
Abstract
Transient forebrain ischemia of 5-min duration causes delayed neuronal death (DND) of vulnerable CA1 neurons in the gerbil hippocampus, which can be prevented by “preconditioning” with a short ischemic stimulus of 2.5-min duration. While a key role of excitatory glutamate receptors for both phenomena has been widely accepted, little is known about the postischemic regulation of central cannabinoid (CB1) receptors. The present study was designed to test whether ischemic preconditioning is associated with specific alterations of protein expression and/or ligand binding of these receptors compared to ischemia severe enough to induce DND. Gerbils were subjected to either a 5-min ischemic period resulting in DND of CA1 neurons, or a 2.5-min period of ischemia usually used for preconditioning. Postischemic hippocampal CB1 receptor protein expression was investigated immunohistochemically, while postischemic ligand binding of [3H]CP 55940 to CB1 receptors was analyzed by quantitative receptor autoradiography in both experimental groups after 24, 48, and 96 h (n=4–5 per time point), respectively, and compared to sham-treated gerbils (n=10). Short-term ischemia of 2.5-min duration caused a transient reduction of hippocampal CB1 receptor protein expression, while receptor binding density was permanently decreased. In contrast, 5-min ischemia did not alter protein expression or ligand binding up to 48 h. Based on these data, postischemic down-regulation of hippocampal CB1 receptors, specifically seen after short-term ischemia usually used for preconditioning, may participate in the mechanisms of endogenous postischemic neuroprotection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transient global ischemia of only 5-min duration causes selective and delayed neuronal death (DND) of hippocampal CA1 neurons 3–4 days after recirculation [19]. DND can be prevented by a short ischemic period or other potentially noxious stimuli close to but below the threshold of damage (for review see Dirnagl et al. [10]). While the crucial role of excitatory glutamate receptors both for DND and tolerance induction has been convincingly demonstrated [4, 18, 30], little is known about the potential participation of cannabinoid receptors. Since the cloning of a central cannabinoid (CB1) receptor from rat and man [12, 24], and the discovery of an endogenous cannabinoid system (for review see Di Marzo et al. [9]), research activities have focused on its therapeutic potentials concerning treatment of a variety of neurological diseases, including in vivo models of excitotoxic brain damage. Enthusiasm roused by earlier studies [28] has been damped by the growing number of data also demonstrating deleterious effects of postlesional cannabinoid receptor activation. Our present study was designed to address the question whether hippocampal CB1 receptors are specifically and differentially regulated after two experimental ischemic paradigms with neuronal death (5-min ischemia) versus neuronal survival (2.5-min ischemia) to further elucidate the role of the endocannabinoid system for the postischemic neuronal fate.
Materials and methods
Animal experiments
Experiments were performed on adult male Mongolian gerbils (Meriones unguiculatus, 70–80 g) obtained from Charles River Deutschland (Sulzfeld, Germany). The animals had free access to food and water prior to experiments. The gerbils were subjected to transient forebrain ischemia by bilateral occlusion of the common carotid artery [20, 35]. Anesthesia was achieved with a mixture of 30% O2, 70% N2O and 1.5% halothane. Two experimental groups of animals were investigated. One group was subjected to a global ischemia of 5-min duration, the other group to a short 2.5-min period of ischemia, usually used for preconditioning. Control gerbils (n=10) were subjected to a sham operation, comprising anesthesia and all surgical procedures except clamping of the carotid arteries. At the determined three time endpoints of the experiment, i.e., 24, 48, and 96 h after reperfusion (n=4–5 per time point), the respective animals were killed. Brains were either perfusion-fixed with 4% paraformaldehyde (PFA) for immunohistochemistry or rapidly removed and frozen for receptor autoradiography. All animal procedures were carried out according to the guidelines of the German animal protection law.
