
Abstract
The abnormal conformation and assembly of proteins in the central nervous system is increasingly thought to be a critical pathogenic mechanism in neurodegenerative disorders such as Creutzfeldt–Jakob disease (CJD) and Alzheimer’s disease (AD). CJD is marked primarily by the buildup of misfolded prion protein (PrPSc) in brain, whereas the accrual of β-amyloid protein (Aβ) and tau protein are characteristic for AD. Prior studies have shown that the ATP-binding cassette transporter P-glycoprotein (P-gp) is a cellular efflux pump for Aβ, and that age-associated deficits in P-gp may be involved in the pathogenesis of Alzheimer’s disease. In the present study, we investigated the relationship between P-gp and idiopathic CJD, and found that CJD, like AD, is associated with a decrease in the expression of cerebrovascular P-gp. In some instances, Aβ and PrP deposits coexist in cases of CJD, suggesting the possibility of pathogenic interactions. Since there is, to date, no evidence that PrP itself is a substrate for P-gp, we hypothesize that the age-related deficits in P-gp could promote the accumulation of PrPSc either by promoting the buildup of Aβ (which could act as a seed for the aggregation of PrPSc), or by overloading the ubiquitin-proteasomal catabolic system, and thereby facilitating the accumulation of PrP. Alternatively, the loss of P-gp could be a non-specific response to neurodegenerative changes in the central nervous system. In either case, dysfunction of this critical toxin-elimination pathway in CJD and AD suggests that selectively increasing cerebrovascular P-gp function could open new therapeutic pathways for the prevention and/or treatment of a number of proteopathic disorders of the central nervous system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Virtually all age-associated neurodegenerative diseases are linked to the abnormal conformation and assembly of proteins in the nervous system [46, 48]. The hallmark of these disorders is the transformation of a normally soluble peptide or protein into insoluble fibrils that are deposited in the brain tissue [6, 9, 25, 26]. The cerebral proteopathies include a range of devastating neurological disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease and Creutzfeldt–Jakob disease (CJD). Age is a factor in the emergence of all of these maladies, although the means by which age influences their expression remains unresolved. While AD is characterized histopathologically by large numbers of senile plaques and neurofibrillary tangles, such lesions also occur, in much smaller numbers, in many non-demented elderly humans [49]. Senile plaques and cerebral β-amyloid angiopathy (CAA) consist primarily of deposits of the protein fragment Aβ, which most often is 40 (Aβ40) or 42 (Aβ42) amino acids in length. In aging humans, parenchymal Aβ42 deposits usually precede the appearance of dense, Aβ40-rich aggregates [47].
Creutzfeldt–Jakob disease belongs to the transmissible spongiform encephalopathies (TSE), or prion diseases, which are characterized by the accumulation of an abnormal isoform of the prion protein (PrPSc). Histopathologically, CJD is diagnosed by the classical triad of spongiform change, neuronal loss, and gliosis, as well as the presence of aggregated PrPSc [5, 12, 14, 43]. In the brains of older CJD patients, Alzheimer-like β-amyloid (Aβ) deposits sometimes can be observed [5, 21], but the presence of such lesions appears to be a non-specific effect of age. However, in these cases, PrP often decorates the periphery of Aβ-plaques, suggesting that Aβ may influence the deposition of PrP [21].
Despite many differences between CJD and AD, both diseases share important pathogenetic similarities that could lead to similar therapeutic strategies [2]. Three broad therapeutic approaches to abrogating the proteopathic cascade are suggested by the commonalities among these disorders: (1) reduce the production of the aggregation-prone proteins, (2) prevent their self-assembly and toxicity, or (3) promote their degradation and removal [46, 48].
In recent years, increasing attention has been devoted to the myriad functions of a class of transport proteins—the so-called ATP-binding cassette transport proteins (ABC transporters)—in the central nervous system. In this regard, P-glycoprotein (P-gp, ABCB1, MDR1) appears to be particularly important. P-gp acts as an efflux pump for several endogenous and exogenous substances and thus plays a critical role in the protection of tissues with excretory and/or barrier function, such as intestine, kidney, and liver. P-gp also is active at the blood-brain barrier (BBB) and blood-cerebrospinal fluid (CSF) barrier [17, 18, 37]. In normal brain, P-gp is expressed by the endothelial cells of blood vessels, especially capillaries [1, 18], and by epithelial cells of the choroid plexus [15].
