing antibodies specific to 3R and 4R isoforms of tau.
Biochemical analysis of sarkosyl-insoluble tau extracted from brains of patients with sporadic FTLD where Pick bodies, or Pick body-like structures, are known to be present has variably shown only 3R-tau [8, 37, 54], a mixture of 3R- and 4R-tau [1, 37, 62, 66] or only 4R-tau [47, 62, 66] to be present. We find here that whenever Pick bodies are present in hippocampus and cerebral cortex in sporadic FTLD cases, these can only be detected using the antibody to 3R-tau. These findings build on an earlier report of ours [10] in which only a single case with Pick bodies was investigated and are in agreement with other immunohistochemical studies which have employed 4R-tau, but not 3R-tau, specific antibodies [1, 26]. Zhukareva and colleagues [66] investigated 14 cases of FTLD by immunohistochemistry using (different to the present study) 3R- and 4R-tau specific antibodies. In 12 of these patients and consistent with present findings, the Pick bodies were reactive only with 3R-tau antibody. However, in seven of these cases 4R-tau species could be detected on western blot, though by immunohistochemistry this seemed to be localized in occasional cells bearing neurofibrillary tangles rather than Pick bodies and in neuropil threads [66]. Nonetheless, the other two cases in this latter study [66] displayed Pick bodies immunoreactive only to 4R-tau species and in these only 4R-tau was detected on western blot. Case #8 in the study by Zhukareva et al. [66] was case #13 in this present study. Consistent with Zhukareva et al. [66], we also found the tau pathology in this particular case reactive only with 4R-tau antibody. However, we are not convinced that these 4R-tau immunoreactive structures are indeed Pick bodies for the following reasons. First, they had a curvilinear, crescent or ring-shaped profile distinct in appearance from the rounded or oval, compact appearance associated with classic Pick bodies [28]. Second, no tau inclusions at all were present in either dentate gyrus granule cells or pyramidal cells of the hippocampus: the presence of Pick bodies in this region is pathognomic. In this present study, case #14 displayed a similar (to case #13) pattern of 4R-tau pathology. Therefore, in this present study, we find that in all cases of sporadic FTLD where unequivocal Pick bodies are present, these contain only 3R isoforms of tau. Nonetheless, it is acknowledged from the literature that there may be (rare) cases of FTLD in which the Pick bodies contain only 4R-tau isoforms [38, 66] and other cases where a few of the Pick bodies may contain 4R-tau while the great majority are only 3R-tau positive [1]. However, Pick bodies containing 4R-tau may better described as Pick-like bodies since, in at least one instance [38], electron microscopy has revealed these to be composed of parallel, long period twisted ribbon-like structures instead of the random, straight filaments typically seen in Pick bodies [28].
Consistent with these findings in sporadic FTLD, we could not detect 4R-tau within the Pick bodies of the frontal cortex of familial FTLD cases associated with MAPT mutations, L266V (case #15) (see also Ref. [23]), Q336R (case #36), E342V (case #37), K369I (case #38) and G389R (case #39), despite western blots showing 4R-tau species [23, 31, 43, 44] to be present in the brains of such patients (where frozen tissues were available for analysis). de Silva and co-workers reported similar findings in two other cases with MAPT G389R mutation [10]. Again this apparent discrepancy can be explained in terms of anatomical compartmentalization in the case of MAPT L266V and E342V mutations at least. In these mutations, tau-immunoreactive astrocytes are prominent and we have shown here that such cells are 4R-tau reactive and probably therefore responsible for the presence of 4R-tau isoforms on western blot. We have not been able to determine the anatomical correlates of 4R-tau isoforms seen on western blot in MAPT K369I and G389R mutations, though in a previous study of ours on one case with MAPT G389R mutation [45] 4R-tau was, on western blotting, a very minor constituent and therefore be undetectable by immunohistochemistry [10]. Similarly, Bronner et al. [4] have recently reported Pick bodies in MAPT G272V mutation to contain only 3R-tau isoforms on immunohistochemistry but because of rare 4R-tau containing NFT, western blotting showed a mix pattern of 3R- and 4R- tau isoforms.
