
Abstract
Alterations in the γ-aminobutyric acid (GABA) neurotransmitter and receptor systems may contribute to vulnerability of hippocampal pyramidal neurons in Alzheimer’s disease (AD). The present study examined the immunohistochemical localization and distribution of GABAB receptor R1 protein (GBR1) in the hippocampus of 16 aged subjects with a range of neurofibrillary tangle (NFT) pathology as defined by Braak staging (I–VI). GBR1 immunoreactivity (IR) was localized to the soma and processes of hippocampal pyramidal cells and some non-pyramidal interneurons. In control subjects (Braak I/II), the intensity of neuronal GBR1 immunostaining differed among hippocampal fields, being most prominent in the CA4 and CA3/2 fields, moderate in the CA1 field, and very light in the dentate gyrus. AD cases with moderate NFT pathology (Braak III/IV) were characterized by increased GBR1-IR, particularly in the CA4 and CA3/2 fields. In the CA1 field of the majority of AD cases, the numbers of GBR1-IR neurons were significantly reduced, despite the presence of Nissl-labeled neurons in this region. These data indicate that GBR1 expression changes with the progression of NFT in AD hippocampus. At the onset of hippocampal pathology, increased or stable expression of GBR1 could contribute to neuronal resistance to the disease process. Advanced hippocampal pathology appears to be associated with decreased neuronal GBR1 staining in the CA1 region, which precedes neuronal cell death. Thus, changes in hippocampal GBR1 may reflect alterations in the balance between excitatory and inhibitory neurotransmitter systems, which likely contributes to dysfunction of hippocampal circuitry in AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over-activation of excitatory amino acid (EAA) receptors leads to excitotoxic neuronal changes that can contribute to neuropathology in Alzheimer’s disease (AD) [3, 4, 13, 30]. Excitotoxicity due to excessive EAA receptor stimulation could be countered by compensatory activation of inhibitory neurotransmission. γ-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the central nervous system [29] and GABA signaling occurs through two major classes of receptors, the ionotropic GABAA type, and the metabotropic GABAB type [20]. Our previous studies demonstrated the relative stability of GABAA receptor subunits in AD hippocampus [24, 25, 26]. The status of GABAB receptors, however, remains to be examined.
GABAB receptor activation increases K+ conductance, hyperpolarizing postsynaptic sites [19, 32] and inhibiting presynaptic Ca2+ conductance, thus suppressing neurotransmitter release and postsynaptic excitatory transmission [31, 37]. GABAB receptors are composed of at least two heteromers, GABABR1 (GBR1) and GABABR2 (GBR2) [17, 18, 36]. Compared to the GBR2, GBR1 immunoreactivity (IR) is more prominent in neuronal soma and proximal dendrites [5], where pathological material is known to accumulate during the formation of neurofibrillary tangles (NFT) in AD. Therefore, immunohistochemical examination of the GBR1 protein in AD brain allows for the assessment of potential neuronal GABAB receptor changes relative to the development of NFT pathology. The present study employed immunohistochemical techniques to examine the cellular localization and density of the GBR1 receptor subunit in the hippocampus of 16 elderly subjects at different stages of NFT pathology progression.
