
Abstract
Three unrelated patients, one girl, one boy, and an adult female, aged 14, 11 and 41 years, respectively, at the time of biopsy, revealed lysosomal glycogen storage, autophagic vacuoles and peculiar globular inclusions of distinct ultrastructure, which were reducing but did not appear like true “reducing bodies” as described in the congenital myopathy “reducing body myopathy”. All three patients had residual activity of acid α-glucosidase in their muscle biopsy samples. Leukocytes in the girl showed normal acid α-glucosidase activity, but in the boy activity was reduced. Molecular genetic analysis of the GAA gene revealed disease-causing mutations in each patient: H568L/R672W, IVS1–13T>G/G615F, and IVS1–13T>G/IVS1–13T>G. Although only one patient with such globular inclusions has been reported up to now, the three patients described here indicate that in the late-onset type of GSD II such inclusions may not be rare.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Among the different types of glycogen storage diseases, type II glycogenosis (GSD II) has a broad clinical spectrum as to involvement of different organs as well as to onset and duration of the disease, allowing clinical distinction of early-onset, i.e. infantile, and delayed/late onset or post-infantile, i.e. juvenile/adult forms, with clinically intermediate variants as well. Owing to a deficiency of lysosomal acid α-glucosidase (or acid maltase, hence also called acid maltase deficiency disease), accumulation of particulate glycogen in the lysosomal compartment was found in almost every type of cell and organ, although striated muscle cells are clinically most severely affected. Apart from lysosomal glycogen and accretion of sarcoplasmic glycogen, autophagic vacuoles may be a prominent feature in older patients. In this report, we describe three patients with unusual “reducing” inclusions in muscle, whose diagnosis of GSD II was established biochemically and molecular genetically.
Clinical data
Two unrelated children, one girl and one boy, showed delay in motor development during early childhood and mild to moderate muscle weakness early in their second decade of life (Table 1). The girl also had large calves, reduced deep tendon reflexes, and elevated creatine kinase (CK) around 1,000 U/L. Electromyography (EMG) revealed a “myopathic” pattern. The third patient was a 41-year-old woman who, for the past 1.5 years, showed reduced sportive achievements, backache, and mild muscle weakness in her iliopsoas muscles. Both thighs appeared somewhat large and deep tendon reflexes were weak. Her EMG showed a “myopathic” pattern and CK was 186 U/L. A maternal great grandmother and a paternal great grandmother were sisters.
Material and methods
Biopsies were performed on the quadriceps muscle in each child between 10 and 14 years of age and in the adult patient (Tables 1, 2). Unfixed frozen tissue was submitted to a standard battery of histological, histochemical, enzyme histochemical and immunohistochemical techniques. Separate small specimens were each obtained for immediate fixation in buffered glutaraldehyde, embedded for electron microscopy and studied in ultrathin sections. The following antibodies were used for immunohistochemical studies: desmin, vimentin, ubiquitin, αB-crystallin, heat-shock protein 72 (hsp72), dys1, dys2, dys3, α-sarcoglycan, merosin 80, and α- and β-dystroglycans. The Thiéry technique of 1% silver protein in distilled water [24] was applied to ultrathin sections.
The glycogen content and the acid α-glucosidase activity were determined using the method described by Shin [23]. Isolated DNAs were analysed for mutations in the α-glucosidase gene (GAA) by direct sequencing.
The entire coding region and the flanking intronic sequences of the 20 exons of the GAA gene were searched for mutations. Genomic DNA was isolated from leukocytes using the QIAmp Blood Kit (Qiagen), according to the manufacturer’s instructions. PCR reactions were performed with modified primers [3] in 25-μl volumes containing 30–40 ng genomic DNA template, 1× PCR buffer, 180 mM of each dNTP, 5 nmol of each primer, and 0.5 U Taq DNA polymerase. The PCR conditions were 95.0°C for 15 min, 30 cycles of 95.0°C for 20 s, 55.0°C for 1 min, and 72.0°C for 1 min, and 72.0°C for 1 min. Direct sequencing was performed by the dsDNA Thermo Sequenase fluorescence-labelled primer cycle sequencing kit (Amersham Pharmacia Biotech) according to established procedures.
