
Abstract
Less neurovirulent strains of rabies virus have been recognized to be stronger inducers of neuronal apoptosis in vitro than more neurovirulent strains, but few studies have clarified whether this also applies in vivo. A comparative study was performed in two-day-old ICR mice inoculated in a hindlimb thigh muscle with recombinant rabies virus vaccine strain SAD-L16 (L16) or SAD-D29 (D29), which contains an attenuating substitution of Arg333 in the rabies virus glycoprotein. Histopathological and immunohistochemical analyses of brains were performed at early daily time points and in moribund animals. Both viruses caused progressive limb weakness; mortality with L16 was 100% at day 7 post-inoculation (p.i.) and 75% at 17 days p.i. for D29 and Kaplan–Meyer survival curves were significantly different. L16 spread to the brain more quickly than D29, and both viruses produced multifocal lesions in the brainstem and cerebellum associated with inflammatory changes and neuronal apoptosis. There was more disseminated involvement of the brain and many more infected neurons in L16 infection, particularly in the neostriatum, hippocampus, and cerebral cortex. Both viruses induced neuronal apoptosis, which was most marked in the brainstem tegmentum and internal granular layer of the cerebellum. In light of the lower burden of infection and smaller number of neurons infected with D29, this less virulent virus was a stronger inducer of neuronal apoptosis than the more virulent L16. These findings support previous in vitro studies indicating that there is an inverse relationship between pathogenicity and apoptosis. Induction of apoptosis, which is an innate mechanism in which the host restricts viral spread, may contribute to severe clinical neurological disease when there is viral invasion into the central nervous system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rabies virus is a highly neurotropic virus that causes fatal encephalomyelitis in humans and animals [7, 12]. The attenuated SAD-B19 vaccine strain, which is widely used in Europe for oral vaccination of foxes [2, 16], is a derivative of the Street Alabama Dufferin strain isolated from a rabid dog in Alabama in 1935, attenuated by multiple passages in baby hamster kidney (BHK) cells, and selected for vaccine production on the basis of its thermostability [2]. A recombinant clone of SAD-B19, SAD-L16 (L16), was generated from a full-length cDNA clone [17]; L16 is highly neurovirulent after intracerebral inoculation of mice and causes extensive neuronal apoptosis with this route of inoculation in young mice [9, 13]. Substitution of the arginine at position 333 of the rabies virus glycoprotein is known to be attenuating in adult mice after different routes of inoculation and it is known that this mutation is much less attenuating in immature mice [6].
We have used the Arg333 mutation to study viral spread and induction of neuronal apoptosis, because we have noted that this mutation produces attenuated disease after peripheral inoculation of neonatal mice. This model will allow assessment of differences in the ability of a virulent and attenuated pair of viruses to induce neuronal apoptosis in the brain. We have compared the infections produced by L16 and SAD-D29 (D29), which contains a substitution of aspartic acid for arginine at position 333 of the glycoprotein [9], after intramuscular inoculation of neonatal mice.
Materials and methods
Viruses
The L16 strain of fixed rabies virus and D29 were obtained from Teshome Mebatsion (Intervet International, B. V., Boxmeer, The Netherlands) [9]. The generation of recombinant rabies virus L16 has been previously described; L16 contains the authentic sequence of the SAD-B19 vaccine strain, which was derived from a full-length cDNA clone, and BSR-T7/5 cells expressing phage T7 RNA polymerase were used to recover infectious virus from cDNA [9, 17]. D29 was generated by substitution of arginine (R333) of the mature rabies virus glycoprotein with aspartic acid (D333) as described [9].
Animals and inoculations
The experimental animal protocol followed the Canadian council on animal care guidelines on animal use and was approved by the Queen’s University Animal Care committee. Two-day-old ICR mice of either sex (Charles River Canada, St. Constant, Quebec, Canada) were used. Mice were inoculated intramuscularly into the right hindlimb thigh muscle with 20 μl containing 1000 focus-forming units of either L16 or D29 diluted in PBS with 4% fetal bovine serum. Uninfected control mice of the same age were inoculated with only the diluent (mock-infected). The mice were anesthetized with isoflurane and perfused with buffered 4% paraformaldehyde. Mice were killed at daily time points between day 1 and 7 p.i. (regardless of their clinical status), when they became moribund, and also at the completion of the observation period on day 17 p.i.