Materials and methods
Subjects
Postmortem brain tissue was obtained from 16 elderly subjects: 12 with a clinical diagnosis of AD (mean age ± SD 77.8±13.9 years) and 4 age-matched cognitively normal (CN) control subjects (mean age 73.3±16.5 years). The mean postmortem interval and brain weight of the cases were 5.3±1.9 h and 1,154±138 g, respectively, with no significant difference between AD and CN groups (Table 1). Clinical diagnosis of CN subjects was based on the absence of dementia, determined through retrospective analysis of medical records as well as interviews with family physicians and immediate family members. All AD subjects were participants in a longitudinal research program maintained by the University of Pittsburgh’s Alzheimer’s Disease Research Center (ADRC). As participants in this program, patients underwent periodic neuropsychological and neurological evaluation. Clinical diagnosis of AD was based on a standardized ADRC evaluation at a Consensus Conference, utilizing DSM-IV [2] and NINCDS/ADRDA [21] criteria. Neuropathological diagnosis was determined by a certified neuropathologist, and was based in part on histological examination of brain tissue sections stained with hematoxylin and eosin, thioflavin-S, and Bielschowsky silver stains. All AD subjects fulfilled CERAD criteria for the diagnosis of “definite” AD [22]. All brains (CN and AD) showed NFT, and dependent on the extent of NFT progression through the entorhinal, hippocampal, and neocortical areas, they were assigned a Braak score, according to neuropathological staging by Braak and Braak [7]. Of the 16 subjects, 4 CN controls were Braak stage I/II with only “mild” hippocampal pathology. Four of the AD patients were in Braak stage III/IV with “moderate” hippocampal pathology, and the remaining eight AD cases were Braak stage V/VI with “severe” hippocampal NFT pathology. Lewy bodies were detected in the cerebral cortex of one moderate case and three severe AD cases, but no Lewy bodies or neurites were detected in the hippocampus of any case. None of the patients included in this study had any confounding neurological or neuropathological disorder, except for isolated old infarcts in the cortex and thalamus of two severe cases (Table 1).
Tissue preparation
Brain tissue was processed according to previously described procedures [24, 26, 27]. The material for this study was obtained from a block of hippocampal tissue cut in the coronal plane at the level of the lateral geniculate body. Tissue was placed in 0.1 M phosphate buffer (PB, pH 7.4) containing 4% paraformaldehyde for 48 h at 4°C, and subsequently cryoprotected in 30% sucrose in PB for several days. Tissue sections for immunohistochemistry were cut at 40 µm on a sliding, freezing microtome. For each case, adjacent sections were stained for Nissl substance to delineate the cytoarchitectural boundaries of hippocampal fields as defined by Duvernoy [10] and Amaral and Insausti [1].
Immunohistochemistry
Tissue sections were processed free-floating for immunohistochemistry of human GABA receptor subunits as described previously [27, 28], using a polyclonal antibody against the receptor subunit GABABR1 (GBR1, AB1531; Chemicon, Temecula, CA). The amino acid sequence is common to both the GABABR1a and GABABR1b receptor isoforms, but this antibody primarily recognizes the GABABR1a [38]. The primary antibody was diluted 1:5,000 in TRIS-saline containing 3% goat serum and 0.25% Triton X-100. At least three sections from each case were immunostained for this study. Sections from all cases were processed together to control for variability in the immunohistochemical procedure. As a control for nonspecific staining, sections were incubated with initial incubation media with the primary antibody omitted, and otherwise processed as described. No positive staining was detectable in these control sections. Control sections for each case were used for background correction in optical density (OD) analyses (described below). Double immunolabeling was performed on each case using GBR1 and MC1 antibodies to investigate alterations of GBR1 in neurons undergoing early NFT changes. MC1 (generously provided by Dr. Peter Davies; used at 1:1,000) is a monoclonal antibody that detects early cytoskeletal alterations involving changes in the conformation of the tau molecule [16]. The sequential double-immunolabeling procedure used diaminobenzidine (DAB) as a chromogen to visualize MC1 immunohistochemistry, with subsequent GBR1 immunohistochemistry using DAB and 2.5% nickel ammonium sulfate, yielding homogeneous light brown (DAB) and granular black reaction products (nickel-conjugated DAB). Additional tissue sections were stained using monoclonal 10D5 (1:3,000; Athena Neurosciences, San Francisco, CA) or polyclonal paired helical filament (PHF) tau (1:10,000; DAKO, Carpinteria, CA) antibodies, to visualize amyloid β (Aβ) plaques and NFT, respectively.
