
Abstract
Although mdx mice share the same genetic defect and lack dystrophin expression as in Duchenne muscular dystrophy (DMD), their limb muscles have a high regenerative capacity that ensures a more benign phenotype and essentially normal function. The cellular pathways responsible for this enhanced regenerative capacity are unknown. We tested the hypothesis that the calcineurin signal transduction pathway is essential for the successful regeneration following severe degeneration observed in the limb muscles of young mdx mice (2–4 weeks old) and that inhibition of this pathway using cyclosporine A (CsA) would exacerbate the dystrophic pathology. Eighteen-day-old mdx and C57BL/10 mice were treated with CsA for 16 days. CsA administration severely disrupted muscle regeneration in mdx mice, but had minimal effect in C57BL/10 mice. Muscles from CsA-treated mdx mice had fewer centrally nucleated fibers and extensive collagen, connective tissue, and mononuclear cell infiltration than muscles from vehicle-treated littermates. The deleterious effects of CsA on muscle morphology were accompanied by a 30–35% decrease in maximal force producing capacity. Taken together, these observations indicate that the calcineurin signal transduction pathway is a significant determinant of successful skeletal muscle regeneration in young mdx mice. Up-regulating this pathway may have clinical significance for DMD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Duchenne muscular dystrophy (DMD), a severe X chromosome-linked myopathy, is caused by mutations in the dystrophin gene. The lack of dystrophin expression is thought to compromise the structural integrity of the muscle membrane and render skeletal muscles more susceptible to contraction-induced injury and degeneration [22]. Muscle regeneration is unable to keep pace with on-going degeneration and there is a progressive loss of skeletal muscle. While sharing the same genotype as in DMD, mdx mice have a more benign dystrophic phenotype. Mdx mice undergo a bout of severe degeneration at 2–4 weeks of age, but a high regenerative capacity ensures almost complete functional and structural recovery of the hindlimb muscles, such that by 10 weeks of age only 10% of the fibers are regenerating [8, 11]. The cellular mechanisms responsible for the enhanced regenerative capacity of mdx hindlimb muscles are not well understood.
The enhanced regenerative capacity in young mdx mice has been attributed, in part, to increased insulin-like growth factor I (IGF-I) levels in blood and muscle [11]. IGF-I is a potent growth factor, essential for satellite cell proliferation and differentiation. It has been shown to be an upstream activator of the calcineurin signal transduction pathway [26, 35]. Calcineurin, a phosphatase enzyme that regulates transcription by sensing changes in intracellular calcium, has been shown to regulate skeletal muscle regeneration [5, 6]. During regeneration, activated calcineurin may promote the transcription of myoregulatory factors, including myogenin, and control satellite cell differentiation and myotube maturation [14]. Calcineurin is a heterodimer composed of calcineurin-A, the catalytic subunit, and calcineurin-B, the regulatory subunit. Calcineurin-B is required for phosphatase activity of calcineurin-A and has been used as an indirect marker of calcineurin activity [25]. Calcineurin-A protein content has been shown to be elevated in regenerating rat muscles for up to 2 weeks following bupivacaine-induced myotoxic injury [30]. Furthermore, inhibition of the calcineurin signaling pathway impairs skeletal muscle regeneration following injury [1, 30]. Whether calcineurin-A and -B protein content is elevated in regenerating muscles of young mdx compared to wild-type (C57BL/10ScSn) mice is unknown.
Studies investigating the role of calcineurin in skeletal muscle regeneration have, to date, been conducted in cell culture or have used non-dystrophic animal models. The contribution of the calcineurin signal transduction pathway to the successful regeneration of mdx hindlimb muscles and the consequences of calcineurin inhibition using cyclosporine A (CsA) are unknown. One study has investigated the effect of CsA administration on regeneration and necrosis in young mdx mice [40]. Thirty days (from day 15 to day 45 postnatum) of CsA administration at a dose of 2 mg/kg per day did not affect necrosis or regeneration in plantaris and soleus muscles from mdx mice. Serum creatine kinase activity was doubled in the CsA-treated mice compared to vehicle-treated controls, suggesting higher levels of injury and possibly impaired regeneration. The results were highly variable among animals, and as such, this finding was not statistically significant [40]. Furthermore, the dose of CsA used in that study was 20-fold lower than that used typically to inhibit calcineurin and to have immunosuppressive effects in mice [12].
We designed a study using an appropriate dose of CsA (30 mg/kg per·day) to test the hypothesis that inhibition of calcineurin activity would impair skeletal muscle regeneration in young mdx mice. We also tested the hypothesis that, due to the on-going muscle degeneration and regeneration and elevated IGF-I levels, calcineurin-A and -B protein content would be greater in extensor digitorum longus (EDL) and soleus muscles from mdx than from C57BL/10 control mice. We report that in young mdx mice calcineurin activity is required for successful skeletal muscle regeneration, since ~16 days of CsA administration inhibited regeneration, compromised muscle growth, increased the dystrophic pathology, and impaired muscle function in mdx mice, but not C57BL/10 mice.
Materials and methods
All experiments were approved by the Animal Experimentation Ethics Committee of The University of Melbourne. Pregnant female wild-type, C57BL/10ScSn (C57BL/10) and dystrophic mdx mice were obtained from the Animal Resource Centre (Nedlands, Western Australia) and maintained on a 12-h-light/12-h-dark cycle, with standard mouse chow and water provided ad libitum. The pups and dams remained in the same cage during the course of the experiment. To minimize the number of animals killed both male and female pups were used.