P-glycoprotein has been shown to actively transport Aβ in vitro [28] and in vivo [11], and deficits in P-gp may be involved in the pathogenesis of Alzheimer’s disease and CAA [44, 45]. Importantly, cerebrovascular P-gp expression decreases with age [45], and could thereby impair the ability of the brain to expel excess proteins. We hypothesize that the resulting accumulation of proteins in cells overwhelms cellular ubiquitin-proteasomal degradation, and thus facilitates the emergence of proteopathic disorders of the nervous system. In the present study, we investigated the relationship between P-gp and prion disease, and found that CJD, like AD and CAA, is associated with a decrease in the expression of cerebrovascular P-gp compared to age-matched control cases. Since P-gp expression can be induced by several drugs, selective enhancement of brain P-gp function might be employed to treat or prevent the abnormal accrual of proteins that characterizes many neurodegenerative disorders.
Materials and methods
Tissue samples of occipital and temporal lobe were obtained at autopsy from ten subjects with histologically, biochemically, and genetically confirmed CJD who died between the ages of 59 and 82 years (mean age 70.3 years), as well as ten age-matched control cases (mean age 70.2 years). Sex and age distribution are shown in Table 1. Brain tissue from all cases was fixed similarly in 4% buffered formalin, inactivated 1 h with 98% formic acid and embedded in paraffin according to the standard guidelines for processing CJD tissues. Sections were subjected to conventional staining and to immunostaining for PrP (3F4) upon hydrolytic autoclaving. Genetic analysis and determination of glycotype were performed as described previously [20].
Immunohistochemistry
For immunohistochemistry, 6 μm-thick tissue sections were cut, mounted on slides and dried overnight at 60°C. Sections were deparaffinized in xylene, rehydrated in a graded series of ethanol and water, and then treated with 10 mM citrate buffer at pH 6.0. Endogenous peroxidase was blocked with 0.3% H2O2. For staining, the biotin-streptavidin immunoperoxidase method with the LSAB®HRP detection system (DAKO, Hamburg, Germany) was used. P-gp was immunolabeled with monoclonal antibody JSB-1 (Alexis, Grünberg, Germany). Aβ was labeled using end-specific monoclonal antibodies to Aβ40 or Aβ42 (Chemicon, Planegg-Muenchen, Germany).
For quantification of P-gp, five consecutive digital pictures were taken of neocortex, white matter and leptomeninges (magnification ×200) from identical regions of each case; for quantification of Aβ40 and Aβ42, four consecutive digital pictures of cortex (magnification ×100) and five consecutive digital pictures of leptomeninges (magnification ×200) were taken (3CCD color camera, Hitachi HV-C20 M, Hitachi Denshi Ltd., Japan, and Axioskop, Zeiss, Jena, Germany). Subsequently, the optical density and the area occupied by immunopositive structures were determined in each photograph using KSRun software (KSRun Version 3.0, Zeiss), revealing a final middle optical density (mod) value for each case.
In selected cases, immunohistochemical double-labeling with antibodies to Aβ (DAKO, clone 6F/3D, dilution 1:50) and PrP (DAKO, clone 3F4, dilution 1:25) was performed. First, Aβ was stained using the EnVision Dual Link System (DAKO) and diaminobenzidine (DAB) as chromogen, yielding a brown color for Aβ. Subsequently, PrP immunostaining was performed with the Chem Mate Detection Kit, Alkaline Phosphatase/RED (DAKO) and fast red as chromogen, staining the PrP deposits red.
Statistical analysis
Statistical analysis of the data was performed using the non-parametric Mann–Whitney U-Test with a two-tailed significance threshold (P<0.05).