OD measurements of neuronal GBR1-IR
Quantitative evaluation of the intensity of immunohistochemical reaction in individual pyramidal neurons was performed by OD measurements using an Olympus AHBT-3 light microscope equipped with a SPOT-2 digital video camera (Diagnostic Instruments, San Jose, CA) and public domain image analysis software (NIH Image, Scion, Frederick, MD). All images were obtained at ×40 magnification under constant illumination and exposure conditions. Densitometric analysis was performed as described previously [23, 34]. Briefly, profiles of individual GBR1-immunoreactive neurons with visible nuclei were outlined with a free-hand marquee to obtain a morphometric mask. In three separate non-adjacent sections from each case, ten neurons were randomly selected for measurements in the pyramidal layer of four hippocampal regions (CA1–4) and the subiculum (Sub). The dentate gyrus (DG) granule cell layer was evaluated by randomly choosing three microscopic fields as regions of interests (ROI), in which all neurons were measured collectively, due to their high packing density. The measurement of the relative concentration of GBR1-immunoreactive material in each ROI was obtained as the gray level (GL), related to the OD for the specimen using the following equation:
where GLspecimen is the gray level of the image delineated by the morphometric mask (individual neurons) and GLbackground is the gray level of the background reference image (obtained by outlining neuronal layers in sections processed in the absence of primary antibody for each case). Specific staining was defined as the difference in immunostaining intensity between sections incubated with and without the primary antisera.
Values presented are means ± standard deviation (SD). Where appropriate, analysis of variance (ANOVA) was used to make comparisons. If differences were detected by ANOVA, individual groups were compared using the Student-Newman-Keuls test. P<0.05 was accepted as statistically significant for all comparisons.
Cell counts
To compare the relative loss of GBR1-immunoreactive compared to Nissl-labeled pyramidal neurons in CA1 hippocampus, cell counts were performed by randomly selecting ten ×40 microscopic fields on three non-adjacent sections in all cases from each Braak group. All neurons on which the entire soma and initial primary dendrite could be identified were counted within each microscopic field, irrespective of neuronal size (i.e., all pyramidal neurons were counted). The total number of neurons within the entire CA1 field was not calculated; instead, numerical densities of GBR1-immunoreactive and Nissl-labeled neurons were obtained in each case and then calculated as means ± SD for each Braak diagnostic group. Correction for laminar shrinkage was not performed, as shrinkage would affect equally GBR1-IR and Nissl-stained cells counted in adjacent tissue sections. However, it is possible that due to laminar shrinkage the reductions in both GBR-1 and Nissl-positive neurons were underestimated in the Braak stage V/VI group (see discussion). Comparisons across Braak groups were made using ANOVA.
Results
GBR1-IR was detected as black, punctate chromogen precipitate that labeled the soma and proximal dendrites of hippocampal neurons and interneurons. GBR1-IR interneurons were infrequently observed, but were present in every case; they were either bipolar or multipolar, with fusiform or round cell bodies and thin dendrites (not shown). Unlike pyramidal neurons, GBR1-IR interneurons were located primarily within the strata radiatum and oriens. Neuropil GBR1 immunostaining was not above background levels.
CN subjects with mild (Braak stage I/II) NFT pathology
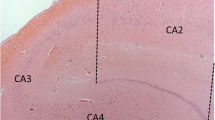
In CN subjects, neuronal GBR1-IR was detected in all hippocampal fields, although the intensity of immunostaining differed between fields (Figs. 1A, D, G, J; 2). Within the DG, light GBR1-IR was observed in granule cells (Figs. 1A, 2). In the CA4 field, moderate GBR1-IR was observed in the soma and proximal dendrites of mossy cells (Fig. 1A). In CA3 and CA2, pyramidal cells showed more intense GBR1-IR, particularly in comparison to CA1 field and DG (Figs. 1, 2). In contrast, GBR1-IR of CA1 and Sub pyramidal cells was considerably less intense (Figs. 1J, 2).

