
Abstract
We report a case displaying upper motor sign, parkinsonism, and behavioral abnormality, with marked degeneration of the precentral cortex, neostriatum and frontotemporal lobes, as well as ubiquitinated neuronal inclusions. The patient was a 66-year-old male at the time of death. At age 57, he noticed progressive difficulties in speaking and swallowing. At age 60, he was severely anarthric and displayed emotional lability and incontinence. Neurologically, very poor movement of tongue was observed, but without atrophy or fasciculation. Deep tendon reflexes were hyperactive. Grasp reflex and snout reflex were also positive. Needle electromyography revealed no abnormalities. A diagnosis of primary lateral sclerosis and character change was made. At age 62, he developed bradykinesia and rigidity of the neck and all extremities. Treatment with carbidopa-levodopa was initiated, but resulted in minimal improvement. At age 65, he was bed-ridden, and had repeated occurrences of aspiration pneumonia; he died of pneumonia. Neuropathological examination revealed marked atrophy of the frontal and temporal lobes with Betz cells completely absent and moderate atrophy of the neostriatum. The spinal cord and nerve roots appeared normal. Immunohistochemically, ubiquitin-positive but tau-negative intraneuronal inclusions were found in the frontal and temporal cortices, including the precentral cortex and the hippocampal dentate gyrus, and the neostriatum. This case could be included with inclusion-associated disorders such as frontotemporal dementia or amyotrophic lateral sclerosis with dementia, and furthermore, predominant upper motor sign and parkinsonism could represent phenotypes of clinical manifestations with such inclusions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary lateral sclerosis (PLS), a progressive neurological disorder selectively affecting the upper motor neuron system, has been considered as representative of one end of a continuous spectrum of amyotrophic lateral sclerosis (ALS) [4]. In ALS with or without dementia, ubiquitinated neuronal inclusions have been found in extramotor structures such as the hippocampal dentate gyrus and frontotemporal cortex [6, 11]. Similar inclusions have also been found in cases of motor neuron disease-type frontotemporal dementia (MND-FTD) [8], referred to as MND-inclusion dementia (MND-ID) [2]. These two diseases are thus very closely linked by the presence of ubiquitinated neuronal inclusions. Recently, an autopsy case, in which the patient presented with a syndrome mimicking PLS without definite lower motor neuron degeneration, was reported to have ubiquitinated neuronal inclusions [7]. The spectrum of disorders associated with ubiquitinated neuronal inclusions needs to be clarified. We report an autopsy case, with ubiquitinated neuronal inclusions, in which the patient presented with predominant upper motor signs mimicking PLS and l-dopa non-responsive parkinsonism in addition to behavioral abnormalities. The extension of clinical manifestations of the condition with such inclusions is discussed.
Case report
A 66-year-old male gradually began to speak slowly, and noticed difficulty in swallowing at age 57. He had no family history of neurological disorders, including parkinsonism, dementia, and motor neuron disease. At 60 years of age, he was admitted to the hospital with marked difficulty in swallowing and speaking. On admission, he was severely anarthric but he could understand conversations. He also displayed emotional lability and incontinence. Neurologically, very poor movement of tongue was observed, but there was no atrophy or fasciculation. Muscle strength of all extremities was normal and muscle tone was normal. Deep tendon reflexes were hyperactive and pathological reflex was negative. Grasp reflex and snout reflex were also positive. Cerebrospinal fluid was normal. Needle electromyography of the tongue and extremities revealed no abnormalities. Brain CT revealed slight atrophy of the frontotemporal lobe. Radiography of the skull and cervical spine was unremarkable. A diagnosis of PLS and character change was made. Tracheostomy was undertaken due to severe pseudobulbar palsy, and tube feeding was initiated. At the age of 61, apathy and decreased activities of daily living were observed, but the patient could still walk unaided. At age 62, he walked with a stooped posture and developed bradykinesia. Myerson’s sign was positive. Rigidity of the neck and all extremities was present but gegenhalten was not present. Deep tendon reflexes were hyperactive and pathological reflexes were positive. Treatment with carbidopa-levodopa was initiated, but resulted in minimal improvement. Results of needle electromyography of the tongue and extremities remained unremarkable. At this stage, the patient was completely anarthric and uncooperative, and cognitive function could not be evaluated. At age 63, he became unable to walk and was admitted to Hatsuishi hospital with pneumonia. Brain CT showed severe atrophy of the frontal and temporal lobes symmetrically on both sides. At age 65, he was bed-ridden with marked disused muscle atrophy. The lower extremities showed contracture in extension and voluntary movements were seen only in the eyes, neck and respiratory muscles with no fasciculations. Forced crying was observed. Aspiration pneumonia occurred repeatedly and the patient died of pneumonia at the age of 66, 9 years after disease onset.