Preparation of tissue sections
Brains were removed and immersion-fixed in the same fixative for 24 h at 4°C. Coronal brain tissue sections (6 μm) were prepared after dehydration and embedding in paraffin. Tissues were stained with cresyl violet for histological examination by light microscopy.
Immunoperoxidase staining for rabies virus antigen and activated caspase-3
Immunoperoxidase staining was performed on mouse brains for rabies virus antigen as previously described [8], and also for activated caspase-3 as previously described [13].
TUNEL staining
Oligonucleosomal DNA fragmentation was assessed in situ in mouse brain sections using the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) method using a TdT-FragEL DNA fragmentation detection kit (Cat# QIA33) (Oncogene Research Products, San Diego, CA, USA) using the manufacturer’s protocol for paraffin-embedded tissue sections.
Results
Clinical observations
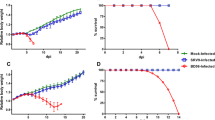
L16—All infected mice developed signs of limb weakness on day 5 post-inoculation (p.i.), which quickly progressed to quadriparesis and growth retardation. All mice became moribund and were killed on day 6 or 7 p.i. (Fig. 1).

Kaplan–Meyer survival curves showing cumulative mortality after inoculation of mice with L16 or D29 through 17 days p.i.
D29—Infected mice initially developed right hindlimb paresis on days 5–7 p.i., which progressed to bilateral hindlimb paresis and quadriparesis with growth retardation. Mice became moribund and were killed between days 9 and 17, with a mortality rate of 75% by day 17 p.i. (Fig. 1). Evaluation of the Kaplan–Meyer survival curves using the log rank test indicated significantly improved survival in D29 infection compared to L16 infection (P<0.0001). In a separate experiment, a minority of mice (3 of 24, 12.5%) was found to have persistent limb weakness and were killed 33 days p.i.
Rabies virus antigen distribution
L16—Rabies virus antigen was first detected in L16 infection in one mouse in neurons in the brainstem tegmentum at 4 days p.i. By 5 days p.i. the distribution became much more widespread in brain neurons with involvement of the brainstem, diencephalon, and cerebellum and, to a lesser extent, of the hippocampus and cerebral cortex. Greater numbers of neurons were noted to contain rabies virus antigen at days 6 and 7 p.i. with heavy staining of brainstem neurons (Fig. 2a), pyramidal neurons of the hippocampus (Fig. 2c), and internal granular layer of the cerebellum (Fig. 2e), and of neurons in the neostriatum and cerebral cortex (Fig. 2g). A few neurons showed staining in the dentate gyrus of the hippocampus. Neurons in the external granular layer of the cerebellum did not show staining.

Immunoperoxidase staining for rabies virus antigen in the midbrain tegmentum (a, b), CA1 region of the hippocampus (c), cerebellum (e, f), cerebral cortex (g), and histology (d) in the CA1 region of the hippocampus 7 days p.i. with L16 and in the cerebellum 10 days p.i. with D29 (g). Many more neurons stain for rabies virus antigen in the midbrain tegmentum in L16 infection (a) than in D29 infection (b). Although many neurons in the pyramidal layer of the hippocampus were infected (c), morphologic changes were not observed (d). In the cerebellum, L16 infected many neurons diffusely in the internal granular layer (e), whereby D29 produced multifocal infection of this layer (f). Many cortical neurons were infected in L16 infection (g). Histopathology in a deep cerebellar region (h) 10 days p.i. with D29 showed infiltration with inflammatory cells and mineralization. a–c, e–g, immunoperoxidase—hematoxylin; d, h, cresyl violet; a, b X 80; c, d X 210; e, f X 45; g X 80; h X 145
D29—Rabies virus antigen was not observed in the brain until day 6 in one mouse with infection observed in Purkinje cells. On day 7 viral antigen was observed in the brainstem tegmentum and cerebellum of all mice. By days 9 and 10, when mice first became moribund, rabies virus antigen had a wide distribution in the brain with involvement of the brainstem (Fig. 2b), diencephalon, cerebellum (deep cerebellar nuclei and multifocal involvement of granule cells, particularly in the internal granular layer) (Fig. 2f), hippocampus, and cerebral cortex. There was a ten-fold or greater number of neurons expressing viral antigen in L16 infection than in D29 infection in all regional areas except in multifocal areas of the internal granular layer of the cerebellum (Fig. 2e, f), and the number of infected neurons in D29 infection was particularly low in the hippocampus and cerebral cortex in comparison to L16 infection. At later time points (day 13–17 p.i.) few neurons showed viral antigen staining, and neurons in the brainstem and cerebellum were predominantly involved.