Photomicrographs showing GBR1 immunohistochemistry in dentate granule cell layer (GL) and in CA4 (A–C), CA3 (D–F), CA2 (G–I) and CA1 (J–L) subfields of controls that are pathologically “mild” (Braak I/II; A, D, G and J), “ moderate” AD (Braak III/IV; B, E, H, K), and “severe” AD (Braak V/VI; C, F, I, L) cases. Compared to mild and severe groups, the overall intensity of neuronal GBR1 immunoreactivity is increased in the moderate cases. In the CA1 subfield of moderate and severe cases, loss of pyramidal cell GBR1 staining is evident (K, L). This illustrates differences in neuronal GBR1 immunostaining intensity, and is not shown as a representative of differences in numbers of GBR1-immunoreactive neurons across Braak stage groups. The latter numbers are displayed in Table 2 (GBR1 GABAB receptor R1 protein, AD Alzheimer’s disease, mod moderate, sev severe). Bar 100 μm

Bar graph showing R.O.D. measurements of neuronal GBR1 immunostaining intensity in five hippocampal fields in control cases with Braak I/II (mild), and AD patients with Braak III/IV (moderate) or Braak V/VI (severe) pathology. Different patterns can be observed in regions more resistant (DG, CA4, CA3/2) versus those more vulnerable (CA1, Sub) to AD. In cases with an onset of hippocampal NFT pathology (Braak III/IV), significant increases of GBR1 staining intensity are observed in CA4 and CA3/2 regions, while a trend for an increase is seen in DG. Asterisks:P<0.01; dagger: P<0.05 (R.O.D. relative optical density, DG dentate gyrus, CA4, CA3/2, CA1 hippocampal CA fields, Sub subiculum, NFT neurofibrillary tangles)
AD subjects with moderate (Braak stage III/IV) and severe (Braak stage V–VI) NFT pathology
In hippocampus of moderate AD cases, GBR1-IR increased markedly (Figs. 1, 2) in DG granule cells and in CA4 and CA3/2 pyramidal cells (Figs. 1B, E, H; 2). In contrast, severe AD cases showed a comparable to decreased cellular GBR1-IR in all hippocampal fields when compared to the mild CN group (Figs. 1C, F, I, L; 2). We next used relative OD (R.O.D.) measurements of neuronal GBR1 immunostaining in individual hippocampal fields to compare GBR1 immunostaining intensity in the three NFT severity groups, and detected significant differences in the CA4 and CA3/2 regions. Moderate AD cases had higher R.O.D. values (Fig. 2) than either mild CN or severe AD groups in CA4 (both P<0.01) or in CA3/2 (P<0.05 and P<0.01). There was a considerable variability in numbers of GBR1-IR and Nissl-stained neurons in the CA1 field of AD cases. Moderate and severe AD cases showed marked loss of GBR1-IR CA1 pyramidal cells compared to the mild CN group (Table 2). Nissl staining of adjacent tissue sections revealed only moderate CA1 pyramidal cell loss across both AD groups, but this is likely an underestimate due to hippocampal atrophy and laminar shrinkage (Table 2; see discussion).
In addition to cellular IR, plaque-like clusters of GBR1-immunoreactive dystrophic neurites were detected in the CA1/Sub (Fig. 3B) and DG molecular layer (not shown) in both moderate and severe AD cases. These clusters were entirely composed of neuritic elements, with no associated neuropil-IR (Fig. 4).