Results
The data for CJD cases regarding the pattern of PrP-immunostaining, PrPSc-type and the methionine/valine polymorphism at codon 129 of the PRNP gene are shown in Table 2. The PrP immunostaining pattern was described as punctate, patchy or plaque-like, respectively. Vascular deposition of PrP was noted in only one case (below). PrPSc-subtypes were divided into two groups (I or II) based on variation at codon 129 of the PrP gene [33]. The cohort included a representative selection of geno- and PrPSc-types in order to minimize potential bias in the analysis.
P-glycoprotein was expressed strongly in endothelial cells of cerebral blood vessels, especially capillaries. Quantitatively, CJD cases had significantly lower P-gp labeling within the cortex (P=0.005), leptomeninges (P=0.003) and white matter (P=0.037) when compared to controls (Fig. 1).

Quantitative immunohistochemical expression of P-gp. CJD cases show significantly lower P-gp expression than controls within the cortex, leptomeninges, (*P<0.01) and white matter (**P<0.05). Mod Middle optical density
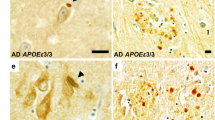
Five out of ten CJD cases had age-associated Aβ42-immunoreactive plaques within the cortex, as well as Aβ42 deposits in the walls of leptomeningeal blood vessels. Two cases also had Aβ42-CAA within the brain parenchyma. Four of the five CJD cases with Aβ42 lesions also showed Aβ40-positive plaques within the cortex, including two cases with intracerebral Aβ40-CAA involvement as well as three cases with leptomeningeal Aβ40-CAA. Regarding the extent of the Aβ burden there were three cases with isolated Aβ plaques only, as is often the case in the non-demented elderly. In contrast, the two cases with intracerebral CAA also showed numerous Aβ plaques, comparable to the lesion density in AD brains. No amyloid deposits were observed in control cases (except one case with a single diffuse plaque).
There was no colocalization of P-gp expression and β-amyloid deposition within the same vessel, i.e., vessels with Aβ deposition in their walls showed no endothelial P-gp labeling, and vice versa (Fig. 2). Generally, prominent vascular Aβ deposition was observed in arterioles and small arteries. There was only one case with Aβ deposits within capillaries, and the absence of P-gp expression was evident in these afflicted vessels as well. Interestingly, this case with capillary β-amyloid angiopathy showed the lowest P-gp expression of all cases analyzed.

Immunohistochemical expression of β-amyloid in a case of CJD (a) and a consecutive section with P-gp immunostaining (b). Note Aβ deposition as plaque and amyloid angiopathy, and the negative expression of P-gp. A control case with strongly detectable P-gp is shown in (c). Bar=50 μm
Because five of the CJD cases also had cerebral Aβ deposition, and because P-gp expression is significantly reduced in blood vessels that manifest CAA, we asked whether the deficits in P-gp expression in CJD cases are influenced by the simultaneous presence of Aβ40 or Aβ42. Comparing CJD cases with or without Aβ40/42 deposition, respectively, there were no differences in P-gp expression (Table 3).
Double immunolabeling often detected colocalization of Aβ and PrP within the same lesions, but the distribution of the two proteins differed. Some senile plaques had an Aβ core and an accumulated rim of PrP (Fig. 3a), whereas some dense PrP plaques were rimmed by Aβ (Fig. 3b). We also observed protein layering in certain plaques, such that a layer of PrP was interposed between an inner core and outer shell of Aβ (Fig. 3c). Interestingly, although PrP was generally absent in vessels, one case with severe leptomenigeal CAA showed occasional small arteries with a colocalisation of Aβ and PrP within the vessel wall (Fig. 3d).