Histopathological changes
Mock-infected mice showed only scattered apoptotic neurons in the regional areas of the brain, likely due to naturally occurring neuronal death during postnatal development.
L16—Mild mononuclear inflammatory infiltrates were noted in the leptomeninges, perivascular regions, and brain parenchyma beginning on day 5 p.i. and they became more marked through day 7 p.i. Multifocal lesions were observed in the brainstem tegmentum (Fig. 3a) and in the cerebellum, including deep cerebellar nuclei and internal granular layer (Fig. 3c). In involved areas there were mononuclear inflammatory changes and neurons showing morphological features of apoptosis, including condensation of the nuclear chromatin and cytoplasmic shrinkage. Mineralization of lesions was not observed. Despite extensive infection (see above), there were few morphological changes of neuronal apoptosis in neurons in the cerebral cortex or hippocampus (Fig. 2d).

Histopathology of L16 infection 7 days p.i. (a and c) and D29 infection 10 days p.i. (b and d) in the midbrain tegmentum (a and b) and in the cerebellum (internal granular layer). In D29 infection there were more neurons with morphologic features of apoptosis, including condensations of nuclear chromatin, in the midbrain (b) and cerebellum (d), than in L16 infection (a and c). cresyl violet; a, b X 150; c, d X 235
D29—Mononuclear inflammatory infiltrates were noted in the leptomeninges, perivascular regions, and brain parenchyma, initially on day 6 p.i. with most prominent involvement of the brainstem and cerebellum. Multifocal lesions with inflammatory changes and associated neuronal apoptosis were observed in the brainstem tegmentum (Fig. 3b) and cerebellum, involving deep cerebellar nuclei and internal granular layer (Fig. 3d), on day 8 p.i. and they became more prominent at later time points. The lesions showed mineralization at day 12 p.i., and the mineralization became more marked at the later time points, including in mice with persistent paresis on day 33 p.i. (Fig. 2h). Pathological changes were not noted in the hippocampus or cerebral cortex.
Quantification of apoptotic cells—Apoptotic cells were counted in the internal granular of the cerebellum in moribund mice in L16 and D29 infections (Fig. 4), and immunoperoxidase staining on adjacent serial sections showed that virtually all granule cells contained rabies virus antigen (data not shown). The mean number of apoptotic cells in L16 infection was 183±69 (SEM) compared with 853±187 in D29 infection. Analysis with an unpaired t test indicated that D29 infection had more apoptotic cells than L16 infection, which was statistically significant with a two-tailed p value of 0.028.

Numbers of apoptotic cells counted in the internal granular layer of the cerebellum in three moribund mice in L16 infection at 7 days p.i. and in three moribund mice in D29 infection at 10 days p.i. The most affected areas were selected for evaluation on masked slides and apoptotic cells were counted in triplicate using a grid at high magnification (40× objective), and the mean numbers of apoptotic cells are shown for each of the mice. The number of apoptotic cells for D29 infection was significantly greater than for L16 infection (p<0.05).
TUNEL staining
In mock-infected mice there was TUNEL staining of scattered neurons in all brain regions, presumably due to developmental neuronal apoptosis. TUNEL staining was observed in neurons showing morphologic features of apoptosis in L16 and D29 infections.