Photomicrographs of the CA1 field in serial tissue sections, processed for Nissl staining (A), GBR1 immunohistochemistry (B) and MC1 immunohistochemistry (C), from an AD case with moderate (Braak III/IV) NFT pathology. Despite unremarkable neuronal loss (A), there is a loss of GBR1 immunoreactivity on pyramidal neurons (B) that coincides with appearance of NFTs (C, arrowheads). Neuritic plaques (arrowheads) are both GBR1 positive (B) and MC1 positive (C). Bar 100 μm

High-power photomicrograph of three GBR1-immunoreactive plaque-like structures in the CA4. GBR1 immunostaining is present in numerous dystrophic neurites, many of which appear as large bulbous swellings (large arrows). Note the punctate GBR1-immunoreactive labeling in nearby neurons (small arrows), contrasting the dark uniform labeling seen in neuritic processes. Bar 75 μm
GBR1 and MC1 co-localization in AD hippocampus
Immunostaining with MC1, a marker of early neurofibrillary changes, revealed a differential distribution of MC1-positive neurons in various hippocampal fields of the three groups of cases, consistent with their pathological classification by Braak stages. CN cases showed infrequent MC1-IR cells in the CA1 region, and no labeled neurons in DG, CA3/2 and CA4 (not shown). These CA1 MC1-IR cells did not co-localize GBR1.
In moderate AD cases, MC1-positive neurons were of substantial numbers in CA1/Sub (Fig. 3C), and only infrequent in other fields. In severe AD cases, there was an abundance of MC1-positive cells in CA1/Sub, and substantial numbers in other hippocampal fields. In both AD groups, only a small percentage of CA1/Sub neurons showed co-localization of MC1 and GBR1. In contrast, the CA4 and CA3/2 regions in these cases showed numerous GBR1/MC1 dual-labeled neurons (Fig. 5).

Photomicrographs showing neuronal double immunolabeling with MC1 (homogeneous brown) and GBR1 (granular purple) in CA4 (A) and CA3 (B) subfields of a severe AD case. Co-localization of MC1 and GBR1 is observed in several pyramidal cells (arrows). In contrast, MC1-stained neuronal processes and threads (arrowheads) are GBR1 negative. Bar 20 μm
GBR1-positive neuritic plaque-like deposits in AD cases also contained MC1-IR neurites (Fig. 3C). There were no correlations between intensity of neuronal GBR1 immunostaining and distribution of Aβ plaques in hippocampal fields.
Discussion
The present study demonstrates alterations in GBR1-IR during the progression of NFT pathology in the hippocampus of AD subjects. Detection of neuronal GBR1-IR in all hippocampal fields in non-demented cases with only mild neurodegenerative changes is consistent with the results of previous immunohistochemical studies of GABAB receptors in hippocampus of aged humans [5, 27]. We detected GABAB-IR exclusively in neuronal soma and proximal dendrites. In contrast, Billinton et al. [5] reported that GBR1-IR is also present in the neuropil of the dentate molecular layer and stratum lacunosum-moleculare of the CA fields. Differences in specificity of the antibodies employed in the two studies could account for the discrepancies between the two studies.
Our results indicate that changes in the expression of hippocampal GBR1 subunit correlate with the progression of neuronal degeneration defined by a general assessment of NFT pathology by Braak and Braak [7], as well as by a marker of early NFT changes (MC1). The GBR1 immunostaining was most robust in CA3/2 regions, where it increased markedly in moderate AD (Braak III/IV) cases. In contrast, numbers of GBR1-immunoreactive neurons decreased in the CA1 region in both moderate and severe AD, and no GBR1 up-regulation was observed in these cells. The more pronounced loss of GBR1-immunoreactive compared to Nissl-labeled pyramidal neurons in the CA1 region suggests that GBR1-expressing neurons may be more sensitive to degeneration than CA1 neurons collectively. Notably, the lack of significant loss of Nissl-labeled pyramidal neurons in CA1 in the severe (Braak V/VI) group in the present study differs from that shown in studies employing unbiased stereological techniques [33, 35]. We recognize potential limitations of our non-stereological cell counting method; the latter inconsistency could be due to laminar shrinkage that likely occurs in end-stage AD hippocampus. However, correcting for laminar shrinkage would further reduce the numbers of both GBR1-IR and Nissl-positive CA1 neurons in severe AD cases. While such correction would bring our Nissl cell counting data in closer agreement with previous stereological investigations, we