Immunohistochemical double-labeling of Aβ (brown) and PrP (red) in Creutzfeldt–Jakob cases. a Plaque with an Aβ core and a PrP shell. b Plaque with a PrP core and an Aβ shell. c Plaque with central Aβ deposition, a layer of PrP and an outer accumulation of Aβ. d Vessel wall with colocalisation of Aβ and PrP (arrow). Bar=50 μm
Discussion
It has been suggested that decreased clearance of peptides (especially Aβ) from the brain at the BBB could contribute to the buildup of proteins, leading to AD and possibly to other neurodegenerative disorders such as CJD [13, 39, 48]. In previous studies we found an inverse relationship between the expression of cerebrovascular P-gp and the deposition of Aβ in the brains of elderly humans, suggesting that a diminution of P-gp function might be involved in the early pathogenesis of AD [44, 45]. Furthermore, P-gp expression declines in the cerebral blood vessels of older humans even in the absence of CAA [45], and could thereby increase the probability of aberant protein accumulation with age. Since there is mounting evidence for pathogenic commonalities in the accumulation of Aβ and PrPSc in diseased tissues [2, 3], we hypothesized that the expression of P-gp might be diminished in cases with idiopathic CJD.
Our present results show that P-gp expression within endothelial cells of brain vessels is significantly lower in cases with CJD than in age-matched control cases. Unlike Aβ, the accumulation of PrPSc in the cerebral vasculature is quite rare [except in a Gerstmann–Straeussler–Scheinker syndrome (GSS) pedigree with a premature stop codon in the PrP gene] [36]. Accordingly, there was almost no immunohistochemically detectable PrP in the blood vessels of the idiopathic CJD cases that we examined. The paucity of demonstrable vascular PrP raises the question of whether down-regulation of the P-gp transporter in the endothelium is the cause or the consequence of the prion disease process. Half of the CJD cases that we analyzed also displayed parenchymal and/or vascular Aβ deposits, as expected in a cohort of older subjects [49]. As in our previous study [45], the presence of Aβ within a particular vessel wall was associated with a deficiency of P-gp within the affected segment. Although there was no statistically significant difference in overall P-gp expression in CJD cases with or without cerebral β-amyloidosis, one CJD case manifesting Aβ-CAA in neocortical capillaries had the lowest P-gp expression of all cases analyzed. Taken together, these findings support the view that CJD is associated with a reduction of P-gp expression independent of the co-existence of Aβ deposits, but further studies are required to determine whether the co-presence of PrP and Aβ is linked to an even greater diminution of transporter expression and function.
There are several potential means by which P-gp function might be related to prion disease. One possibility is that P-gp could be down-regulated in the brain as a result of the disease process, i.e., that P-gp expression loss is a consequence rather than a cause of PrP accumulation. Longitudinal studies of P-gp expression in PrP-transgenic mice, which do not deposit Aβ with age, would help to establish whether P-gp function declines before, or after, the onset of disease phenotype.
Another possibility is that the loss of P-gp is a causative factor in the accumulation of PrP in brain. The age-associated loss of P-gp function [45] suggests one means whereby age might influence the risk of CJD. To date, there is no evidence that PrP itself is a substrate for P-gp, but there are two general, alternative pathways through which P-gp reduction might stimulate the accumulation and subsequent toxicity of PrPSc.
First, the loss of P-gp function could facilitate the buildup of Aβ, which then acts as a heterologous seed for the aggregation of PrPSc. Aβ deposition frequently is found in TSE, including CJD [8], although at present it is uncertain whether Aβ accumulation is pathogenically linked to prion disease, or whether it is an independent (e.g., age-related) process. Interestingly, the levels of Aβ42 are decreased in the CSF of patients with CJD, similar to the reduction seen in patients with AD [32, 50]. Other studies have reported a colocalization of Aβ and PrP in the same plaques in CJD cases, suggesting that preexisting β-amyloid might augment PrP accumulation by promoting the aggregation of one amyloidogenic protein (PrPSc) onto a core composed of the other (Aβ) [21, 27]. Our results confirm that PrP can decorate the periphery of Aβ plaques. We also observed the converse, i.e., that Aβ can deposit on PrP plaque cores. Occasionally, there were even multilayered plaques, with central Aβ deposition, a rim of PrP and an outer accumulation of Aβ. Furthermore, there were rare blood vessels with mural colocalisation of Aβ and PrP. These results suggest that both proteins interact complementarily, and that each may act as a seed for the accumulation of the other. This mechanism could be evaluated in dual PrP-βAPP transgenic mice.