CsA administration
From days 17–18 postnatum, mice were injected with either vehicle or CsA (30 mg/kg per·day; kindly provided by Novartis Pharmaceuticals Australia Pty) intraperitoneally once daily for 16 days. This dose has been shown to suppress calcineurin activity in skeletal muscle [25] and was well tolerated by the mice for the duration of the experiment. The vehicle consisted of 7.8% cremphor (C5135, Sigma-Aldrich, Castle Hill, Australia) and 4% ethanol in sterile PBS. A saline only vehicle control group was not included in the experimental design because previous studies have indicated that the cremphor vehicle does not affect skeletal muscle contractile function in vitro [18].
Assessment of skeletal muscle function
At the conclusion of treatment, mice were anesthetized with pentobarbital sodium (Nembutal; Rhone Merieux, Pikenba, Australia; 40 mg/kg) such that they were unresponsive to tactile stimuli. Isometric contractile properties of the EDL (fast-twitch) and the soleus (slow-twitch) muscles from 12 mice per group were evaluated in vitro, as described in detail previously [12, 13, 22]. Briefly, muscles were tied at the proximal and distal tendons with braided surgical silk, excised surgically, and transferred to a custom built plexiglas bath filled with Krebs Ringer (137 mM NaCl, 24 mM NaHCO3, 11 mM d-glucose, 5 mM KCl, 2 mM CaCl2, 1 mM NaH2PO4·H2O, 1 mM MgSO4, 0.025 mM d-tubocurarine chloride), bubbled with Carbogen (5% CO2 in oxygen; BOC Gases; Preston, Australia), and thermostatically maintained at 25°C. The distal tendon of the muscle was tied to an immovable pin and the proximal tendon was attached to the lever arm of a dual mode servomotor/force transducer (300B-LR; Aurora Scientific, Ontario, Canada). The EDL and soleus muscles were stimulated by supramaximal square wave pulses of 350 ms and 1,200 ms in duration, respectively, which were amplified (Ebony EP500B Audio Assemblers, Campbellfield, Victoria, Australia) and delivered via two platinum electrodes that flanked the length of the muscle to produce a maximum isometric contraction [18, 21]. All stimulation parameters and contractile responses were controlled and measured using custom built applications (D.R. Stom Inc., Ann Arbor, MI) of LabView software (National Instruments, Austin, TX) driving a personal computer with an onboard controller (PCI-MIO-16XE-10, National Instruments) interfaced with the transducer-servomotor control/feedback hardware (Aurora Scientific) [16, 21]. Optimum muscle length (Lo) was determined from micromanipulations of muscle length and series of isometric twitch contractions. Maximum isometric tetanic force (Po) production was determined from the plateau of the frequency-force relationship. To assess fatigability, Po was determined once every 5 s for a duration of 4 min. Recovery of Po was determined 5, 10, and 15 min following the fatigue protocol. Following testing, the muscles were removed from the bath, trimmed of their tendons and non-muscle tissue, weighed on an analytical balance, and stored at −70°C. Whole muscle cross-sectional area was determined by dividing the muscle mass by the product of optimum fiber length (Lf) and muscle density (1.06 mg/mm3) [21, 22]. Lf was determined by multiplying Lo by previously determined muscle length to fiber length ratios, 0.44 for the EDL and 0.71 for the soleus [21, 22]. Since Po is dependent upon muscle size, Po values were normalized for cross-sectional area (Po was divided by the calculated total muscle cross-sectional area) and expressed as specific force (sPo; kN/m2) [22].
Histology and immunohistochemistry
The contralateral EDL and soleus muscles were surgically excised, mounted in embedding medium at approximately Lo, frozen in thawing isopentane, and stored at −70°C. Four 5-μm-thick sections were cut from the mid-belly region of each muscle on a cryostat at −20°C. The sections were air dried overnight and stored at −70°C. The first section was stained with hematoxylin and eosin (H&E) for determination of general muscle architecture and fiber cross-sectional area, the second with Van Gieson’s stain for determining collagen content, and the remaining two sections were allocated for immunohistochemical analysis (n=8–10 different mice per muscle per group). Colored, digitized images of H&E and Van Gieson’s sections were captured with a digital camera (Spot, v2.2, Diagnostic Instruments, Sterling Heights, MI) mounted to an upright microscope (BX-51, Olympus, Tokyo, Japan), at 200× and 400× magnification, respectively, and analyzed using a computerized image analysis system (AIS, v6.0, Imaging Research Inc., St. Catherines, Ontario, Canada). Mean fiber cross-sectional area was calculated by determining the area of 127±15 individual muscle fibers from each muscle section. The area occupied by connective tissue and cellular infiltrate was quantified and expressed relative to total muscle section area to determine the extent of muscle degeneration. Collagen content was quantified by determining the percentage of the cross-section stained pink, indicating collagen infiltration. The Van Gieson’s-stained sections were analyzed twice with the coefficient of variation being less than 10%.
Immunohistochemical techniques were used to determine the number of macrophages and myogenin-positive nuclei in each section. Negative control sections were included in all analyses and a section of mouse spleen was used as an additional positive control for the macrophage analysis. The sections were fixed in cold acetone for 15 min, then incubated with 1.5% H2O2 in 0.5 M TRIS-buffered saline (TBS) for 5 min, and blocked with 1% normal goat serum for 15 min. The primary antibody was diluted in 1% normal goat serum and positive sections were incubated for 60 min, following which all slides were incubated for 30 min with a secondary antibody. Primary antibody dilutions were as follows: myogenin (sc-576, Santa Cruz Biotechnology Inc., Santa Cruz, CA) at 1:60 and F4/80 for macrophages (MCA497R, Serotech, Oxford, UK) at 1:60 [14]. The secondary antibodies used were the Dako Envision+ anti-rabbit antibody (K4003, Dako Corporation, Botany, Australia) for myogenin staining, and the goat anti-rat STAR72 antibody (Serotech, Oxford, UK) for macrophage staining. The AEC+ substrate chromogen (K3468, Dako Corporation) was used for color development.