Consistent with the many reports showing, by western blot, that patients with familial FTLD associated with exon 10 coding and splice site mutations contain only mutated 4R-tau isoforms [30] or selective increases in the proportion of wild-type 4R-tau [2, 6, 9, 19, 24, 25, 35, 40, 46, 60] in their brains, we found that the neuronal and glial tau pathology in MAPT N279K, N296H, P301L, S305S and the MAPT exon 10 +16 (see also Refs. [10] and [43] for exon 10 +3 mutation) mutations was exclusively associated with 4R-tau, except in two of the MAPT P301L mutation cases (cases #27 and 28) in whom rare 3R-tau immunoreactive cells were present and which might reflect the coincidental presence of minor Alzheimer-type pathology involving PHF, immunoreactive with both 3R- and 4R-tau antibodies because all six tau isoforms are equally represented. Some β-amyloid plaques were also present in these two MAPT P301L cases, again suggestive of additional Alzheimer-type pathology. Only in MAPT R406W mutation did we find the numerous NFT and neuropil threads [49, 63] immunoreactive with both 3R- and 4R-tau antibodies. Western blotting of the sarkosyl-insoluble tau extracted from the brains of patients with this particular mutation shows all six tau isoforms, similar to Alzheimer’s disease, to occur [24, 63]. Indeed, ultrastructurally, the NFT in MAPT R406W mutation are in the form of paired helical filaments identical to those in AD [49]. However, it is not clear whether the same tangle in MAPT R406W mutation cases is both 3R- and 4R-tau immunoreactive, or whether separate populations of tangle bearing cells are involved, some containing exclusively 3R-tau, others 4R-tau, but the finding [10] in AD that NFT cells are doubly immunoreactive for 3R- and 4R-tau would make this unlikely. Notably, MAPT V337M mutation shares a very similar neurofibrillary pathology as MAPT R406Wmutation [57, 61] with all six tau isoforms being represented on western blot [57]. It might be inferred therefore that here too the tangles would be both 3R- and 4R-tau immunoreactive, though to our knowledge such investigations have not as yet been performed.
Although some studies [31, 37] have corralled cases of CBD and PSP, as tauopathies, under the umbrella of FTLD, as allowed under McKhann criteria [34], we did not include such cases within the present investigation. This was partly because we have already reported [10] that neurofibrillary tangles in PSP are stained only or mostly with antibody to 4R-tau, as is consistent with tau biochemical findings of mostly or only 4R-tau isoforms being present in sarkosyl insoluble fractions of tau [10, 37]. Furthermore, Mott et al. [37] have also shown that sarkosyl insoluble fractions of tau in CBD are likewise composed mostly of 4R-tau and a similar pattern of 4R-tau immunoreactivity would therefore be anticipated. Indeed this was so in a single case of CBD we have so far studied (Unpublished data).
In conclusion, we have shown here that whenever classic Pick bodies are present, be these in cases of sporadic FTLD or familial cases with MAPT mutations (L266V, Q336R, E342V, K369I), only 3R-tau isoforms are present in such structures. The presence of 4R-tau isoforms on western blot in MAPT L266V and E342V mutations relates to their presence in glial cells and not Pick bodies. We have therefore not been able to confirm previous reports of the presence of 4R-tau isoforms in Pick bodies [66] and given the structural differences between these and classic Pick bodies [38], it might be better for the moment to consider the latter as Pick body-like structures pending further investigation. Neurofibrillary and glial cell tangles in cases of familial FTLD with MAPT exon 10 mutations (i.e., N279K, N296H, P301L, S305S and exon 10 +16 mutations) were usually or exclusively composed of 4R-tau, consistent with biochemical findings of mostly or only 4R-tau isoforms within sarkosyl-insoluble fractions from brains of such cases. The use of such tau isoform-specific antibodies may help to further refine the pathological criteria underpinning FTLD.