Immunohistochemistry
At the determined endpoints of the experiment, the animals were deeply anesthetized and perfused through the ascending aorta with saline for 2 min followed by 4% PFA in 0.1 M phosphate buffer for 10 min. The brains were removed, postfixed overnight in the same fixative, and then transferred to 0.5% PFA/0.1 M phosphate buffer until further processing. Immunohistochemistry was performed on coronal free-floating 50-μm vibratome sections with an antibody against cannabinoid receptor 1, obtained from Calbiochem (Schwalbach, Germany), generated in rabbits immunized with a synthetic peptide corresponding to amino acid residues 1–14. The antibody is well characterized and the specificity of antisera has previously been demonstrated [17, 27]. After pretreatment with methanol 70%/1% H2O2 for 10 min, free aldehyde moieties were blocked by a 10-min incubation in phosphate-buffered saline (PBS)/0.4% borohydride. Sections were then incubated in normal swine serum (10% in PBST) for 30 min, followed by the primary antisera for 72 h at 4°C. The primary antibody was diluted 1:200. Immunoreactivity (IR) was visualized by the avidin-biotin complex method (Vectastain, Vector Laboratories, USA). Sections were developed in 0.02% diaminobenzidine with 0.02% H2O2. The reaction product was intensified by adding 0.02% cobalt chloride and nickel ammonium sulfate. Omitting the primary antisera in a subset of control slides resulted in no immunostaining at all (not shown).
For semiquantitative analysis of receptor protein expression, hippocampi were investigated by optical densitometry as previously described [34], with minor modifications. All sections were examined at a final magnification of ×40 using a Zeiss Axiophot microscope (Jena, Germany). Images were scanned in equal light conditions with a digital camera (Roper, Ottobrunn/Munich, Germany). From hippocampal subfields CA1 and CA3, the pyramidal layer and both dendritic layers were selected on the monitor. Optical density was automatically determined using the AIS imaging research software (Imaging Research Inc., St. Catharines, Ontario, Canada). The optical density of the corpus callosum was used as reference value for background staining and subtracted from the total optical density, resulting in specific immunolabeling. Values (given in %) are expressed as mean ± SEM. Statistical analysis was performed using the General statistics module of Analyse-it for Microsoft Excel (Analyse-it Software, Ltd., Leeds, UK). Significant group effects were confirmed by analysis of variance (ANOVA) and Bonferroni error protection with a significance level at P<0.05.
Receptor autoradiography
For receptor autoradiography, the gerbils were decapitated, their brains rapidly removed, frozen in isopentane at –30°C for 10 min, and stored at –80°C until analysis. Coronal cryostat sections of 12-µm thickness were serially cut at –20°C at the level of the dorsal hippocampus and mounted on TESPA-coated slides. Quantitative in vitro receptor autoradiography studies were performed using [3H]CP 55940, purchased from NENTM Life science Products Inc. (Boston, USA), as ligand for CB1 receptors. Labeling of the CB1 receptor was performed according to the protocol of Herkenham et al. [16]. Briefly, slides were incubated with 10 nM [3H]CP 55940 (50 mM Tris-HCl buffer, pH 7.4, containing 5% BSA) for 90 min at 22°C. Incubation was terminated by washing in ice-cold buffer containing 1% BSA for 4 h. Unspecific binding was determined by co-incubation of alternating sections with [3H]CP 55940 and an excess of appropriate unlabeled CP 55940. After the final rinsing procedure, slides were carefully dried in a stream of warm air. Air-dried, 3H-labeled sections were co-exposed with 3H-labeled plastic standards (Microscales®; Amersham, Braunschweig, Germany) and brain-paste standards to a 3H-sensitive film (Kodak BioMaxMR, Kodak Scientific Imaging Film) for 8 weeks. Developed films were scanned in equal light conditions with a DMC video camera (Polaroid, Offenbach, Germany) and digitized with the AIS image analysis system (Imaging Research Inc., St. Catharines, Ontario, Canada). Gray value images of the co-exposed plastic standards were used to compute a non-linear calibration curve, which defined the relationship between gray values in the autoradiographic brain slices and concentrations of radioactivity. Plastic standards were calibrated to tissue standards with known concentrations of radioactivity. Final values were normalized to sham-operated controls (100%) and expressed as mean ± SEM, as described by Hansen et al. [13]. Quantitative analysis of radioactivity was performed in the hippocampal formation. Subfields CA1 and CA3 were marked on the monitor and the gray values automatically assessed by the imaging software. In all cases, nonspecific binding was just above background labeling or not visible at all. Therefore, background density could be used as an estimate of unspecific binding and subtracted from total binding. Statistical analysis was performed using the General statistics module of Analyse-itTM for Microsoft Excel. Ligand binding was analyzed by calculating mean concentration values for each reperfusion time point and hippocampal region. Significant group effects were confirmed by analysis of variance (ANOVA) and Bonferroni error protection. A P value <0.05 was considered statistically significant.