L16—In the brainstem tegmentum there were small numbers of TUNEL positive neurons. In the cerebellum there was staining of a moderate number of neurons in the internal granular layer (Fig. 5a). These staining neurons were distributed diffusely throughout this layer. There was also staining of a relatively small number of neurons in the external granular layer of the cerebellum. There was staining of a small number of neurons in the diencephalon and neostriatum, and a few neurons in the pyramidal layer and dentate gyrus of the hippocampus. In the cerebral cortex there was staining in a few neurons, which was most prominent in outer pyramidal layer (layer III) and, less prominently, in the outer granule cell layer (layer II).

TUNEL staining in the internal granular layer of the cerebellum (a, b) and activated caspase-3 immunostaining in the internal granular layer of the cerebellum (c, d, e) and in the midbrain tegmentum (f, g, h) for L16 infection at 7 days p.i. (a, c, g), D29 infection at 10 days p.i. (b, d, h), and mock-infected at 7 days p.i. (e, f). TUNEL staining is present in a greater number of neurons in the internal granular layer of the cerebellum in D29 infection (b) than in L16 infection (a). More neurons show activated caspase-3 immunostaining in D29 infection in the internal granular layer of the cerebellum (d) and midbrain tegmentum (h) and than in L16 infection in these regions (c, g). a, b, TUNEL staining—methyl green; c–h, immunoperoxidase—hematoxylin; a, b X 185; e–h X 280
D29—In the brainstem tegmentum there was staining of a moderate number of neurons with a diffuse distribution. In the cerebellum there was prominent staining of many neurons in a multifocal distribution in the internal granular layer and mild staining in a more diffuse distribution in the same layer (Fig. 5b). Only small numbers of neurons showed staining in the diencephalon and neostriatum. A few neurons showed staining in the hippocampus and cerebral cortex, which involved layers III and VI.
Activated caspase-3 staining
Immunohistochemical staining for activated caspase-3 was observed in scattered neurons in mock-infected mice. There was staining in neurons with morphological features of apoptosis, including neurons in the brainstem and cerebellum (deep cerebellar nuclei and internal granular layer), and also in neurons in the cerebral cortex that showed relatively few morphological changes of neuronal apoptosis. In general, the pattern of caspase-3 staining was similar to TUNEL staining. Background staining was low (Fig. 5e, f).
L16—There was staining of a mild to moderate number of neurons in the internal granular layer of the cerebellum with a diffuse distribution (Fig. 5c), whereas fewer cells showed staining in the external granular layer. Only a small number of neurons showed staining in the brainstem tegmentum with a diffuse distribution (Fig. 5g). A small number of neurons showed staining in the diencephalon, neostriatum, and cerebral cortex (especially layer III). Staining was not observed in the hippocampus.
D29—In the cerebellum there was prominent staining of many neurons in a multifocal distribution in the internal granular layer and mild staining in a more diffuse distribution in the same layer (Fig. 5d). In the brainstem tegmentum there was staining of a moderate number of neurons with a diffuse distribution (Fig. 5h). Only small numbers of neurons showed staining in the diencephalon, neostriatum, and cerebral cortex (especially layer III), and staining was not observed in the hippocampus.
Quantification of activated caspase-3 expression—Cells expressing activated caspase-3 were counted in the midbrain tegmentum and in the internal granular layer of the cerebellum in three moribund mice in L16 infection at 7 and at 10 days p.i. in D29 infection. Staining cells were counted on masked slides in triplicate on a grid in the midbrain and internal granular layer of the cerebellum. The mean number of staining cells in the midbrain in L16 infection was 42.5±15.2 (SEM) compared with 237±42 in D29 infection, which was statistically greater for D29 infection by analysis with an unpaired t test with a two-tailed p value of 0.021. L16 infection showed 69.9±11.5 stained cells per high power field (SEM) compared with 214±37 in D29 infection in the internal granular layer of the cerebellum, which was also statistically greater in D29 infection with a p value of 0.012.