The number of macrophages and myogenin-positive nuclei in each section were counted and expressed as number of positive cells per mm2 of muscle. Entire muscle cross-sections were captured with a digital camera at 100× magnification and the whole section area was quantified using a computerized image analysis system.
Muscle homogenization
To ensure adequate protein content, two soleus or EDL muscles were pooled prior to analysis (n=8 samples per group). Frozen muscles were homogenized in ice-cold buffer (1:20 w:v; 50 mM KCl, 10 mM KH2PO4, 2 mM MgCl2·6H2O, 0.5 mM EDTA, 2 mM DTT) [32], then centrifuged at 1000 g for 10 min at 5°C. The supernatant (cytosolic fraction) was used for determination of calcineurin-A and -B protein content [37]. The pellet was re-suspended in ice-cold buffer (1:20) w:v; 50 mM KCl, 10 mM KH2PO4, 2 mM MgCl2·6H2O, 0.5 mM EDTA, 2 mM DTT, 1% Triton X-100) [32] and allocated for myosin heavy chain (MyHC) isoform analysis. Protein content of the pellet and supernatant fractions was determined using the Bradford Assay (500-0006, Bio-Rad Laboratories, Regents Park, Australia), with 0.6 μg protein loaded per lane for MyHC isoform analysis and 30 μg protein loaded per lane for calcineurin analysis [37]. Samples were diluted to appropriate concentrations with 4× sample buffer and storage buffer (60 mM KCl, 130 mM imidazole, 2 mM MgCl2·6H2O, 1 mM DTT) [32]. The MyHC isoform samples were run the same day, and the calcineurin samples were stored at −70°C until analysis.
Western blotting for calcineurin-A and -B
All samples were run in duplicate and each gel contained EDL and soleus muscle samples from both vehicle and CsA-treated C57BL/10 and mdx mice, a pre-stained molecular weight protein standard (161-0372, Bio-Rad Laboratories), and a control sample consisting of a mixture of EDL and soleus muscle homogenate. Cytosolic proteins were separated by SDS-PAGE on a 12% acrylamide gel, transferred electrophoretically to a polyvinylidene difluoride (PVDF) membrane (162-0177, Bio-Rad), and blocked for 1 h in 9% cold fish gelatin (G7765, Sigma-Aldrich, Castle Hill, Australia) in TRIS-buffered saline with 1% Tween-20 (TTBS). The PVDF membrane was cut in half at the 50-kDa mark, the bottom half probed with a primary antibody against calcineurin-B (1:3,000 in TTBS, 07-069, Upstate Biotechnology Inc., Waltham, MA) and the top half with a primary antibody against calcineurin-A (1:3,000 in TTBS, sc7090, Santa-Cruz Biotechnologies, Santa Cruz, CA). Membranes were incubated at 4°C overnight, then washed extensively with TTBS prior to incubation with a secondary goat, anti-rabbit antibody conjugated with biotinylated-streptavidin alkaline phosphatase (1:3,000 in TTBS, sc2020, Santa-Cruz Biotechnologies) for 1.5 h. Blots were developed using an alkaline phosphatase conjugate substrate kit with 5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazolium (BCIP/NBT; 170-6432, Bio-Rad). Blots were digitized using a scanner and a computerized image analysis system was used to determine the band density and molecular weight. Calcineurin-A and -B protein content was expressed relative to the protein content of the control sample and averaged between the two duplicate blots.
SDS-PAGE for MyHC isoform analysis
MyHC isoforms were separated on 9% glycerol acrylamide gel according to methods described by Talmadge and Roy [38] with the following modifications: 0.08% 2-mercaptoethanol was added to the upper and lower running buffer to improve MyHC isoform resolution [32] and proteins were visualized using a commercially available silver stain kit (LC6100, Invitrogen Corporation, Melbourne, Australia). All gels were run and analyzed in duplicate. The region of gels containing the MyHC isoforms was scanned and a computerized image analysis system was used to determine the proportion of each MyHC isoform.
Statistics
Values presented in all graphs and tables are expressed as means ± SEM. All statistical analyses were performed using a statistics software package (Minitab Inc., State College, PA). With regards to the contractile properties, MyHC isoform proportions, whole muscle and fiber cross-sectional area data differences between EDL and soleus muscles have been well characterized, thus these data were analyzed using a two-way ANOVA with the factors being strain and treatment. Body and muscle mass data were also analyzed using a two-way ANOVA. The immunohistochemical data, proportion of fibers with central nuclei, proportion of total muscle area undergoing degeneration, collagen content, and calcineurin protein content data were analyzed using a three-way ANOVA with the factors being muscle (EDL vs soleus), strain, and treatment. Three-way ANOVA was performed on these results because muscle specific differences were not known. Tukey’s post-hoc test was used to locate pair-wise significant differences, where appropriate. For all comparisons, a P<0.05 was considered significant.