Results
Immunohistochemistry
Ischemia for 5 min
Hippocampal CB1 receptor-like IR in sham controls was prominent in the neuropil of CA1 and CA3 pyramidal layers. Strata radiatum and oriens were stained to a lesser degree. Some strongly labeled interneurons were visible within the pyramidal cell layer but also in dendritic layers. After a 5-min period of global ischemia, IR was slightly, but not significantly, reduced in CA1 and CA3. Significant lower levels of CB1 receptor staining intensity were found 96 h after ischemia, especially in the dendritic strata oriens and radiatum of CA1 hippocampal subregion (Figs. 1, 2).

Representative photomicrographs from hippocampal CB1 receptor protein expression (left) and [3H]CP 55940 ligand binding (right) after a short 2.5-min ischemia, usually used for preconditioning. Hippocampal CB1 receptor IR in the vulnerable CA1 subfield is maintained up to 48 h, and then declines in association with delayed neuronal death. In contrast, ligand binding to CB1 receptors is decreased in CA1 and CA3 from 24 h up to 96 h of reperfusion (CB1 receptor cannabinoid type 1 receptor, IR immunoreactivity). Bar 100 μm

Semiquantitative analysis of hippocampal CB1 receptor IR after 5- and 2.5-min ischemia. After 5-min ischemia, CB1 receptor IR in CA1 is slightly, but not significantly, decreased after 24 and 48 h. In association with neuronal death after 96 h, immunostaining is significantly lowered. Similarly, in CA3, IR is only slightly reduced. In contrast, a 2.5-min period of ischemia usually used for preconditioning leads to a transient decrease of CB1 receptor IR in CA1 and CA3 between 24 and 48 h (so stratum oriens, sp stratum pyramidale, sr stratum radiale). *Significant; P<0.05, ANOVA with Bonferroni error protection
Ischemia for 2.5 min
A 2.5-min period of ischemia caused a significant reduction of CB1 receptor-like IR, by about 40%, of sham-operated animals after 24 and 48 h in all layers of hippocampal CA1 and CA3 subfields. At 96 h after reperfusion, CB1 receptor IR returned to control levels (Figs. 1, 2).
Receptor autoradiography
Ischemia for 5 min
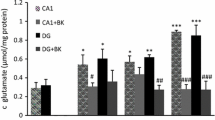
In sham-operated control gerbils, dense [3H]CP 55940 ligand binding was visible in hippocampal subfields CA1 and CA3 (Fig. 1). [3H]CP 55940 ligand binding levels in the vulnerable CA1 subfield remained widely unchanged after a 5-min period of ischemia, up to 96 h after reperfusion (Fig. 3). In CA3, a marked but not significant increase of [3H]CP 55940 binding values to about 60% compared to that of sham-operated gerbils was detectable after 96 h Fig. 3).

Quantitative analysis of hippocampal [3H]CP 55940 ligand binding after 5- and 2.5-min ischemia. A 2.5-min period of ischemia usually used for preconditioning causes significant down-regulation of CB1 receptor binding density in CA1 and CA3 from 24 h onwards, while after a 5-min period of ischemia ligand binding is widely unchanged at any time point investigated. *Significant; P<0.05, ANOVA with Bonferroni error protection
Ischemia for 2.5 min
A 2.5-min period of ischemia caused a significant and persistent reduction of [3H]CP 55940 binding levels by about 30% of sham-operated control animals (Figs. 1, 3). Similarly, [3H]CP 55940 binding density was also significantly reduced in CA3, 24, 48, and 96 h, respectively, after reperfusion (Figs. 1, 3).