A second possibility is that the accumulation of aberrant proteins (including Aβ) due to diminished P-gp activity could overwhelm the protein-degrading enzymes of the ubiquitin-proteasomal system (UPS) and thereby facilitate the accumulation and conversion of PrP. The UPS plays a fundamental part in many basic cellular processes by degrading a wide range of specific cellular proteins, including mutated and misfolded proteins [10]. Dysfunction of the UPS contributes to the accumulation of proteins in neurodegenerative diseases, such as α-synuclein in PD [34], or Aβ [29] and tau [31] in AD. PrP also is degraded via the UPS [35, 52]. Inhibition of the UPS, e.g., due to aging or drug treatment, causes an accumulation of normal PrPC in the cytoplasm, where it can be spontaneously converted into a PrPSc-like species because it is not promptly degraded by the UPS [30]. Interestingly, there are recent hints that P-gp interacts actively with the proteasome complex [4].
It cannot be excluded that, in CJD, there are additional mechanisms that damage the integrity of the BBB and, secondarily, reduce the expression of P-gp. However, since P-gp acts as a critical detoxifying system in the brain [42], we favor the hypothesis that the age-associated decline in P-gp function elevates the levels of toxic proteins such as PrP in brain, thereby increasing the likelihood that they will accumulate to pathogenic levels via permissive templating [22].
Additionally, P-gp might play a part in the pathogenesis of neurodegenerative diseases by one or more indirect mechanisms. Diminished P-gp expression could cause a pronounced influx of exogenous, neurotoxic compounds, leading to the damage and loss of neurons that is believed to promote the development of some cases of idiopathic parkinsonism [16, 19]. What is more, P-gp could act as a neuroprotective factor by suppressing the activation of caspases [40, 41] involved in apoptosis.
Similarities in the pathogenesis of aberrant protein deposits in CJD and AD have been suggested in the context of the concept of protein misfolding diseases, or proteopathies. Since these neurodegenerative disorders may be amenable to similar therapeutic principles [2, 48], selectively augmenting cerebral P-gp expression represents a novel therapeutic strategy to forestall the accumulation of insoluble proteins in the brain. P-gp activity can be modulated by a variety of substances such as verapamil or cyclosporin A (inhibition) [51], as well as rifampin or St. Johns wort (induction) [17, 23]. Because P-gp plays an important part in a variety of tissues, the challenge for drug discovery will be to identify an agent that selectively enhances P-gp function in the brain.
Finally, it has been shown that the MDR1 gene, which codes for P-gp, is highly polymorphic [7, 24], and that P-gp expression can be influenced by these polymorphisms [24, 38]. Thus, it is conceivable that variations in the MDR1 gene might influence the risk of developing certain neurodegenerative proteopathies, including CJD, an issue that warrants further research.
Conclusion
The results of our study show that the cerebrovascular expression of the multifaceted transporter P-gp is down-regulated in CJD. We propose that diminution of P-gp expression with age reduces the expulsion of toxic proteins by the BBB. The buildup of these proteins within cells overwhelms the protein degradation machinery, thereby further promoting the accumulation of aberrant proteins such as PrP. Our findings suggest that selectively augmenting cerebrovascular P-gp function could open new therapeutic pathways for the prevention and/or treatment of prionoses and other proteopathic disorders of the central nervous system.