References
Arai T, Ikeda K, Akiyama H, Shikamoto Y, Tsuchiya K, Yagishita S, Beach T, Rogers J, Schwab C, McGeer PL (2001) Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 101:167–173
Arima K, Kowalska A, Hasegawa M, Mukoyama M, Watanabe R, Kawai M, Takahashi K, Iwatsubo T, Tabira T, Sunohara N (2000) Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology 54:1787–1795
Bird TD, Nochlin D, Poorkaj P, Cherrier M, Kaye J, Payami H, Peskind E, Lampe TH, Nemens E, Boyer P, Schellenberg GD (1999) A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 122:741–756
Bronner IF, ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, Ravid R, Heutink P, van Swieten JC (2005) Hereditary Pick’s disease with the G272V tau mutation shows a predominant three-repeat tau pathology. Brain 128:2645–2653
Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B (1999) Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol 58:667–677
Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D’Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC (1998) Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA 95:13103–13107
Constantinidis RJ, Richard J, Tissot R (1974) Pick’s disease: histological and clinical correlations. Eur Neurol 11:208–217
Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y (1998) Vulnerable neuronal subsets in Alzheimer’s and Pick’s disease are distinguished by their tau isoform distribution and phosphorylation. Ann Neurol 43:193–204
Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, Soulages X, Mohr M, Calvas P, Ghetti B (1999) A mutation at codon 279 (N279K) in exon 10 of the tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol 98:62–77
de Silva R, Lashley T, Gibb G, Hope A, Reid A, Bandopadhyay R, Utton M, Strand C, Jowett T, Khan N, Anderton B, Wood N, Holton J, Revesz T, Lees A (2003) Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule-binding repeat domains as demonstrated by new specific monoclonal antibodies. Neuropathol Appl Neurobiol 29:288–302
D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD (1999) Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA 96:5598–5603
Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D’Amato CJ, Gilman S (1997) Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol 41:706–715
Fujino Y, Wang DS, Thomas N, Espinoza M, Davies P, Dickson DW (2005) Increased frequency of argyrophilic grain disease in Alzheimer disease with 4R tau-specific immunohistochemistry. J Neuropathol Exp Neurol 64:209–214
Ghetti B, Murrell JR, Zolo P, Spillantini MG, Goedert M (2000) Progress in hereditary tauopathies: a mutation in the tau gene (G389R) causes a Pick disease-like syndrome. Ann N Y Acad Sci 920:52–62
Goedert M (2005) Tau gene mutations and their effects. Mov Disord 20(Suppl 12):S45–S52
Goedert M, Jakes R, Crowther RA, Six J, Lubke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VM (1993) The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci USA 90:5066–5070
Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Petersen PB, Gambetti P (1999). Tau gene mutation in familial progressive subcortical gliosis. Nat Med 5:454–457
Grover A, DeTure M, Yen SH, Hutton M (2002) Effects on splicing and protein function of three mutations in codon N296 of tau in vitro. Neurosci Lett 323:33–36
Halliday GM, Song YJC, Creasey H, Morris JG, Brooks WS, Kril JJ (2006) Neuropathology in the S305S tau gene mutation. Brain (in press)
Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M (1999) FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett 443:93–96
Hayashi S, Toyoshima Y, Hasegawa M, Umeda Y, Wakabayashi K, Tokiguchi S, Iwatsubo T, Takahashi H (2002) Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol 51:525–530
Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, Kril JJ, Halliday GM (2004) Clinicopathological correlates in frontotemporal dementia. Ann Neurol 56:399–406
Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP, Herzog LL, Weintraub S, Mesulam MM, LaPointe NE, Gamblin TC, Berry RW, Binder LI, de Silva R, Lees A, Espinoza M, Davies P, Grover A, Sahara N, Ishizawa T, Dickson D, Yen SH, Hutton M, Bigio EH (2003) The L266V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy. Acta Neuropathol 106:323–336
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon MJ, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski JQ, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JBJ, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705
Iseki E, Matsumura T, Marui W, Hino H, Odawara T, Sugiyama N, Suzuki K, Sawada H, Arai T, Kosaka K (2001) Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol 102:285–292