Results
Morphology
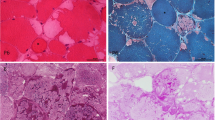
Each of the three different muscle specimens displayed similar myopathological features. While variation in fibre diameters was mild, without necrosis and regeneration or ragged red fibres, numerous scattered muscle fibres displayed vacuoles, partly of the autophagic type, with conspicuous histochemical activity of acid phosphatase (Figs. 1, 3) and distinct globules inside many of them (Table 2). These globules were round or oval in shape, greenish or reddish in the modified Gomori trichrome preparation (Figs. 1, 3) or bluish in the haematoxylin-eosin preparation (Fig. 2). They were also reducing in that, when the substrate in the menadione-linked α-glycerophosphate dehydrogenase (MAG) preparation was omitted, the bodies showed a bluish hue of formazan deposition, i.e. they were “reducing” (Fig. 1).

Patient 1. a Distinct globules within autophagic vacuoles (arrows); modified Gomori trichrome stain, x640. b Strong acid phosphatase activity in vacuoles, x320. c The globules are “reducing”, i.e. appear bluish-grey within muscle fibres; MAG reaction without substrate, ×1,040. d Accumulation of αB-crystallin within muscle fibres; immunohistochemistry, ×480. e Accumulation of heat-shock protein 72 in muscle fibres (arrows); immunohistochemistry, ×240. f Focal accretion (arrows) of desmin within muscle fibres; immunohistochemistry, ×135. g Focal accumulation (arrows) of dystrophin within muscle fibres distant to normal subsarcolemmal expression; immunohistochemistry, ×200. h Focal accumulation (arrows) of spectrin within muscle fibres distant to normal transsarcolemmal expression; immunohistochemistry, ×160. i Two demarcated very electron-dense globules surrounded by granular debris within a large autophagic vacuole, ×58,320. j Glycogen identified by the Thiéry technique as particulate glycogen granules is not present within globules, ×22,500

Patient 2. a Globules (arrows) within autophagic vacuoles of a muscle fibre; haematoxylin-eosin stain, ×65. b There are several distinct electron-dense globules (arrows) within an autophagic vacuole, ×6,500
The globular lesions did not show activity of oxidative enzymes and ATPase. Immunohistochemically, they did not react with antibodies against desmin, α-actinin, actin, vimentin, ubiquitin, merosin, α-sarcoglycan or α-dystroglycan, but did so with antibodies against αB-crystallin, hsp72, β-dystroglycan, dystrophin, and spectrin (Figs. 1, 3).

Patient 3. a Intravacuolar globules (arrows) within muscle fibres; modified Gomori trichrome stain, ×640. b Vacuoles (arrows) show activity of acid phosphatase, ×320. c Focal accretion (arrows) of α-B crystallin within muscle fibres; immunohistochemistry, ×200. d Globular inclusion, ×34,020
Ultrastructurally, the vacuoles contained particulate glycogen and/or autophagic debris including myelin-like figures. The distinct globules appeared rather homogeneously electron dense (Figs. 1, 2, 3) without any further special features recognizable therein (Table 2). Using the Thiéry technique, which identifies granular and fibrillar glycogen, particulate glycogen was labelled, whereas the globular inclusions remained unlabelled (Fig. 1).
Biochemistry
Reduced acid α-glucosidase activity and elevated glycogen content were found in muscles of each patient (Table 3). The adult female patient had the highest residual acid maltase activity and lowest content of glycogen in her muscle. In leukocytes, the young female patient showed a normal acid maltase activity, similar to that of her mother, while the male patient showed a decreased acid maltase activity.
Molecular analysis
The young female patient showed two different missense mutations, an apparently novel 1703 transversion, which predicts an amino acid exchange H568L (http://www.pompecenter.nl) and a previously published R672W mutation. The male patient was also compound heterozygous for two known mutations, G615E and IVS1–13T>G, having inherited the latter mutation from his father. The adult female patient was homozygous for the IVS1–13T>G mutation (Table 3), each inherited from her parents.