Results
Effect of CsA treatment on body mass
All mice tolerated the CsA and vehicle injections well and no decrease in body mass was observed following the onset or during of treatment. At the conclusion of treatment, however, CsA-treated mdx mice weighed ~25% less than the vehicle-treated mdx mice and ~13–20% less than CsA- and vehicle-treated C57BL/10 mice (P<0.05). Vehicle-treated C57BL/10 and mdx mice gained on average 7.0±0.4 g in body mass during the course of the experiment and the CsA-treated C57BL/10 mice gained 5.6±0.4 g, whereas the CsA-treated mdx mice gained only 2.6±0.5 g (Table 1). CsA treatment also compromised growth of C57BL/10 mice, since the overall weight gain tended to be less than in vehicle-treated littermates, although this was not significant (P=0.08) (Table 1). At the end of the treatment period, body mass of vehicle-treated mdx mice was greater than the vehicle-treated C57BL/10 mice (P<0.05). This finding is characteristic of the mdx phenotype and indicative of the overcompensation of the muscle regenerative response following the peak period of muscle degeneration [39].
Effect of CsA treatment on muscle mass
The mass of all muscles (EDL, soleus, plantaris, and tibialis anterior) examined was lower in CsA-treated mice than vehicle-treated littermates (P<0.05) and this effect was greater in mdx mice than C57BL/10 mice (Table 1). Since body mass of vehicle-treated mdx mice was greater than C57BL/10 mice, EDL, soleus, plantaris, and tibialis anterior (TA) muscle mass was also greater (P<0.05).
Maximum force producing capacity
Maximum isometric force (Po) of EDL and soleus muscles from mdx mice was lower than that from C57BL/10 mice (P<0.05), indicative of the extent of the dystrophic pathology at this age. Po of the EDL and soleus muscles from CsA-treated mdx and C57BL/10 mice was less that from vehicle-treated littermates (P<0.05), and this effect was more pronounced in mdx than C57BL/10 mice. Compared with vehicle-treated mdx mice, Po of EDL and soleus muscles from CsA-treated littermates was ~30–35% lower (Table 1). Specific force (sPo) of EDL and soleus muscles was lower in mdx than C57BL/10 mice. sPo of soleus muscles from CsA-treated mdx mice was lower than that of vehicle-treated littermates, whereas sPo of soleus muscles from CsA and vehicle-treated C57BL/10 mice was not different. In contrast, no difference in sPo of EDL muscles from CsA- and vehicle-treated mdx and C57BL/10 mice was observed (Table 1).
Muscle fatigue
Following the 4-min fatigue protocol, Po of EDL muscles was reduced to approximately 40% of initial Po but recovered to 65–75% of initial values following 5 min of rest, and to 75–90% following 10–5 min of recovery. The EDL muscles from CsA-treated C57BL/10 mice demonstrated the greatest resistance to fatigue and greatest recovery Po compared with the EDL muscles of mice from the other groups (P<0.05). The Po of soleus muscles was reduced to approximately 50% of initial Po following the fatigue protocol and recovered to 80–95% of initial Po values following 5 min of rest, but was not altered following an additional 10 min and 15 min of rest. Unlike EDL muscles, Po of soleus muscles during fatigue or recovery was not statistically different in any of the experimental groups (Fig. 1).

Relative change in force output of EDL (A) and soleus (B) muscles following 4 min of intermittent determination of Po and 15 min of recovery. CsA administration did not decrease the resistance to fatigue of EDL or soleus muscles nor did it impede recovery of force. EDL muscles from CsA-treated C57BL/10 mice had the highest Po after fatigue and in recovery compared with the other groups (*P<0.05) (EDL extensor digitorum longus, CsA cyclosporine A, veh vehicle-treated)
Muscle morphology
CsA administration did not affect the general architecture of muscles from C57BL/10 mice (see Fig. 4a, b). Indicative of impaired regeneration, in CsA-treated mdx mice increased connective tissue and cellular infiltrate, and a loss of viable muscle fibers was observed in all hindlimb muscles examined (Fig. 2a–h). In EDL and soleus muscles from CsA-treated mdx and C57BL/10 mice, overall muscle cross-sectional area and mean fiber area were significantly smaller than in vehicle-treated mice (P<0.05, Table 1). In CsA-treated mdx mice, the proportion of muscle cross-section occupied by connective tissue and cellular infiltrate was greater than both vehicle-treated littermates and C57BL/10 mice (P<0.05). The proportion of degenerating muscle fibers was ~300% greater in EDL and ~400% greater in soleus muscles from CsA-treated mdx mice than from vehicle-treated littermates (see Fig. 4f). Associated with the observed degeneration was a 200% greater collagen content in muscles from CsA-treated mdx mice (P<0.05, see Fig. 4g). In mdx mice, the area occupied by connective tissue and cellular infiltrate in soleus muscles from CsA- and vehicle-treated mice was 2.5–4 times greater than in EDL muscles. Furthermore, the collagen content in soleus muscles from CsA- and vehicle-treated mdx mice was ~3 times greater than in EDL muscles (P<0.05).

Representative cross-sections of hindlimb muscles stained with H&E from vehicle-treated (A, C, E, G) and CsA-treated (B, D, F, H) mdx mice; A, B soleus, C, D EDL, E, F tibialis anterior, G, H plantaris muscles. Note the increase in cellular infiltrate and loss of viable muscle fibers in the CsA-treated muscles. Representative soleus muscle cross-sections stained with Van Gieson’s; I from a vehicle-treated mdx mouse and J from a CsA-treated mdx mouse. Muscle tissue is orange, connective tissue pink and nuclei were counter-stained with hematoxylin (blue) (H&E hematoxylin and eosin). Original magnification ×200
Skeletal muscle regeneration: myogenin-positive nuclei and centrally nucleated fibers
EDL and soleus muscles from CsA-treated mdx mice contained four and two times more myogenin-positive nuclei, respectively, than muscles from vehicle-treated littermates (P<0.05). In contrast, myogenin-positive nuclei counts were not different between the muscles from treated and untreated C57BL/10 mice. Muscles from vehicle-treated mdx mice had a similar number of myogenin-positive nuclei as muscles from vehicle-treated C57BL/10 mice (Fig. 3a–c). In contrast, the proportion of centrally nucleated fibers was lower in CsA-treated mdx mice compared with their vehicle-treated littermates (P<0.05), such that muscles from CsA-treated mdx mice had a similar number of centrally nucleated fibers as muscles from C57BL/10 mice (Fig. 4c–e).