Ishizawa K, Ksiezak-Reding H, Davies P, Delacourte A, Tiseo P, Yen S-H, Dickson DW (2000) A double-labelling immunohistochemical study of tau exon 10 in Alzheimer’s disease, progressive supranuclear palsy and Pick’s disease. Acta Neuropathol 100:235–244
Ishizawa T, Ko LW, Cookson N, Davies P, Espinoza M, Dickson DW (2002) Selective neurofibrillary degeneration of the hippocampal CA2 sector is associated with four repeat tauopathies. J Neuropathol Exp Neurol 61:1040–1047
Kertesz A, McGonagle P, Blair M, Davidson W, Munoz DG (2005) The evolution and pathology of frontotemporal dementia. Brain 128:1996–2005
Kobayashi T, Ota S, Tanaka K, Ito Y, Hasegawa M, Umeda Y, Motoi Y, Takanashi M, Yasuhara M, Anno M, et al (2003) A novel L266V mutation of the tau gene causes frontotemporal dementia with a unique tau pathology. Ann Neurol 53:133–137
Lippa CF, Zhukareva V, Kawarai T, Uryu K, Shafiq M, Nee LE, Grafman J, Liang Y, St George-Hyslop PH, Trojanowski JQ, Lee VM (2000) Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol 48:850–858
Lipton AM, White CL III, Bigio EH (2004) Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol 108:379–385
Mann DMA (2005) The genetics and molecular pathology of frontotemporal lobar degeneration. In: Burns A, O’Brien J, Ames D, Arnold H (eds) Dementia, 3rd edn. , London, pp 689–701
Mann DMA, South PW, Snowden JS, Neary D (1993) Dementia of frontal lobe type: neuropathology and immunohistochemistry. J Neurol Neurosur Ps 56:605–614
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ (2001) Workgroup on Frontotemporal dementia and Picks disease; Clinical and pathological diagnosis of frontotemporal dementia: report of the workgroup on Frontotemporal dementia and Picks disease. Arch Neurol 59:1203–1204
Mirra SS, Murrell JR, Gearing M, Spillantini MG, Goedert M, Crowther RA, Levey AI, Jones R, Green J, Shoffner JM, Wainer BH, Schmidt ML, Trojanowski JQ, Ghetti B (1999) Tau pathology in a family with dementia and a P301L mutation in tau. J Neuropathol Exp Neurol 58:335–345
Miyasaka T, Morishima-Kawashima M, Ravid R, Kamphorst W, Nagashima K, Ihara Y (2001) Selective deposition of mutant tau in the FTDP-17 brain affected by the P301L mutation. J Neuropathol Exp Neurol 60:872–884
Motoi Y, Iwamoto H, Itaya M, Kobayashi T, Hasegawa M, Yasuda M, Mizuno Y, Mori H (2005) Four-repeat tau positive Pick body-like inclusions are distinct from classic Pick bodies. Acta Neuropathol 110:431–433
Mott RT, Dickson DW, Trojanowski JQ, Zhukareva V, Lee VM, Forman M, van Deerlin V, Ervin JF, Wang DS, Schmechel DE, Hulette CM (2005) Neuropathologic, biochemical, and molecular characterization of the frontotemporal dementias. J Neuropathol Exp Neurol 64:420–428
Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M (1999) Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol 58:1207–1226
Nasreddine ZS, Loginov M, Clark LN, Lamarche J, Miller BL, Lamontagne A, Zhukareva V, Lee VM, Wilhelmsen KC, Geschwind DH (1999) From genotype to phenotype: a clinical pathological, and biochemical investigation of frontotemporal dementia and parkinsonism (FTDP-17) caused by the P301L tau mutation. Ann Neurol 45:704–715
Neary D, Snowden JS, Mann DMA (2005) Frontotemporal dementia. Lancet Neurol 4:771–780
Neumann M, Schulz-Schaeffer W, Crowther RA, Smith MJ, Spillantini MG, Goedert M, Kretzschmar HA (2001) Pick’s disease associated with the novel Tau gene mutation K369I. Ann Neurol 50:503–513
Neumann M, Mittelbronn M, Simon P, Vanmassenhove B, de Silva R, Lees A, Klapp J, Meyermann R, Kretzschmar HA (2005) A new family with frontotemporal dementia with intronic 10+3 splice site mutation in the tau gene: neuropathology and molecular effects. Neuropathol Appl Neurobiol 31:362–373
Papasozomenos SC (1989) Tau protein immunoreactivity in dementia of the Alzheimer type: II. Electron microscopy and pathogenetic implications. Effects of fixation on the morphology of the Alzheimer’s abnormal filaments. Lab Invest 60:375–389
Pickering-Brown SM, Baker M, Yen S-H, Liu W-K, Hasegawa M, Cairns NJ, Lantos PL, Rossor M, Iwatsubo T, Davies Y, Allsop D, Furlong R, Owen F, Hardy J, Mann DMA, Hutton M (2000) Pick’s disease is associated with mutations in the tau gene. Ann Neurol 48:859–867
Pickering-Brown SM, Richardson AM, Snowden JS, McDonagh AM, Burns A, Braude W, Baker M, Liu WK, Yen SH, Hardy J, Hutton M, Davies Y, Allsop D, Craufurd D, Neary D, Mann DMA (2002) Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain 125:732–751