Discussion
Our present study demonstrates for the first time that a 2.5-min period of ischemia, usually used to induce ischemic tolerance, is associated with a decrease of hippocampal CB1 receptor density as shown both by receptor autoradiography and immunohistochemistry (Figs. 1, 2, 3). Global ischemia of 5-min duration followed by DND of vulnerable CA1 neurons fails to alter ligand binding to these receptors (Figs. 2, 3). Immunohistochemically, decreased CB1 receptor immunoreactivity in CA1 is seen only in association with neuronal death 96 h after reperfusion. These data suggest that down-regulation of CB1 receptors may participate in endogenous ischemic resistance.
Originally, elevation of endocannabinoids after brain injury and subsequent activation of CB1 receptors was thought to be part of an autocrine protection system of the CNS [25]. Therefore, activation of CB1 receptors should be protective and their blockade deleterious [22, 23, 28, 31, 32, 33, 40]. However, a growing body of evidence rebuts a general neuroprotective role of cannabinoids and the endocannabinoid system in response to excitotoxic injuries. Tetrahydrocannabinol (THC), the major psychoactive component of marijuana, has been shown to induce apoptotic cell death in hippocampal slices [6]. Furthermore, THC could not prevent hippocampal neuronal death in a rat forebrain ischemia model [22]. Transgenic mice lacking the enzyme fatty acid amide hydrolase, which is necessary to degrade endocannabinoids, exhibited increased seizure susceptibility and increased neuronal loss of hippocampal CA1 and CA3 neurons in bicuculline and kainate seizure models [7]. On the other hand, pharmacological blockade of CB1 receptor function was able to prevent disseminating brain damage in a neonatal rat model following NMDA-induced excitotoxicity [14]. In a rat model of focal ischemia, a significant decrease of infarct volumes by application of the cannabinoid receptor antagonist SR141716A has convincingly been demonstrated by Berger et al. [3]. These controversial results may be due to the diverse cellular distribution of CB1 receptors, which are not only expressed by neurons but also—and obviously of more importance—by vascular smooth muscle cells and endothelial cells [11, 15, 41]. In fact, in the heart, with CB1 and CB2 receptors restricted to the endothelium, pharmacological blockade of these receptors leads to impaired vasodilatation and loss of the preconditioning effect [5]. Since changes in the cerebral blood flow do not represent the biological basis for preconditioning in the brain [1], our findings demonstrating down-regulation of these receptors in the same situation is not per se contradictory, but indicates that CB1 receptors are only a first step in a multitude of intracellular pathways.
The kinetics of [3H]CP 55940 binding after a 2.5-min ischemia, with persistent decrease between 24 and 96 h, paralleled reduced CB1 receptor immunoreactivity in the preconditioned CA1 subfield between 24 and 48 h. After 96 h, however, CB1 receptor protein expression returned to control levels in contrast to persistently lowered [3H]CP 55940 binding density at this time point (Figs. 1, 2, 3). It could be argued that this reduction of CB1 receptor protein expression is an epiphenomenon due to inhibition of protein synthesis, which also occurs after a short period of ischemia and, eventually, recovers to near normal 4 days after recirculation [2, 29]. However, after a 5-min period of ischemia with irreversible inhibition of protein synthesis [29, 38], CB1 receptor immunoreactivity persists up to 48 h, suggesting that the observed reduction after the short ischemic period is an active process. Persistently decreased [3H]CP 55940 ligand binding to CB1 receptors 96 h after a priming 2.5-min ischemia, the time point when the second ischemic insult is superimposed in our model of ischemia tolerance induction [35, 36, 37], indicates that despite retained abundance of receptor protein the endocannabinoid system is still down-tuned, which may confer neuroprotection. An alternative explanation for the transient decrease of CB1 receptor immunoreactivity could be an increased amount of internalized receptors as a consequence of enhanced release and binding of endocannabinoids upon transient ischemia [3, 8]. However, pre-treatment of slides with Triton should also detect internalized receptors.