Representative muscle cross-sections reacted with an antibody against myogenin; soleus muscles from a vehicle-treated mdx mouse (A) and a CsA-treated mdx mouse (B). Note the greater number of myogenin-positive nuclei (arrow) in the soleus from the CsA-treated compared to the vehicle-treated mdx mouse. C Number of myogenin-positive nuclei in EDL and soleus muscles from C57BL/10 and mdx mice. Muscles from CsA-treated mdx mice have a greater number of myogenin-positive nuclei than their vehicle-treated littermates or the C57BL/10 mice (*P<0.05). D, E Representative muscle cross-sections react with an antibody against macrophages (F4/80); D soleus from a vehicle-treated C57BL/10 mouse, and E EDL from a vehicle-treated mdx mouse. Macrophages (arrow) are rarely detected in the C57BL/10 muscles, but are prevalent in mdx muscles due to the on-going degeneration and regeneration. Note the presence of centrally nucleated fibers in the mdx EDL muscle (arrowhead). F Number of macrophages in EDL and soleus muscles from C57BL/10 and mdx mice. Macrophage infiltration is greater in muscles from mdx mice than C57BL/10 mice (*P<0.05). A, B, D, E ×400

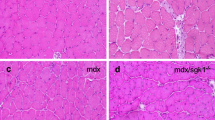
Histological characteristics of EDL and soleus muscles from C57BL/10 and mdx mice. Each cross-section is stained with H&E. A EDL from a C57BL/10 vehicle-treated mouse; B EDL from a C57BL/10 CsA-treated mouse. C EDL from a vehicle-treated mdx mouse; note the presence of centrally nucleated fibers (arrows). D EDL from a CsA-treated mdx mouse; note the absence of centrally nucleated fibers and the infiltrating mononuclear cells. E The proportion of fibers with central nuclei in EDL and soleus muscles from C57BL/10 and mdx mice. Muscles from vehicle-treated mdx mice have the greatest proportion of centrally nucleated fibers (*P<0.05). CsA treatment reduces the number of fibers with central nuclei in EDL and soleus muscles from mdx mice to similar levels seen in muscles from C57BL/10 mice. F Proportion of muscle infiltrated by mononuclear cells and connective tissue in EDL and soleus muscles. CsA administration increases skeletal muscle degeneration in mdx but not C57BL/10 muscles († P<0.05). Soleus muscles from mdx mice show more degeneration than EDL muscles from mdx mice, and EDL and soleus muscles from C57BL/10 mice (*P<0.05). G Proportion of collagen in EDL and soleus muscles. CsA administration increased the collagen content in muscles from mdx mice but not C57BL/10 mice († P<0.05). Soleus muscles from mdx mice contained the greatest amount of collagen when compared to EDL muscles from mdx mice, and EDL and soleus muscles from C57BL/10 mice (*P<0.05)
Inflammatory response: macrophage infiltration
Due to the on-going degeneration and regeneration within the skeletal muscles of mdx mice, the number of infiltrating macrophages was greater in both the soleus and EDL muscles of mdx compared with C57BL/10 mice (P<0.05) (Fig. 3d–f). Macrophage numbers were not different in EDL and soleus muscles from CsA- and vehicle-treated mdx mice, indicating that CsA administration did not affect macrophage infiltration.
Calcineurin-A and -B protein content
The calcineurin-A or calcineurin -B protein content of skeletal muscle was similar in vehicle- and CsA treated mice. C57BL/10 mice had greater calcineurin-A protein content in their EDL muscles than mdx mice. Calcineurin-A protein content also was greater in EDL than soleus muscles of C57BL/10 mice, but was not different in mdx mice. In both mdx and C57BL/10 mice, calcineurin-B protein content was greater in EDL muscles compared with soleus muscles (Fig. 5a–c).

A Representative Western blots for calcineurin-A and -B protein. Lane 1: mdx CsA soleus; lane 2: mdx vehicle soleus; lane 3: BL10 CsA soleus; lane 4: BL10 vehicle soleus; lane 5: mdx CsA EDL; lane 6: mdx vehicle EDL; lane 7: BL10 CsA EDL; lane 8: BL10 vehicle EDL; lane 9: control sample; lane 10: molecular weight marker. B Calcineurin-A protein content is greater in EDL muscles from C57BL/10 mice than mdx mice (*P<0.05). Fiber type specific differences in calcineurin A protein content are detected in C57BL/10 mice, but not mdx mice († P<0.05). C Calcineurin-B protein content is greater in the fast EDL muscles than slow soleus muscles. D, E Proportion of MyHC isoforms in EDL (D) and soleus (E) muscles from mdx and C57BL/10 mice. D EDL muscles from mdx mice contain a greater proportion of the type IIa MyHC isoform than from C57/BL10 mice (*P<0.05). E The type IIx MyHC isoform is detected in the soleus muscles of mdx mice only and these muscles contain a greater proportion of the type I MyHC isoform than C57BL10 soleus († P<0.05). Sixteen days of CsA administration does not alter MyHC isoform proportions in either the EDL (D) or soleus muscles (E)
MyHC isoforms
CsA administration did not alter the proportion of MyHC isoforms in either the EDL or soleus muscles from mdx or C57BL/10 mice. The type IIx MyHC isoform was detected in the soleus muscles of mdx but not C57BL/10 mice. The soleus muscles from mdx mice also had a greater proportion of the type I MyHC isoform than soleus muscles from C57BL/10 mice. EDL muscles from mdx mice had a greater proportion of the type IIa MyHC isoform than EDL muscles from C57BL/10 mice (Fig. 5d, e).