Methods
The left hemisphere of the brain was fixed in formalin, and embedded in paraffin. Serial coronal sections of the brain were examined macroscopically. Sections were stained using hematoxylin-eosin (HE), Klüver-Barrera, and modified Bielschowsky and Gallyas-Braak methods for histological examination. The immunostaining was performed using the rabbit polyclonal antibodies against ubiquitin (Z 0458; Dako, Glostrup, Denmark, 1:250) and cystatin C (A 0451; Dako, 1:1,000) and a mouse monoclonal antibody against phosphorylated tau (AT8, Innogenetics, Ghent, Belgium, 1:10,000). Sections were deparaffinized and incubated with primary antibodies overnight at 4°C, then visualized by the avidin-biotin-peroxidase complex method using diaminobenzidine tetrahydrochloride as the chromogen.
Results
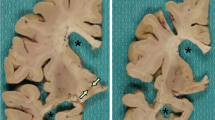
The brain weighed 1,000 g before fixation. There was marked atrophy of the frontal and temporal lobes including the precentral and superior temporal gyri (Fig. 1A). Coronal sections showed moderate atrophy of the neostriatum (caudate nucleus and putamen) and a marked reduction in volume of the white matter with a dilatation of the lateral ventricle. In the brainstem, pigment was moderately reduced in the substantia nigra and there was marked atrophy of the cerebral peduncle and medullary pyramid (Fig. 1B). The spinal cord and nerve roots appeared unremarkable. Microscopic examination revealed widespread neuronal loss and gliosis in the frontal and temporal cortices, being more accentuated in the superior temporal cortex and precentral cortex with marked microvacuolation in cortical layer II (Fig. 2A). Betz cells were completely absent. The white matter under the frontotemporal cortices showed myelin pallor and gliosis. The hippocampus and dentate gyrus were relatively preserved. Moderate neuronal loss and gliosis with microvacuolation were noted in the neostriatum. The substantia nigra showed moderate to severe neuronal loss with gliosis. The hypoglossal nucleus appeared preserved. The pyramidal tract showed marked degeneration with gliosis through the internal capsule to medulla oblongata. The lateral and anterior corticospinal tracts of the spinal cord showed marked degeneration (Fig. 1C, D). The anterior horn at the level of C8 was slightly atrophic with neuronal cells well preserved (Fig. 1B). Immunohistochemically, ubiquitin-positive but tau-negative intraneuronal inclusions were found in the frontal and temporal cortices, including the precentral cortex and the hippocampal dentate gyrus, and the basal ganglia, including the amygdala, putamen, and caudate (Fig. 2B–E), but not in the brainstem and spinal cord. Bunina bodies and skein-like inclusions were not detected. No Lewy bodies, Pick bodies, neurofibrillary tangles, or senile plaques were present.

Frontotemporal atrophy and pyramidal tract degeneration. A Brain showing remarkable frontotemporal lobe atrophy. B Severe atrophy in the cerebral peduncle of the midbrain. C, D Myelin pallor in the anterior and lateral corticospinal tracts in the cervical cord (C8) (C) and in the lateral corticospinal tract in the lumbar cord (L4) (D). Klüver-Barrera

Microscopic findings. A Severe neuronal loss and gliosis of the precentral cortex with marked microvacuolation. Hematoxylin-eosin. B, C Higher magnification of the precentral cortex (B) and frontal cortex (C) showing microvacuolation and the presence of ubiquitinated inclusions in the residual small neurons. Immunostaining. D, E ubiquitinated inclusions in the granular cells of the dentate gyrus (D) and in the neurons of the putamen (E). Immunostaining. Bar A 200 μm; B 20 μm; C–E 10 μm
Discussion
ALS with dementia, MND-FTD and MND-ID display ubiquitinated neuronal inclusions in the frontotemporal cortex and hippocampal dentate gyrus [2, 8, 11]. These conditions may be closely linked as different manifestations of the clinical spectrum of diseases associated with such inclusions. ALS with dementia, characterized by progressive motor neuronal and psychiatric symptoms, is a variant of ALS that was proposed by Mitsuyama as a new entity [5]. Lower motor symptoms predominate with little upper motor involvement and no parkinsonism, although ubiquitinated neuronal inclusions have recently been reported as present in the neostriatum and substantia nigra of ALS with dementia [1, 10]. Conversely, MND-FTD, characterized by behavioral abnormalities and dementia with or without lower motor symptoms, displays late parkinsonism but no cortical bulbar or spinal deficits. The latter, in fact, serve to exclude diagnosis of MND-FTD [8]. In the present case, characterized by ubiquitinated neuronal inclusions in the frontotemporal cortex, including the precentral gyrus and neostriatum, in addition to the hippocampal dentate gyrus, severe degeneration of the precentral gyrus was present throughout the pyramidal tract and neostriatum, corresponding to the clinical features of predominant upper motor neuron signs mimicking PLS and l-dopa non-responsive parkinsonism, respectively. Predominant upper motor neuron sign is usually absent in MND-FTD, and has only been reported in three cases [3, 7, 9] with marked degeneration of the pyramidal tract and the absence of definite lower motor neuron degeneration, two of which manifested as PLS [3, 7] and one as frontal Pick’s disease [9]. Parkinsonism is absent in ALS with dementia [5], and occurs as a late but not prominent sign in MND-FTD [8]. In line with these findings, conditions associated with ubiquitinated neuronal inclusions may represent multisystem degeneration involving the frontotemporal lobe, neostriatum, and pyramidal tract, in addition to the lower motor system, and should be noted to include clinical phenotypes such as MND-FTD, ALS with dementia, PLS, and l-dopa non-responsive parkinsonism.