Discussion
While slow progression of mild to moderate muscle weakness, the accumulation of lysosomal glycogen within skeletal muscle fibres and capillary walls with absence of cardiac abnormalities and profound biochemical reduction in α-glucosidase activities in skeletal muscle biopsy material are compatible with a post-infantile or delayed/late onset type of GSD II, our patients show a few unusual features, especially the globular inclusions. There were distinct, sharply demarcated inclusions, often close to or within autophagic vacuoles, but distant to nuclei. Glycogen, however, could not be well ascertained in these inclusions, either by inspection or using the Thiéry technique [24], which identifies particulate and fibrillar glycogen [7]. Based on the original technique that identified reducing bodies in reducing body myopathy [4], the globular bodies had a reducing capacity, but both the location in autophagic vacuoles and their electron density, even with some electron-lucent spots within these globular inclusions, differed from the granulotubular appearance and perinuclear site of the original “reducing” bodies or those occasionally observed in combination with desmin deposits [2, 8]. Review of the literature revealed a single report on a 2.5-year-old boy with acid maltase deficiency who had similar “reducing body”-like inclusions in his muscle fibres [15]. This observation was the only report found so far; however, our results here suggest that these globular inclusions may not necessarily be an exceptional feature in the post-infantile forms of GSD II. The origin and nature of these globular inclusions appear unclear. They may be of nuclear origin; however, nuclei of muscle fibres, both in GSD II and other instances, are not reducing and nuclei usually are not present in autophagic vacuoles. Autophagic vacuoles as components in post-infantile GSD-II can be added to the enlarging list of neuromuscular conditions marked by such non-specific autophagic or rimmed vacuoles [21]. Although reducing bodies, when viewed at higher magnification, are composed of granular and sometimes even tubular ultrastructure, they may appear similar to our “reducing” inclusions in GSD II. This has, indeed, been observed earlier in reducing body myopathy reports when the combination of autophagic vacuoles and true reducing bodies in reducing body myopathy were presented [16, 17]. However, reducing bodies, occasionally forming reducing rings around nuclei within muscle fibres, have also been encountered [16, 17]. The similarity, although not complete identity, of true reducing bodies and our GSD II reducing inclusions may suggest, within the context of autophagic vacuoles and their contents, that other inclusions in autophagic vacuoles in other neuromuscular—and other—disorders may also show “reducing” capacity when applying the “MAG without substrate” technique.
Another remarkable feature was aggregation of several proteins within muscle fibres, foremost the chaperone proteins αB-crystallin and heat-shock proteins, but also few transsarcolemmal proteins. The diversity of accrued proteins suggests both accretion of transsarcolemmal proteins and, perhaps, up-regulation of chaperone proteins, enhancing extralysosomal protein degradation within the respective muscle fibres. Thus, GSD II—and, perhaps, other lysosomal disorders—may belong to the increasing group of primary and secondary protein aggregate myopathies (PAM) [6], primary PAM being those associated with mutations in muscle fibre protein-encoding genes, secondary PAM being of different aetiology. Accumulation of transsarcolemmal proteins has been reported in GSD II [22]. Differential diagnosis to delayed/late-onset GSD II is lysosome-associated membrane protein-2 (LAMP-2) deficiency or Danon disease, also called “lysosomal glycogenosis with normal acid maltase deficiency” (the latter being a misnomer because lysosomal glycogen accumulation is confined to muscle fibres in Danon disease though ubiquitously present in GSD II). Clinically, Danon disease differs markedly from delayed/late-onset GSD II in that it is an X-linked disorder, often associated with mental retardation, severe cardiac problems and mutations in the LAMP-2 gene.
Another important feature is the merely reduced, although not absent residual activity of acid α-glucosidase/acid maltase in skeletal muscles of all three patients. The normal acid maltase activity in leukocytes of the girl could be due to the neutral and/or renal enzymes which are active at acid pHs. This selected organ deficiency had been documented in the juvenile type of disease [12, 15] as well as in the adult type [20], and may be due to the fact that the biochemical assay may distinguish between different isoforms of acid α-glucosidase, i.e. the one present in skeletal muscle versus the one present in leukocytes [11]. In line with the notion that the level of residual enzyme activity determines the clinical course, the highest level of reduced activity of acid maltase was found in the adult patient, who had not only late onset but also mild clinical symptoms and very slow progression.