In spite of decreased CB1 receptor immunoreactivity in CA1 96 h after global ischemia of 5-min duration in association with severe neuronal loss [34, 36], [3H]CP 55940 binding was not significantly reduced in CA1 at any time point investigated (Fig. 3). A similar phenomenon was observed in a model of focal ischemia in the infarct core 5 h after ischemia (unpublished observation). One possible explanation for this persistence of near-normal levels of [3H]CP 55940 binding in postischemic CA1 is a delayed degradation of CB1 receptor-binding sites. Alternatively, [3H]CP 55940 may bind to ischemia-resistant GABAergic interneurons [39] or to astrocytes, which also possess CB1 receptors [26] and proliferate after the ischemic insult [21]. However, in this case, immunolabeling of CB1 receptors should also be maintained.
In conclusion, survival of vulnerable CA1 neurons after a short ischemic period, usually used for tolerance induction, is associated with a decrease of CB1 receptor density in hippocampal subfields CA1 and CA3, whereas an ischemic period of 5-min duration followed by DND of hippocampal CA1 principal neurons fails to reduce CB1 receptors in these regions. Thus, our data strongly suggest that down-regulation of central CB1 receptors may participate in endogenous postischemic neuroprotection of vulnerable hippocampal neuronal subpopulations.
References
Alkayed NJ, Goyagi T, Joh HD, Klaus J, Harder DR, Traystman RJ, Hurn PD (2002) Neuroprotection and P450 2C11 upregulation after experimental transient ischemic attack. Stroke 33:1677–1684
Araki T, Kato H, Inoue T, Kogure K (1990) Regional impairment of protein synthesis following brief cerebral ischemia in the gerbil. Acta Neuropathol 79:501–505
Berger C, Schmid PC, Schäbitz W-R, Wolf M, Schwab S, Schmid HHO (2004) Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem 88:1159–1167
Bond A, Lodge D, Hicks CA, Ward MA, O’Neill MJ (1999) NMDA receptor antagonism, but not AMPA receptor antagonism attenuates induced ischaemic tolerance in the gerbil hippocampus. Eur J Pharmacol 380:91–99
Bouchard JF, Lepicier P, Lamontagne D (2003) Contribution of endocannabinoids in the endothelial protection afforded by ischemic preconditioning in the isolated rat heart. Life Sci 72:1859–1870
Chan GC-K, Hinds TR, Impey S, Storm DR (1998) Hippocampal neurotoxicity of Δ9-tetrahydrocannabinol. J Neurosci 18:5322–5332
Clement AB, Hawkins EG, Lichtman AH, Cravatt BF (2003) Increased seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J Neurosci 23:3916–3923
Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, Irving AJ (2001) Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci 21:2425–2433
Di Marzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, Zimmer AM, Zimmer A, Martin BR (2000) Levels, metabolism and pharmacological activity of anandamide in CB1 cannabionoid receptor knockout mice: evidence for non-CB1, non-CB2 receptor-mediated actions of anandamide in mouse brain. J Neurochem 75:2434–2444
Dirnagl U, Simon RP, Hallenbeck JM (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 26:248–254
Gebremedhin D, Lange AR, Campbell WB, Hillard CJ, Harder DR (1999) Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am J Physiol 276:H2058–2093
Gérard CM, Mollereau C, Vassart G, Parmentier M (1991) Molecular cloning of a human cannabinoid receptor shich is also expressed in testis. Biochem J 279:129–134
Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, Manzanares J, Ikonomidou C, Schmid HH, Fernandez-Ruiz JJ, Hansen HS (2001) Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem 78:1–14
Hansen HH, Azcoitia I, Pons S, Romero J, Garcia-Segura LM, Ramos JA, Hansen HS, Fernández-Ruiz J (2002) Blockade of cannabinoid CB1 receptor function protect against in vivo disseminating brain damage following NMDA-induced excitotoxicity. J Neurochem 82:154–158
Herkenham M, Lynn AB, Little MD, Johnson R, Melvin LS, Costa BR de, Rice KC (1990) Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA 87:1932–1936
Herkenham M, Lynn AB, Johnson MR, Melvin LS, Costa BR de, Rice KC (1991) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11:563–583
Howlett AC, Song C, Berglund BA, Wilken GH, Pigg JJ (1998) Characterization of CB1 cannabinoid receptors using receptor peptide fragments and site-directed antibodies. Mol Pharmacol 53:504–510