Reference
Al-Sarraj S, Maekawa S, Kibble M, Everall I, Leigh N (2002) Ubiquitin-only intraneuronal inclusion in the substantia nigra is a characteristic feature of motor neurone disease with dementia. Neuropathol Appl Neurobiol 28:120–128
Jackson M, Lennox G, Lowe J (1996) Motor neuron disease-inclusion dementia. Neurodegeneration 5:339–350
Konagaya M, Sakai M, Matsuoka Y, Konagaya Y, Hashizume Y (1998) Upper motor neuron predominant degeneration with frontal and temporal lobe atrophy. Acta Neuropathol 96:532–536
Le Forestier N, Maisonobe T, Piquard A, Rivaud S, Crevier-Buchman L, Salachas F, Pradat PF, Lacomblez L, Meininger V (2001) Does primary lateral sclerosis exist? A study of 20 patients and a review of the literature. Brain 124:1989–1999
Mitsuyama Y (1984) Presenile dementia with motor neuron disease in Japan: clinico-pathological review of 26 cases. J Neurol Neurosurg Psychiatry 47:953–959
Okamoto K, Hirai S, Yamazaki T, Sun XY, Nakazato Y (1991) New ubiquitin-positive intraneuronal inclusions in the extra-motor cortices in patients with amyotrophic lateral sclerosis. Neurosci Lett 129:233–236
Tan CF, Kakita A, Piao YS, Kikugawa K, Endo K, Tanaka M, Okamoto K, Takahashi H (2003) Primary lateral sclerosis: a rare upper-motor-predominant form of amyotrophic lateral sclerosis often accompanied by frontotemporal lobar degeneration with ubiquitinated neuronal inclusions? Report of an autopsy case and a review of the literature. Acta Neuropathol 105:615–620
The Lund and Manchester Groups (1994) Consensus statement. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry 57:416–418
Tsuchiya K, Ikeda K, Haga C, Kobayashi T, Morimatsu Y, Nakano I, Matsushita M (2001) Atypical amyotrophic lateral sclerosis with dementia mimicking frontal Pick’s disease: a report of an autopsy case with a clinical course of 15 years. Acta Neuropathol 101:625–630
Wakabayashi K, Piao YS, Hayashi S, Kakita A, Yamada M, Takahashi H (2001) Ubiquitinated neuronal inclusions in the neostriatum in patients with amyotrophic lateral sclerosis with and without dementia-a study of 60 patients 31 to 87 years of age. Clin Neuropathol 20:47–52
Wightman G, Anderson VE, Martin J, Swash M, Anderton BH, Neary D, Mann D, Luthert P, Leigh PN (1992) Hippocampal and neocortical ubiquitin-immunoreactive inclusions in amyotrophic lateral sclerosis with dementia. Neurosci Lett 139:269–274
Acknowledgement
This study was supported by the University of Tsukuba Research Projects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mochizuki, A., Komatsuzaki, Y., Iwamoto, H. et al. Frontotemporal dementia with ubiquitinated neuronal inclusions presenting with primary lateral sclerosis and parkinsonism: clinicopathological report of an autopsy case. Acta Neuropathol 107, 377–380 (2004). https://doi.org/10.1007/s00401-003-0818-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0818-7