Pickering-Brown SM, Baker M, Nonaka T, Ikeda K, Sharma S, MacKenzie J, Simpson SA, Moore JW, Snowden JS, de Silva R, Revesz T, Hasegawa M, Hutton M, Mann DM (2004) Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain 127:1415–1426
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825
Reed LA, Grabowski TJ, Schmidt ML (1997) Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol 42:564–572
Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, Xuereb JH, Crowther RA, Spillantini MG (2000) Tau gene mutation K257T causes a tauopathy similar to Pick’s disease. J Neuropathol Exp Neurol 59:990–1001
Rizzu P, van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P (1999) High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 64:414–421
Rosso SM, van Herpen E, Deelen W, Kamphorst W, Severijnen LA, Willemsen R, Ravid R, Niermeijer MF, Dooijes D, Smith MJ, Goedert M, Heutink P, van Swieten JC (2002) A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick’s disease. Ann Neurol 51:373–376
Rosso SM, Landweer EJ, Houterman M, Donker Kaat L, van Duijn CM, van Swieten JC (2003) Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case-control study. J Neurol Neurosur Ps 74:1574–1576
Sergeant N, Wattez A, Delacourte A (1999) Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively exon 10 isoforms. J Neurochem 72:1243–1249
Shi J, Shaw CL, Du Plessis D, Richardson AMT, Bailey K, Julien C, Neary D, Snowden JS, Mann DMA (2005) Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol 110:501–512
Snowden JS, Neary D, Mann DMA (1996) Fronto-temporal lobar degeneration: Fronto-temporal dementia, progressive aphasia, semantic dementia. Churchill Livingstone, Edinburgh, pp 1–227
Spillantini MG, Crowther RA, Goedert M (1996) Comparison of the neurofibrillary pathology in Alzheimer’s disease and familial presenile dementia with tangles. Acta Neuropathol 92:42–48
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:7737–7741
Spillantini MG, Yoshida H, Rizzini C, Lantos PL, Khan N, Rossor MN, Goedert M, Brown J (2000) A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol 48:939–943
Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, Morris JG, Fulham MJ, Schofield PR (2000) Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain 123:880–893
Sumi SM, Bird TD, Nochlin D, Raskind MA (1992) Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and degeneration of the amygdala. Neurology 42:120–127
Taniguchi S, McDonagh AM, Pickering-Brown SM, Umeda Y, Iwatsubo T, Hasegawa M, Mann DM (2004) The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol Appl Neurobiol 30:1–18
van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, de Koning I, Kamphorst W, Ravid R, Spillantini MG, Niermeijer M, Heutink P (1999) Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol 46:617–626
Wszolek ZK, Pfeiffer RF, Bhatt MH, Schelper RL, Cordes M, Snow BJ Rodnitsky RL, Wolters EC, Arwert F, Calne DB (1992) Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 32:312–320
Zarrantz JJ, Ferrer I, Lezcano E, Forcadas MI, Eizaguirre B, Atares B, Puig B, Gomez-Esteban JC, Fernandez-Maiztegui C, Rouco I, Perez-Concha T, Fernandez M, Rodruigez O, Rodriguez-Martinez AB, Martinez de Pancorbo M, Pastor P, Perez-Tur J (2005) A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology 64:1578–1585
Zhukareva V, Mann DMA, Uryu K, Shuck T, Shah K, Grossman M, Miller BL, Hulette CM, Feinstein S, Trojanowski JQ, Lee VM-Y (2002) Sporadic Pick’s disease: a tauopathy characterised by a spectrum of gray and white matter pathological tau isoforms. Ann Neurol 51:730–739
Acknowledgements
AMS was supported by a Wolfson Scholarship and Alzheimer’s Research Trust Alzheimer’s Disease Research Centre Grant to DMAM. TL is supported by the Parkinson’s Disease Society. This work was supported by the Reta Lila Weston Trust for Medical Research (RdS, AL, TL) and the PSP (Europe) Association (KS), which also support the Queen Square Brain Bank. The authors thank the many other people who were involved in collecting and characterising the familial FTLD cases with MAPT mutations and the other sporadic FTLD cases and by doing so making this multicentre collaborative study possible. The authors would also like to thank the patients and their families, without whose generous support none of this research would have been possible.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Silva, R., Lashley, T., Strand, C. et al. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol 111, 329–340 (2006). https://doi.org/10.1007/s00401-006-0048-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-006-0048-x