Discussion
To our knowledge, this is the first report to demonstrate that successful regeneration of hindlimb skeletal muscles in mdx mice is dependent on the calcineurin signal transduction pathway and that inhibition of calcineurin with CsA impairs regeneration of dystrophic muscle. In mdx mice, CsA administration dramatically impaired whole body and muscle growth. Muscle masses of treated mdx mice were ~25% lower than in untreated mdx mice. EDL and soleus muscle cross-sectional area and mean muscle fiber area were ~25% smaller in CsA-treated mdx mice than in vehicle-treated littermates. Furthermore, EDL and soleus muscles from CsA-treated mdx mice had 4 and 20 times fewer centrally nucleated fibers, respectively, and 2 to 4 times more collagen, connective tissue, and mononuclear cell infiltration than vehicle-treated littermates. All of these factors had a deleterious impact on force producing capacity. Taken together, these findings provide strong support that CsA treatment impairs muscle regeneration and growth in mdx mice.
Calcineurin may regulate the early stages of myotube formation in regenerating skeletal muscle [13]. Inhibition of the calcineurin signaling pathway using CsA has been shown to inhibit or delay myotube formation and maturation in regenerating skeletal muscle [1]. Sakuma et al. [30] administered CsA to wild-type mice for 2 weeks following intramuscular injection of bupivacaine to the tibialis anterior (TA) muscle to cause myotoxic injury. They reported that the mass of the injured TA muscles from CsA-treated mice was lower, mean fiber cross-sectional area was smaller, and the TA had a greater number of small diameter fibers compared with injured TA muscles from vehicle-treated mice. However, when CsA was administered to wild-type mice 3 days following bupivacaine-induced myotoxic injury to soleus muscles, mean muscle fiber area was not different at 10 days post injury from vehicle-treated controls [13], because by day 3 myotube formation would have already taken place [36]. Experimental data on the effect of calcineurin inhibition on muscle regeneration has not been consistent. Irintchev et al. [18] reported increased cross-sectional area of cryo-injured soleus muscles from mice treated with CsA for 4–7 weeks compared with injured soleus muscles from vehicle-treated mice. They suggested that, while CsA inhibits myoblast differentiation, it may simultaneously promote proliferation. Thus, the prolonged period of differentiation, which may appear as “impaired” regeneration initially, produced a greater number of myogenic cells prior to maturation and myotube fusion [18].
CsA administration may also have inhibited the growth of non-regenerating muscle fibers. In wild-type mice, growth of soleus muscles following hindlimb unloading-induced atrophy has been shown to depend upon satellite cell activation and fusion with existing myofibers, events apparently under the control of the calcineurin signal transduction pathway [25]. Over the course of the present experiment, body mass of vehicle-treated C57BL/10 mice doubled, likely due to muscle hypertrophy via increased contractile protein synthesis and myonuclear accretion [23]. The CsA-treated C57BL/10 mice gained ~20% less body mass and their hindlimb muscles weighed ~10% less (except for the plantaris); the EDL and soleus muscle cross-sectional area was ~5% and ~15% less, respectively, and mean fiber cross-sectional area in EDL and soleus muscles was ~15% and ~25% less, respectively, than their vehicle-treated littermates. Although not statistically significant, the magnitude and consistency of the values lends support to the hypothesis that the calcineurin signal transduction pathway may regulate muscle growth in young wild-type mice. It is unlikely that the decrease in body and muscle mass and muscle and fiber cross-sectional area observed in mdx and C57BL/10 mice was due to the toxic side effects of CsA, since no animals lost weight at the onset of treatment.
Whether the calcineurin signal transduction pathway regulates muscle growth and hypertrophy is contentious [15]. Skeletal muscles of mice over-expressing an activated form of calcineurin do not exhibit a greater muscle mass (hypertrophied muscles) than wild-type mice [27]. Reports from experiments using CsA (or other pharmacological inhibitors of calcineurin) are also conflicting, and dependent on the experimental setting [33]. In contrast, experiments where hypertrophy is dependent on myoblast maturation and fusion [35] or the addition of myonuclear number [25], the involvement of the calcineurin signal transduction pathway is more definitive.
Absolute Po and sPo were lower in muscles from 32-day-old mdx than age-matched C57BL/10 mice, likely due to the existence of degenerating and regenerating fibers, which have a reduced functional capacity than mature muscle fibers [7]. The reduced functional capacity observed in muscles of CsA-treated mdx mice (compared to vehicle-treated littermates), was associated with an increase in collagen, connective tissue and cellular infiltrate, and a loss of viable fibers (P<0.05). In contrast, no pathology was observed in the muscles of C57BL/10 mice.