References
Abbott NJ, Khan EU, Rollinson CM, Reichel A, Janigro D, Dombrowski SM, Dobbie MS, Begley DJ (2002) Drug resistance in epilepsy: the role of the blood-brain barrier. Novartis Found Symp 243:38–47
Aguzzi A, Haass C (2003) Games played by rogue proteins in prion disorders and Alzheimer’s disease. Science 302:814–818
Armstrong RA, Lantos PL, Cairns NJ (2001) The spatial patterns of prion protein deposits in Creutzfeldt–Jakob disease: comparison with beta-amyloid deposits in Alzheimer’s disease. Neurosci Lett 298:53–56
Begley GS, Horvath AR, Taylor JC, Higgins CF (2005) Cytoplasmic domains of the transporter associated with antigen processing and P-glycoprotein interact with subunits of the proteasome. Mol Immunol 42:137–141
Budka H (2003) Neuropathology of prion diseases. Br Med Bull 66:121–130
Carrell RW, Lomas DA (1997) Conformational disease. Lancet 350:134–138
Cascorbi I, Gerloff T, Johne A, Meisel C, Hoffmeyer S, Schwab M, Schaeffeler E, Eichelbaum M, Brinkmann U, Roots I (2001) Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther 69:169–174
Chapman J, Cervenakova L, Petersen RB, Lee HS, Estupinan J, Richardson S, Vnencak-Jones CL, Gajdusek DC, Korczyn AD, Brown P, Goldfarb LG (1998) APOE in non-Alzheimer amyloidoses: transmissible spongiform encephalopathies. Neurology 51:548–553
Chiti F, Calamai M, Taddei N, Stefani M, Ramponi G, Dobson CM (2002) Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc Natl Acad Sci USA 99(Suppl 4):16419–16426
Ciechanover A, Brundin P (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40:427–446
Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM (2005) P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 115:3285–3290
Collinge J (1998) Human prion diseases: aetiology and clinical features. In: Growdon JH, Rossor M, Newton MA (eds) The dementias. Butterworth-Heinemann, Oxford, pp 113–148
Deane R, Wu Z, Zlokovic BV (2004) RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 35(Suppl 1):2628–2631
DeArmond SJ, Prusiner SB (1995) Etiology and pathogenesis of prion diseases. Am J Pathol 146:785–811
Demeule M, Regina A, Jodoin J, Laplante A, Dagenais C, Berthelet F, Moghrabi A, Beliveau R (2002) Drug transport to the brain: key roles for the efflux pump P-glycoprotein in the blood-brain barrier. Vascul Pharmacol 38:339–348
Drozdzik M, Bialecka M, Mysliwiec K, Honczarenko K, Stankiewicz J, Sych Z (2003) Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson’s disease. Pharmacogenetics 13:259–263
Fromm MF, Kauffmann HM, Fritz P, Burk O, Kroemer HK, Warzok RW, Eichelbaum M, Siegmund W, Schrenk D (2000) The effect of rifampin treatment on intestinal expression of human MRP transporters. Am J Pathol 157:1575–1580
Fromm MF (2004) Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci 25:423–429
Furuno T, Landi MT, Ceroni M, Caporaso N, Bernucci I, Nappi G, Martignoni E, Schaeffeler E, Eichelbaum M, Schwab M, Zanger UM (2002) Expression polymorphism of the blood-brain barrier component P-glycoprotein (MDR1) in relation to Parkinson’s disease. Pharmacogenetics 12:529–534
Glatzel M, Rogivue C, Ghani A, Streffer JR, Amsler L, Aguzzi A (2002) Incidence of Creutzfeldt–Jakob disease in Switzerland. Lancet 360:139—141
Hainfellner JA, Wanschitz J, Jellinger K, Liberski PP, Gullotta F, Budka H (1998) Coexistence of Alzheimer-type neuropathology in Creutzfeldt–Jakob disease. Acta Neuropathol 96:116–122
Hardy J (2005) Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: permissive templating as a general mechanism underlying neurodegeneration. Biochem Soc Trans 33:578–581
Hennessy M, Kelleher D, Spiers JP, Barry M, Kavanagh P, Back D, Mulcahy F, Feely J (2002) St Johns wort increases expression of P-glycoprotein: implications for drug interactions. Br J Clin Pharmacol 53:75–82
Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmoller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, Brinkmann U (2000) Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA 97:3473–3478
Kakizuka A (1998) Protein precipitation: a common etiology in neurodegenerative disorders? Trends Genet 14:396–402
Koo EH, Lansbury PT Jr, Kelly JW (1999) Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc Natl Acad Sci USA 96:9989–9990
Kovacs GG, Budka H (2002) Aging, the brain and human prion disease. Exp Gerontol 37:603–605
Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB (2001) beta-Amyloid efflux mediated by P-glycoprotein. J Neurochem 76:1121–1128
Lopez Salon M, Pasquini L, Besio Moreno M, Pasquini JM, Soto E (2003) Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp Neurol 180:131–143
Ma J, Lindquist S (2002) Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science 298:1785–1788
Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM (2004) Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43:321–332
Otto M, Esselmann H, Schulz-Shaeffer W, Neumann M, Schroter A, Ratzka P, Cepek L, Zerr I, Steinacker P, Windl O, Kornhuber J, Kretzschmar HA, Poser S, Wiltfang J (2000) Decreased beta-amyloid1–42 in cerebrospinal fluid of patients with Creutzfeldt–Jakob disease. Neurology 54:1099–1102
Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H (1999) Classification of sporadic Creutzfeldt–Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233
Petrucelli L, Dawson TM (2004) Mechanism of neurodegenerative disease: role of the ubiquitin proteasome system. Ann Med 36:315–320
Rane NS, Yonkovich JL, Hegde RS (2004) Protection from cytosolic prion protein toxicity by modulation of protein translocation. EMBO J 23:4550–4559
Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (2003) Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 62:885–898
Schinkel AH, Jonker JW (2003) Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev 55:3–29
Schwab M, Eichelbaum M, Fromm MF (2003) Genetic polymorphisms of the human MDR1 drug transporter. Annu Rev Pharmacol Toxicol 43:285–307
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. (2000) Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106:1489–1499
Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW (1998) The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc Natl Acad Sci USA 95:7024–7029
Tainton KM, Smyth MJ, Jackson JT, Tanner JE, Cerruti L, Jane SM, Darcy PK, Johnstone RW (2004) Mutational analysis of P-glycoprotein: suppression of caspase activation in the absence of ATP-dependent drug efflux. Cell Death Differ 11:1028–1037
Terasaki T, Ohtsuki S (2005) Brain-to-blood transporters for endogenous substrates and xenobiotics at the blood-brain barrier: an overview of biology and methodology. NeuroRx 2:63–72
Unterberger U, Voigtlander T, Budka H (2005) Pathogenesis of prion diseases. Acta Neuropathol 109:32–48
Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert-Keil C, Walker LC, Warzok RW (2002) Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 12:535–541
Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, Siegmund W, Walker LC, Pahnke J (2004) The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 1:121–125
Walker LC, LeVine H (2000) The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly. Mol Neurobiol 21:83–95
Walker LC, Pahnke J, Madauss M, Vogelgesang S, Pahnke A, Herbst EW, Stausske D, Walther R, Kessler C, Warzok RW (2000) Apolipoprotein E4 promotes the early deposition of Abeta42 and then Abeta40 in the elderly. Acta Neuropathol 100:36–42
Walker LC, LeVine H III (2002) Proteopathy: the next therapeutic frontier? Curr Opin Investig Drugs 3:782–787
Warzok RW, Kessler C, Apel G, Schwarz A, Egensperger R, Schreiber D, Herbst EW, Wolf E, Walther R, Walker LC (1998) Apolipoprotein E4 promotes incipient Alzheimer pathology in the elderly. Alzheimer Dis Assoc Disord 12:33–39
Wiltfang J, Esselmann H, Smirnov A, Bibl M, Cepek L, Steinacker P, Mollenhauer B, Buerger K, Hampel H, Paul S, Neumann M, Maler M, Zerr I, Kornhuber J, Kretzschmar HA, Poser S, Otto M (2003) Beta-amyloid peptides in cerebrospinal fluid of patients with Creutzfeldt–Jakob disease. Ann Neurol 54:263–267
Yang ZY, Liu GQ (2004) Effect of P-glycoprotein inhibitor combinations on drug efflux from rat brain microvessel endothelial cells. Pharmazie 59:952–956
Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A (2001) Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J 20:5383–5391
Acknowledgments
We thank S. Uffmann, A. Wolter, and C. Mueller for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vogelgesang, S., Glatzel, M., Walker, L.C. et al. Cerebrovascular P-glycoprotein expression is decreased in Creutzfeldt–Jakob disease. Acta Neuropathol 111, 436–443 (2006). https://doi.org/10.1007/s00401-006-0042-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-006-0042-3