would still observe relatively lower numerical density of GBR1-immunoreactive neurons compared to Nissl-labeled neurons in AD. The CA1 region is particularly vulnerable to AD pathology, while CA3/2 is relatively resistant [11, 12]; thus, elevations in GBR1-IR in CA3/2 pyramidal neurons could reflect a compensatory response to increased excitatory neurotransmission, and render these cells more resistant to excitoxic insults. The GABAB receptor is known to modulate postsynaptic excitatory transmission [31, 37], and when excitation increases, GABAB-mediated slow inhibition is recruited to offset excessive neuronal excitation [6]. Thus, up-regulation of GBR1 in pyramidal cells could serve to reduce excessive (toxic) stimulation of these cells by increasing inhibitory tone. Thus, transient up-regulation of the GBR1 in pyramidal cells, which occurs at the onset of hippocampal NFT pathology (i.e., Braak stage III/IV), could function to reduce excitotoxic neuronal damage in this region. Previous studies reported that excitotoxicity mediated via calcium-permeable α-amino-3 hydroxy-5-methyl-4-isoazolepropionate (AMPA) and N-methyl-d-aspartate (NMDA) types of glutamate receptors may contribute, at least in part, to the development of neurodegenerative changes in the hippocampus [13, 14] as well as in other vulnerable regions [3, 15] of AD brains. In accordance with these studies, we have shown previously that loss of the GluR2 AMPA receptor subunit precedes hippocampal NFT formation [13]. However, it is possible that not only increased excitatory signal, but also reduced GABA compensatory (inhibitory) mechanisms contribute to NFT formation and cell death during the progression of AD. Alternatively, metabolic alterations due to neurofibrillary tangle pathology may impede synthesis of the receptor. Previous studies suggest that GABA receptor subunits can be substantially and selectively affected in AD. For example, radiolabeled ligand binding experiments in the hippocampus [8] and frontal cortex [9] from AD subjects have demonstrated significant reductions in GABAB receptors, while GABAA receptors were relatively preserved. Furthermore, GABAA receptor density was reduced only in stratum pyramidale of CA1 subfield, while reductions in GABAB receptor density were more extensive [8]. In agreement with these studies, our previous work demonstrated that GABAA receptor alpha and beta 2/3 protein and beta 2 mRNA signals were well preserved in the DG, even in cases with severe AD pathology, [24, 25, 26]. Collectively, these results suggest that pathological processes might differentially affect the stability of GABA receptor subunits, with GABAB being more vulnerable to AD.
We observed that in CA1, an area particularly affected by the development of mature NFT and cell death in AD, neurons expressing an early marker of neurofibrillary changes (MC1) did not co-localize GBR1. In contrast, in the CA3/2 and CA4 regions many of the MC1-immunoreactive neurons also expressed GBR1. Because neurons in the CA1/Sub region are known to develop NFT sooner in the progression of AD than those in any other field in the hippocampus, loss of GBR1 subunits from this cell population might contribute to their propensity to transform into NFT. In contrast, pyramidal neurons from CA3/2 and CA4 regions are resistant to the conversion into NFT, and this might be due to their ability to up-regulate, or sustain, functional GBR1. Thus, early in the course of neurofibrillary change, up-regulation of GBR1 in the CA2/3–4 regions may render these neurons more resistant to the progression of neurofibrillary pathology, whereas in CA1,this compensatory change is either not occurring, or is ineffective. Alternatively, neurons in CA2/3–4 may be more resistant than those in CA1 to the effects of NFT formation, and thereby retain a greater level of GBR1-IR synthesis.
In conclusion, our results indicate that there is a transient increase in GBR1-IR in the hippocampus during the progression of neurofibrillary pathology in the course AD. This potentially compensatory change is not sustainable, as GBR1-IR in severe AD cases is comparable to subjects with only mild NFT pathology. Additionally, there is considerable intersubject variability in AD, with loss of CA1 GBR1-IR in several cases of moderate and severe AD. Thus, changes in GABAB receptor subunit expression in AD hippocampus could mark a compensatory response to the onset of regional neurodegenerative changes, while loss of such a response might facilitate or be due to further progression of NFT pathology.