The impaired contractile function following CsA treatment in mdx mice was not attributed to impaired mitochondrial metabolism [4, 18, 31] or to direct effects of CsA on muscle contractility [29]. CsA administration did not impair the fatigue resistance of EDL or soleus muscles from C57BL/10 or mdx mice nor did it impede recovery (Fig. 3). This provides strong evidence that mitochondrial function in the muscles of the treated mice was not impaired. Convincing evidence does exists that CsA has adverse effects on cardiac muscle contractility in vivo and in vitro [2, 19, 29]. However, this does not seem to be the case in skeletal muscles since sPo values of EDL and soleus muscles from vehicle and CsA-treated C57BL/10 mice were similar. This finding is further supported by reports that CsA does not alter the properties of the sarcoplasmic reticulum (SR) calcium-release channel and SR calcium ATPase activity in fast or slow twitch skeletal muscle [29]. However, since muscle function was evaluated in vitro, we cannot exclude the possibility that CsA may have adverse effects on muscle function in vivo. In rats, 4 weeks of CsA administration at an immunosuppressive dose decreased capillarity in EDL muscles, which might impact upon muscle fatigability and force producing capacity in vivo [4]. In organ transplant patients, CsA administration has been associated with myopathy and reduced exercise tolerance [6]. However, it is unknown whether compromised muscle function following organ transplantation is due to direct effects of CsA or due to detraining and/or disuse muscle atrophy.
We quantified myogenin-positive nuclei and the proportion of centrally nucleated fibers to determine the effects of calcineurin inhibition on muscle regeneration. Satellite cells express myogenin once they are committed to differentiation [13]. Therefore, myogenin expression is an early marker of muscle regeneration and precedes cell cycle withdrawal, myoblast fusion, and myotube formation [13, 34]. In cell culture, inhibition of calcineurin down-regulates myogenin expression and delays myoblast maturation, whereas activation of calcineurin up-regulates myogenin expression and enhances myoblast maturation [10, 13]. The appearance of centrally nucleated fibers is a marker of myotube formation and successful muscle regeneration [5]. Contrary to our initial hypothesis, CsA-treated mdx mice had approximately 2–4 times more myogenin-positive cells per mm2 of muscle tissue than their vehicle-treated littermates or the C57BL/10 mice (P<0.05). Myogenin expression is controlled by three intracellular signal transduction pathways: calcineurin, p38 mitogen-activated protein kinase, and calcium-calmodulin-dependent protein kinase. These pathways act in parallel, but non-redundantly to control myogenin expression [41]. In cell culture, inhibition of any one of these pathways will decrease or completely inhibit myogenin expression and myoblast differentiation [41]. Our results indicate that this does not occur in vivo and that other factors play a role.
CsA administration decreased the proportion of centrally nucleated fibers in mdx mice. The EDL and soleus muscles from vehicle-treated mdx mice contained ~30 and ~4 times more centrally nucleated fibers, respectively, than the EDL and soleus muscles from CsA-treated littermates. It has been hypothesized that inhibition of the calcineurin signal transduction pathway does not “impair” regeneration but rather “delays” it [18]. However, since we are unable to determine whether the reduced numbers of centrally nucleated fibers and the increased numbers of myogenin-positive nuclei seen in muscles from CsA-treated mdx mice represent a “delay” in regeneration, rather than an “impairment”, we feel that regeneration was impaired as indicated by the severity of the dystrophic pathology in muscles from CsA-treated mdx mice. The reduced number of centrally nucleated fibers in muscles from CsA-treated mdx mice suggests that calcineurin may also regulate myoblast fusion. It is the alignment and fusion of myoblasts during regeneration that gives muscle fibers their characteristic centrally nucleated appearance [5]. The mechanisms by which muscle fibers regulate the fusion of myoblasts are not well characterized; however, the NFATc2 transcription factor, a downstream target of calcineurin, has been implicated [17].
Due to ongoing degeneration and regeneration, macrophage infiltration was greater in muscles of mdx than C57BL/10 mice. No difference in macrophage numbers in muscles from vehicle and CsA-treated mdx mice was observed. CsA should not affect phagocytic cell function nor should it prevent granulocyte and macrophage migration [24], although in experimental settings, the effect of CsA on macrophages has not been consistent [20].
We hypothesized that due to on-going regeneration, calcineurin-A and -B protein content would be greater in muscles from mdx mice than from C57BL/10 mice. Contrary to our hypothesis, calcineurin-A protein content was greater in EDL muscles from C57BL/10 than mdx mice, but not different in soleus muscles. The observed differences in calcineurin-A protein content in slow and fast twitch muscles in C57BL/10 mice is consistent with previous reports [25, 37]. No difference in calcineurin-B protein content in muscles from C57BL/10 and mdx mice was observed. Unfortunately, there was insufficient muscle tissue for us to assess calcineurin activity directly. It is clear that further studies are needed to directly compare calcineurin activity in these muscles between C57BL/10 and mdx mice.
It is well established that calcineurin controls the specification of slow skeletal muscle fibers [3, 9, 28, 33, 36]. However, 16 days of CsA administration did not alter MyHC isoform proportions in soleus or EDL muscles from C57BL/10 or mdx mice (Fig. 5), and this was consistent with the absence of any deleterious changes in fatigability and force recovery. The absence of an effect of CsA treatment on MyHC isoform proportions may be attributed to the shorter duration of treatment employed in this study compared with other investigations [9, 36].
This study is the first to provide evidence that the enhanced regenerative capacity of mdx hindlimb muscles is regulated by the calcineurin signal transduction pathway. Inhibition of this pathway with CsA compromised growth, structure, and function of muscles from mdx mice, but had minimal effects on muscles from C57BL/10 mice. In mdx mice, CsA administration impaired the formation and maturation of myotubes, as evidenced by the lack of centrally nucleated fibers and the increased proportion of muscle infiltrated by collagen. It remains to be determined whether the calcineurin signal transduction pathway is impaired in DMD and whether increased activity of calcineurin can ameliorate dystrophic pathology of skeletal muscles.