Discussion
As previously reported after intracerebral [13] and peripheral [14] inoculation of 7-day-old mice, L16 was found to be highly neurovirulent in 2-day-old mice after peripheral inoculation. There was widespread infection of brain neurons in L16 infection. Pathological changes included multifocal inflammatory lesions in the brainstem and cerebellum with associated neuronal apoptosis. The severe brainstem involvement may have played an important role in the fatal outcome of the infection.
Substitution of the arginine at position 333 of the rabies virus glycoprotein is known to convert neurovirulent CVS to an avirulent virus phenotype, which is normally assessed after intracerebral inoculation of 1000 plaque-forming units of virus in adult mice [4, 18]. This mutation was selected by resistance to neutralization in vitro using an anti-rabies virus glycoprotein monoclonal antibody [3, 5], and the mutation results in inefficient viral spread in cell culture and in animals; mice recover from the infection with participation of the immune response. The Arg333 replacement, however, does not abolish neurovirulence in immature mice; viruses with the Arg333 mutation remain fully neurovirulent in young mice after intracerebral inoculation. There is also evidence of reversion of the virus isolated from neonatal mouse brains to full pathogenicity in adult mice with susceptibility to neutralization with the monoclonal antibody used for selection of the original mutant virus [5].
After peripheral inoculation in a hindlimb muscle with D29, we have found that the clinical disease developed more slowly and there was a minority of survivors. D29 spread more slowly to the brain than L16; brain infection was first noted on day 4 for L16 and on day 6 for D29. D29 infected a smaller number of brain neurons than L16. Both viruses induced an inflammatory response in the brain. Histopathological lesions were most prominent in the brainstem and cerebellum in both viral infections, and there was subsequent mineralization of the lesions. Spinal cord involvement may have also played a significant role in the clinical disease, but this site was not assessed in this study. A comparison of the histopathology in the midbrain tegmentum and the internal granular layer of the cerebellum indicated that there were many more neurons showing apoptosis in proportion to the number of infected neurons in the less virulent D29 infection than in the more virulent L16 infection. In addition, despite widespread infection of neurons in the cerebral cortex and hippocampus in L16 infection, relatively few neurons showed apoptotic changes or biochemical markers of apoptosis (TUNEL staining or activated caspase-3 immunostaining) in these regions. These observations strongly support previous in vitro data indicating that there is an inverse relationship between pathogenicity and apoptosis in primary neuron cultures [11]. These results are also supported by an in vivo study comparing infections produced by the more virulent CVS-11 and the less neuroinvasive Pasteur virus strain 4 (PV4) in mouse spinal cords that showed more neuronal apoptosis in PV4 infection [1]. Another in vivo study by Sarmento et al. [15] using a variety of rabies virus strains and recombinants with different pathogenic properties, showed that neuronal apoptosis may serve a protective function by restricting viral spread from the spinal cord to the brain. In these cases, the “protective” responses were not restricted to the peripheral nervous system, which Mori et al. [10] recently speculated to be the neuroanatomical site of protective neuronal apoptosis. In the case of D29 infection in the current model, induction of neuronal apoptosis was not sufficient to prevent invasion of the virus into the brain and protection from the development of fatal disease. Once the attenuated D29 invaded the CNS, induction of apoptosis, which particularly involved brainstem and cerebellar neurons, may have contributed to the development of severe clinical neurological disease with a high mortality rate. Hence, under certain circumstances, a “protective” host response to a highly attenuated rabies virus strain may produce major neuropathological injury and contribute to the fatal outcome of the infection. In older mice, this protective response is much more efficient with neuroattenuated rabies virus strains, including L16 (and SAD-B19), and prevents viral invasion into the nervous system, resulting in a good clinical outcome.