References
Amaral DG, Insausti R (1990) Hippocampal formation. In: Paxinos G (ed) The human nervous system. Academic Press, New York, pp 711–756
American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders, 4th edn. American Psychiatric Association, Washington, D.C.
Armstrong DM, Ikonomovic MD, Sheffield R, Wenthold RJ (1994) AMPA-selective glutamate receptor subtype immunoreactivity in the entorhinal cortex of non-demented elderly and patients with Alzheimer’s disease. Brain Res 639:207–216
Armstrong DM, Ikonomovic MD, Mizukami K, Mishizin A, Sheffield R, Wolfe BB (1998) Glutamatergic mechanisms in Alzheimer’s disease. Advances in Neurodegenerative Disorder: In Marwah J, Teitelbaum H (eds) Alzheimer’s and Aging, vol 2, Prominent Press, Scottsdale, pp 71–97
Billinton A, Ige AO, Wise A, White JH, Disney GH, Marshall FH, Waldvogel HJ, Faull RLM, Emson PC (2000) GABAB receptor heterodimer-component localization in human brain. Mol Brain Res 77:111–124
Bonard LS (1995) N-Methyl-d-aspartate transmission modulates GABAB-mediated inhibition of rat hippocampal pyramidal neurons in vitro. Neuroscience 68:637–643
Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82:239–259
Chu DC, Penney JB Jr, Young AB (1987) Quantitative autoradiography of hippocampal GABAB and GABAA receptor changes in Alzheimer’s disease. Neurosci Lett 82:246–252
Chu DCM, Penny JB, Young AB (1987) Cortical GABAB and GABAA receptors in Alzheimer’s disease: a quantitative autoradiographic study. Neurology 37:1454–1459
Duvernoy HM (1998) The human hippocampus, 2nd edn. Springer, Berlin
Flood DG, Guarnaccia M, Coleman PD (1987) Dendritic extent in human CA2–3 hippocampal pyramidal neurons in normal aging and senile dementia. Brain Res 409:88–96
Hof PR (1996) Morphology and neurochemical characterics of the vulnerable neurons in brain aging and Alzheimer’s disease. Eur Neurol 37:71–81
Ikonomovic DM, Mizukami K, Davies P, Hamilton RL, Sheffield R, Armstrong DM (1997) The loss of GluR2(3) immunoreactivity precedes neurofibrillary tangle formation in the entorhinal cortex and hippocampus of Alzheimer brains. J Neuropathol Exp Neurol 56:1018–1027
Ikonomovic MD, Mizukami K, Warde D, Sheffield R, Hamilton R, Wenthold RJ, Armstrong DM (1999) Distribution of glutamate receptor subunit NMDAR1 in the hippocampus of normal elderly and patients with Alzheimer’s disease. Exp Neurol 160:194–204
Ikonomovic MD, Nocera R, Mizukami K, Armstrong DM (2000) Age-related loss of the AMPA receptor subunits GluR2/3 in the human nucleus basalis of Meynert. Exp Neurol 166:363–375
Jicha GA, Bozer R, Kazam IG, Davies P (1997) Alz-50 and MC1, a new monoclonal antibody raised to paired helical filaments, recognize confirmational epitopes on recombinant tau. J Neurosci Res 48:128–132
Jones KA, borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, huang LY, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C (1998) GABAB receptors function as a heterometric assembly of the subunits GABABR1 and GABABR2. Nature 396:674–679
Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, mosbacher J, bichoff S, Kulik A, Shigemoto R, Karschin A, Bettler B (1998) GABAB-receptor subtypes assemble into functional heterometric complexs. Nature 396:683–687
Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA (1997) G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19:687–695
Macdonald RL, Olsen RW (1994) GABAA receptor channels. Annu Rev Neurosci17:569–602