Our molecular data revealed one novel mutation [10], which has not been hitherto reported according to the current data base of GAA mutations (http://www.pompecenter.nl, last update: 20 December 2004). Two of our three patients were genetically compound heterozygotes, in line with the many other patients studied in earlier series, i.e. 11/12 [11], 11/11 [1], 1/21 patients [19], 7/29 [10] including patients with infantile GSD II in whom six new mutations were described that were completely different from any seen in our patients [5]. Apparently, neither of the two mutations of the GAA gene, IVS1–13T>G and R672W, which permit a trace of residual enzyme activity, gives rise to a severe form of GSD II with early onset and early death. The “leaky splice” IVS1−13T>G mutation is known to be the most frequent one among late-onset GSD II patients of Northern European origin [3, 10, 13, 18, 19]. However, homozygosity for this insertion has been a rather rare event in a large series of patients [1, 10, 19]. Concerning the “leaky splice” IVS1–13T>G mutation (not associated with the early-onset form), transcription from a GAA allele carrying a T-to-G transversion at base IVS1–13 (intron 1) results in RNA skipping of exon 2 and a reduced level of correctly spliced mRNA (10–15% of normal).
Finally, both calves were large in the young girl, adding calf hypertrophy as a rare sign to the clinical spectrum of childhood GSD II. As most biopsies had been performed only in quadriceps muscles, including in our young female patient, the morphological equivalent of these large calves remains unknown. A paravertebral pseudotumour has been identified in juvenile acid maltase deficiency [14], and our adult patient was noted to have somewhat large thighs. Our three patients apparently had a mild form of delayed/late-onset GSD II, compared to a large cohort of recently published 54 Dutch patients (described to have late-onset Pompe disease, although the designation “Pompe disease” should be confined to the infantile form which was originally described by Pompe) as our three patients were neither wheelchair-bound nor dependent on artificial ventilation or respiratory support. This is possibly due to the fact that the duration of their disease up to the time of biopsy was rather short. Proximal muscle weakness was the main symptom and affected movements as amply described in this recent cohort of 54 Dutch patients [9], such as running, walking staircases, rising from sitting or lying positions, dressing, etc. Details in such clinical findings are important to assess therapeutic success or failure, the basis for this large-scale study by questionnaire [9].
References
Angelini C, Cenacchi G, Nascimbeni AC, Fulizio L (2003) Morphological changes in late onset acid maltase deficiency patients with splicing gene mutation. Acta Myol 22:90−96
Bertini E, Salviati G, Apollo F, Ricci E, Servidei S, Broccolini A, Papacci M, Tonali P (1994) Reducing body myopathy and desmin storage in skeletal muscle: morphological and biochemical findings. Acta Neuropathol 87:106−112
Boerkoel CF, Exelbert R, Nicastri C, Nichols RC, Miller FW, Plotz PH, Raben N (1995) Leaky splicing mutation in the acid maltase gene is associated with delayed onset of glycogenosis type II. Am J Hum Genet 56:887−897
Brooke MH, Neville HE (1972) Reducing body myopathy. Neurology 22:829−840
Fernandez-Hojas R, Huie ML, Navarro C, Dominguez C, Roig M, Lopez-Coronas D, Teijeira S, Anyane-Yeboa K, Hirschhorn R (2002) Identification of six novel mutations in the acid alpha-glucosidase gene in three Spanish patients with infantile onset glycogen storage disease type II (Pompe disease). Neuromuscul Disord 12:159−166
Goebel HH, Borchert A (2002) Protein surplus myopathies and other rare congenital myopathies. Semin Pediatr Neurol 9:160−170
Goebel HH, Shin YS, Gullotta F, Yokota T, Alroy J, Voit T, Haller P, Schulz A (1992) Adult polyglucosan body myopathy. J Neuropathol Exp Neurol 51:24−35