References
Baloul L, Lafon M (2003) Apoptosis and rabies virus neuroinvasion. Biochimie 85:777–788
Blancou J, Meslin F-X (1996) Modified live-virus rabies vaccines for oral immunization of carnivores. In: Meslin F-X, Kaplan MM, Koprowski H (eds) World health organization, laboratory techniques in rabies, 4th edn. World Health Organization, Geneva, pp 324–337
Coulon P, Rollin P, Aubert M, Flamand A (1982) Molecular basis of rabies virus virulence. I. Selection of avirulent mutants of the CVS strain with anti-G monoclonal antibodies. J Gen Virol 61:97–100
Dietzschold B, Wiktor TJ, Trojanowski JQ, Macfarlan RI, Wunner WH, Torres-Anjel MJ, Koprowski H (1985) Differences in cell-to-cell spread of pathogenic and apathogenic rabies virus in vivo and in vitro. J Virol 56:12–18
Dietzschold B, Wunner WH, Wiktor TJ, Lopes AD, Lafon M, Smith CL, Koprowski H (1983) Characterization of an antigenic determinant of the glycoprotein that correlates with pathogenicity of rabies virus. Proc Natl Acad Sci USA 80:70–74
Jackson AC (2002) Pathogenesis. In: Jackson AC, Wunner WH (eds) Rabies, Academic Press, San Diego, pp 245–282
Jackson AC (2002) Human disease. In: Jackson AC, Wunner WH (eds) Rabies, Academic Press, San Diego, pp 219–244
Jackson AC, Ye H, Phelan CC, Ridaura-Sanz C, Zheng Q, Li Z, Wan X, Lopez-Corella E (1999) Extraneural organ involvement in human rabies. Lab Invest 79:945–951
Mebatsion T (2001) Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. J Virol 75:11496–11502
Mori I, Nishiyama Y, Yokochi T, Kimura Y (2004) Virus-induced neuronal apoptosis as pathological and protective responses of the host. Rev Med Virol 14:209–216
Morimoto K, Hooper DC, Spitsin S, Koprowski H, Dietzschold B (1999) Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J Virol 73:510–518
Niezgoda M, Hanlon CA, Rupprecht CE (2002) Animal rabies. In: Jackson AC, Wunner WH (eds) Rabies, Academic Press, San Diego, pp 163–218
Rasalingam P, Rossiter JP, Jackson AC (2005) Recombinant rabies virus vaccine strain SAD-L16 inoculated intracerebrally in young mice produces a severe encephalitis with extensive neuronal apoptosis. Can J Vet Res 69:100–105
Rasalingam P, Rossiter JP, Mebatsion T, Jackson AC (2005) Comparative pathogenesis of the SAD-L16 strain of rabies virus and a mutant modifying the dynein light chain binding site of the rabies virus phosphoprotein in young mice. Virus Res 111:55–60
Sarmento L, Li X, Howerth E, Jackson AC, Fu ZF (2005) Glycoprotein-mediated induction of apoptosis limits the spread of attenuated rabies viruses in the central nervous system of mice. J Neurovirol 11:571–581
Schneider LG (1995) Rabies virus vaccines. Dev Biol Stand 84:49–54
Schnell MJ, Mebatsion T, Conzelmann KK (1994) Infectious rabies viruses from cloned cDNA. EMBO J 13:4195–4203
Seif I, Coulon P, Rollin PE, Flamand A (1985) Rabies virulence: effect on pathogenicity and sequence characterization of rabies virus mutations affecting antigenic site III of the glycoprotein. J Virol 53:926–935
Acknowledgments
The authors are grateful for recombinant rabies virus vaccine strain SAD-L16 and SAD-D29 from Teshome Mebatsion (Intervet International) and for monoclonal antibody 5DF12 from Alexander I. Wandeler (Centre for Rabies Expertise, Canadian Food Inspection Agency, Nepean, Ontario). This work was supported by a research contract with Intervet International, B.V., Canadian Institutes of Health Research grant MOP-64376, and the Queen’s University Violet E. Powell Research Fund (all to A. C. Jackson).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jackson, A.C., Rasalingam, P. & Weli, S.C. Comparative pathogenesis of recombinant rabies vaccine strain SAD-L16 and SAD-D29 with replacement of Arg333 in the glycoprotein after peripheral inoculation of neonatal mice: less neurovirulent strain is a stronger inducer of neuronal apoptosis. Acta Neuropathol 111, 372–378 (2006). https://doi.org/10.1007/s00401-005-0006-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-005-0006-z