McKhann G, Drachman D, Folstein M, Katzman, R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of Health and Human Services Task Force on Alzheimer’s disease. Neurology 34:939–944
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Mize RR (1994) Quantitative image analysis for immunocytochemistry and in situ hybridization. J Neurosci Methods 54:219–237
Mizukami K, Ikonomovic MD, Grayson DR, Rubin RT, Warde D, Sheffield R, Hamilton RL, Davies P, Armstrong DM (1997) Immunohistochemical study of GABAA receptor β2/3 subunits in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Exp Neurol 147:333–345
Mizukami K, Grayson DR, Ikonomovic MD, Sheffield R, Armstrong DM (1998) GABAA receptor β2 and β3 subunits mRNA in the hippocampal formation of aged human brain with Alzheimer-related neuropathology. Mol Brain Res 56:268–272
Mizukami K, Ikonomovic MD, Grayson DR, Sheffield R, Armstrong DM (1998) Immunohistochemical study of GABAA receptor α1 subunit in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Brain Res 799:148–155
Mizukami K, Sasaki M, Ishikawa M, Iwakiri M, Hidaka S, Shiraishi H, Iritani S (2000) Immunohistochemical localization of GABAB receptor in the hippocampus of subjects with schizophrenia. Neurosci Lett 283:101–104
Mizukami K, Ishikawa M, Hidaka S, Iwakiri M, Sasaki M, Iritani S (2002) Immunohistochemical localization of GABAB receptor in the entorhinal cortex and inferior temporal cortex of schizophrenic brain. Prog Neuropsychopharmacol Biol Psychiatry 26:393–396
Möhler H, Benke D, Benson J, Lüscher B, Rudolph U, Fritschy JM (1997) Diversity in structure, pharmacology, and regulation of GABAA receptors. In: Enna SJ, Bowery NG (eds) GABA receptors, 2nd edn. Humana Press, Clifton, pp 11–36
Palmer AM, Gershon S (1990) Is the neuronal basis of Alzheimer’s disease cholinergic or glutamatergic? FASEB J 4:2745–2752
Pfrieger FW, Gottmann K, Lux HD (1994) Kinetics of GABAB receptor-mediated inhibition of calcium currents and excitatory synaptic transmission in hippocampal neurons in vitro. Neuron 12:97–107
Premkumar LS, Gage PW (1994) Potassium channels activated by GABAB agonists and serotonin in cultured hippocampal neurons. J Neurophysiol 71: 2570–2575
Rössler M, Zarski R, Bohl J, Ohm TG (2002) Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer’s disease. Acta Neuropathol 103:363–369
Smolen AJ (1990) Image analytic techniques for quantification of immunohistochemical staining in the nervous system. In: Conn PM (ed) Methods in Neurosciences, vol 3. Academic Press, New York, pp 208–229
West MJ (1993). Regionally specific loss of neurons in the aging human hippocampus. Neurobiol Aging 14:287–293
White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH (1998) Heterodimerization is required for the formation of a functional GABAB receptor. Nature 396:679–682
Wu LG, Saggau P (1995) GABAB receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J Physiol 485:649–657
Yung KKL, Ng TKY, Wong CKC (1999) Subpopulations of neurons in the rat neotriatum display GABABR1 receptor immunoreactivity. Brain Res 830:345–352
Acknowledgements
This work was supported by the NIH grant AG05133, and the Research Grant for Longevity Sciences (14C-4) from the Ministry of Health, Labor and Welfare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Iwakiri, M., Mizukami, K., Ikonomovic, M.D. et al. Changes in hippocampal GABABR1 subunit expression in Alzheimer’s patients: association with Braak staging. Acta Neuropathol 109, 467–474 (2005). https://doi.org/10.1007/s00401-005-0985-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-005-0985-9