Goebel HH, Halbig LE, Goldfarb L, Schober R, Albani M, Neuen-Jacob E, Voit T (2001) Reducing body myopathy with cytoplasmic bodies and rigid spine syndrome: a mixed congenital myopathy. Neuropediatrics 32:196−205
Hagemans MLC, Winkel LPF, Van Doorn PA, Hop WH, Loonen MCB, Reuser AJJ, Van der Ploeg AT (2005) Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 128:671−677
Hermans MM, Leenen D van, Kroos MA, Beesley CE, Van Der Ploeg AT, Sakurba H, Wevers R, Kleijer W, Michelakakis H, Kirk EP, et al (2004) Twenty-two novel mutations in lysosomal α-glucosidase gene (GAA) underscores the genotype-phenotype correlation in glycogen storage disease type II. Hum Mutat 23:47−56
Hirschhorn R, Reuser AJJ (2001) Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 3389−3420
Hübner G, Pongratz D (1982) Granularkörpermyopathie (sog. reducing body myopathy). Pathologe 3:111−113
Huie ML, Chen AS, Tsujino S, Shanake S, DiMauro S, Engel AG, Hirschhorn R (1994) Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1(-13T>G) mutation in a majority of patients and a novel IVS10(+1GT>CT) mutation. Hum Mol Genet 3:2231−2236
Iancu TC, Lerner A, Shiloh H, Bashan N, Moses S (1988) Juvenile acid maltase deficiency presenting as paravertebral pseudotumour. Eur J Pediatr 147:372−376
Jay V, Christodoulou J, Mercer-Connolly A, McInnes RR (1992) “Reducing body”-like inclusions in skeletal muscle in childhood-onset acid maltase deficiency. Acta Neuropathol 85:111−115
Kiyomoto BH, Murakami N, Kishibayashi J, Sunohara N, Nonaka I (1995) Reducing bodies in distal myopathy with rimmed vacuole formation. Acta Neuropathol 89:109−111
Kiyomoto BH, Murakami N, Kobayashi Y, Nihei K, Tanaka T, Takeshita K, Nonaka I (1995) Fatal reducing body myopathy: ultrastructural and immunohistochemical observations. J Neurol Sci 128:58−65
Kroos MA, Van der Kraan M, Van den Boogaard MJ, Ausems MGEM, Ploos van Amstel HK, Poenaru L, Nicolino M, Wevers RA, Diggelen OP van, Kleijer WJ, Reuser AJJ (1995) Glycogen storage disease type II: the frequency of three common mutant alleles and their associated clinical phenotypes studied in 121 patients. J Med Genet 32:836−837
Lafôret P, Nicolino M, Eymard B, Puech JP, Caillaud C, Poenaru L, Fardeau M (2000) Juvenile and adult-onset acid maltase deficiency in France: genotype-phenotype correlation. Neurology 55:1122−1128
Minato S, Kobayashi T, Tanaka K, Kitaguchi T, Goto I (1988) Adult Pompe disease with normal acid α-glucosidase activity in leukocytes (in Japanese). Rinsho Shinkeigaku 28:413−416
Neudecker S, Krasnianski M, Bahn E, Zierz S (2004) Rimmed vacuoles in facioscapulohumeral muscular dystrophy: a unique ultrastructural feature. Acta Neuropathol 108:257−259
Radojevic V, Humm AM, Rösler KM, Lauterburg T, Burgunder J-M (2003) Abnormal trafficking of sarcolemmal proteins in α-glucosidase deficiency. Acta Neuropathol 105:373−380
Shin YS (1990) Diagnosis of glycogen storage disease. J Inherit Metab Dis 13:419−434
Thiéry JP (1967) Mise en evidence des polysaccharides sur coupes fines en microscopie electronique. J Microsc 6:987−1018
Acknowledgements
We gratefully acknowledge a fellowship to Dr. M.C. Sharma from the Deutsche Forschungsgemeinschaft (DFG−INSA), the technical support of Ms. M. Schlie and Ms. I. Warlo, photographic assistance from Mr. W. Wagner and editorial help of Ms. A. Wöber.
Conflict of interest:
No information supplied
Author information
Authors and Affiliations
Corresponding author
Additional information
Note added in proof.
After this paper went to print, both parents of patient 3 were found to be heterozygous for a IVS1-13t>g mutation in intron 1 of the GAA gene for GSD II.
Rights and permissions
About this article
Cite this article
Sharma, M.C., Schultze, C., von Moers, A. et al. Delayed or late-onset type II glycogenosis with globular inclusions. Acta Neuropathol 110, 151–157 (2005). https://doi.org/10.1007/s00401-005-1026-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-005-1026-4