Kato H, Liu Y, Araki T, Kogure K (1992) MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett 139:118–121
Kirino T (1982) Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239:57–69
Kirino T, TsujitaY, Tamura A (1991) Induced tolerance to ischemia in gerbil hippocampal neurons. J Cereb Blood Flow Metab 11:299–307
Kitagawa K, Matsumoto M, Ohtsuki T, Kuwabara K, Mabuchi T, Yagita Y, Hori M, Yanagihara T (2000) Extended neuronal protection induced after sublethal ischemia adjacent to the area with delayed neuronal death. Neuroscience 96:141–146
Louw DF, Yang FW, Sutherland GR (2000) The effect of δ-9-tetrahydrocannabinol on forebrain ischemia in rat. Brain Res 857:183–187
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B (2003) CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302:84–88
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
Mechoulam R, Lichtman AH (2003) Neuroscience. Stout guards of the central nervous system. Science 302:65–67
Moldrich G, Wenger T (2000) Localization of the CB1 cannabinoid receptor in the rat brain. An immunohistochemical study. Peptides 21:1735–1742
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA (1999) Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 19:2987–2995
Nakagomi T, Kirinio T, Kanemitsu H, Tsujita Y, Tamura A (1993) Early recovery of protein synthesis following ischemia in hippocampal neurons with induced tolerance in the gerbil. Acta Neuropathol 86:10–15
Nellgard B, Wieloch T (1992) Postischemic blockade of AMPA but not NMDA receptors mitigates neuronal damage in the rat brain following transient severe cerebral ischemia. J Cereb Blood Flow Metab 12:2–11
Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R,Shohami E (2001) An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413:527–531
Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E (2005) CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab 25:477–484
Parmentier-Battuer S, Jin K, Mao XO, Xie L, Greenberg DA (2002) Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci 22:9771–9775
Sommer C, Kiessling M (2002) Ischemia and ischemia tolerance induction differentially regulate protein expression of GluR1, GluR2 and AMPA receptor binding protein (ABP) in the gerbil hippocampus: GluR2 (GluR-B) reduction does not predict neuronal death. Stroke 33:1093–1100
Sommer C, Gass P, Kiessling M (1995) Selective c-JUN expression in CA1 neurons of the gerbil hippocampus during and after acquisition of an ischemia-tolerant state. Brain Pathol 5:135–144
Sommer C, Fahrner A, Kiessling M (2002) [3H]Muscimol binding to GABAA receptors is upregulated in CA1 neurons of the gerbil hippocampus in the ischemia tolerant state. Stroke 33:1698–1705
Sommer C, Fahrner A, Kiessling M (2003) Postischemic neuroprotection in the ischemia-tolerant state gerbil hippocampus is associated with increased binding to inhibitory GABAA receptors. Acta Neuropathol 105:197–202
Thilmann R, Xie Y, Kleihues P, Kiessling M (1986) Persistent inhibition of protein synthesis precedes delayed neuronal death in postischemic gerbil hippocampus. Acta Neuropathol (Berl) 71:88–93
Tsou K, Mackie K, Sanudopena MC, Walker JM (1999) Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing gabaergic interneurons in the rat hippocampal formation. Neuroscience 93:969–975
Van der Stelt M, Veldhuis WB, Haaften GW van, Fezza F, Bisogno T, Bar PR, Veldink GA, Vliegenthart JF, Di Marzo V, Nicolay K (2001) Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J Neurosci 21:8765–8771
Wagner JA, Jarai Z, Kunos G (2001) Hemodynamic effects of cannabinoids: coronary and cerebral vasodilation mediated by cannabinoid CB(1) receptors. Eur J Pharmacol 423:203–210
Acknowledgements
The authors greatly acknowledge the excellent technical assistance of Mr. Stephan Hennes. Ms. Astrid Wöber kindly gave editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
The first two authors contributed equally
Rights and permissions
About this article
Cite this article
Schomacher, M., Müller, H.D. & Sommer, C. Short-term ischemia usually used for ischemic preconditioning down-regulates central cannabinoid receptors in the gerbil hippocampus. Acta Neuropathol 111, 8–14 (2006). https://doi.org/10.1007/s00401-005-1109-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-005-1109-2