References
Abbott KL, Friday BB, Thaloor D, Murphy TJ, Pavlath GK (1998) Activation and cellular localization of the cyclosporine A-sensitive transcription factor NF-AT in skeletal muscle cells. Mol Biol Cell 9:2905–2916
Banijamali HS, Keurs MHC ter, Leendert PC, Keurs HEDJ ter (1993) Excitation-contraction coupling in rat heart: influence of cyclosporine A. Cardiovasc Res 27:1845–1854
Bigard X, Sanchez H, Zoll J, Mateo P, Rousseau V, Veksler V, Ventura-Clapier R (2000) Calcineurin co-regulates contractile and metabolic components of slow muscle phenotype. J Biol Chem 275:19653–19660
Biring MS, Fournier M, Ross DJ, Lewis MI (1998) Cellular adaptations of skeletal muscle to cyclosporine. J Appl Physiol 84:1967–1975
Bischoff R (1994) The satellite cell and muscle regeneration. In: Engel AG (ed) Myology: basic and clinical. McGraw-Hill, New York, pp 97–118
Breil M, Chariot P (1999) Muscle disorders associated with cyclosporine treatment. Muscle Nerve 22:1631–1636
Brooks SV, Faulkner JA (1990) Contraction-induced injury: recovery of skeletal muscles in young and old mice. Am J Physiol 258:C436–C442
Chambers RL, McDermott JC (1996) Molecular basis of skeletal muscle regeneration. Can J Appl Physiol 21:155–184
Chin ER, Olson EN, Richardson JA, Yang Q, Humphries C, Shelton JM, Wu H, Zhu W, Bassel-Duby R, Williams RS (1998) A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev 12:2499–2509
Delling U, Tureckova J, Lim HW, De Windt LJ, Rotwein P, Molkentin JD (2000) A calcineurin-NFATc2-dpendent pathway regulates skeletal muscle differentiation and slow myosin heavy-chain expression. Mol Cell Biol 20:6600–6611
De Luca A, Pierno S, Camerino C, Cocchi D, Conte Camerino D (1999) Higher content of insulin-like growth factor I in dystrophic mdx mouse: potential role in the spontaneous regeneration through an electrophysiological investigation of muscle function. Neuromuscul Disord 9:11–18
Dunn SE, Burns JL, Michel RN (1999) Calcineurin is required for skeletal muscle hypertrophy. J Biol Chem 274:21908–21912
Friday BB, Harsley V, Pavlath GK (2000) Calcineurin activity is required for the initiation of skeletal muscle differentiation. J Cell Biol 149:657–665
Gersch C, Dewald O, Zoerlain M, Michael LH, Entman ML, Frangogoannis NG (2002) Mast cells and macrophages in normal C57BL/6 mice. Histochem Cell Biol 118:41–49
Glass DJ (2003) Signaling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol 5:87–90
Gregorevic P, Plant DR, Leeding KS, Bach LA, Lynch GS (2002) Improved contractile function of the mdx dystrophic mouse diaphragm muscle following IGF-I administration. Am J Pathol 161:2263–2272
Horsley V, Friday BB, Matteson S, Miller Kegley K, Gephart J, Pavlath GK (2001) Regulation of the growth of multinucleated muscle cells by an NFATc2-dependent pathway. J Cell Biol 153:329–338
Irintchev A, Zweyer M, Cooper RN, Butler-Browne GS, Wernig A (2002) Contractile properties, structure, and fiber phenotype of intact and regenerating slow-twitch muscles treated with cyclosporin A. Cell Tissue Res 308:143–156
Janssen PML, Zeitz O, Keweloh B, Siegel U, Maier LS, Brackhausen P, Pieske B, Prestle J, Lehnart SE, Hasenfuss G (2000) Influence of cyclosporine A on contractile function, calcium handling, and energetics in isolated human and rabbit myocardium. Cardiovasc Res 47:99–107
Losa G, Rodriguez MF, Lopez JA, Arellano PJL (1996) Action of cyclosporine A on mononuclear phagocytes. J Invest Allergol Clin Immunol 6:222–231
Lynch GS, Cuffe SA, Plant DR, Gregorevic P (2001) IGF-I treatment improves the functional and contractile properties of fast- and slow-twitch skeletal muscle from dystrophic mice. Neuromuscul Disord 11:260–268
Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV, Faulkner JA (2001) Force and power output of fast and slow skeletal muscles from mdx mice 6–28 month old. J Physiol 535:591–600
McCall GE, Allen DL, Linderman JK, Grindeland RE, Roy RR, Mukku V, Edgerton VR (1998) Maintenance of myonuclear domain size in rat soleus after overload and growth hormone/IGF-I treatment. J Appl Physiol 84:1407–1412
Mehta M (2001) Physicians Desk Reference Database. Medical Economics Company, Montvale
Mitchell PO, Mills ST, Pavlath GK (2002) Calcineurin differentially regulates maintenance and growth of phenotypically distinct muscles. Am J Physiol 282:C984–C992
Musaro A, McCullagh KAJ, Naya FJ, Olson EN, Rosenthal N (1999) IGF-I induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature 400:581–585
Naya FJ, Mercer B, Shelton J, Richardson JA, Williams RS, Olson EN (2000) Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem 275:4545–4548
Olson EN, Williams RS (2000) Remodeling muscles with calcineurin. BioEssays 22:510–519
Park KS, Kim TK, Kim DH (1999) Cyclosporine A treatment alters characteristics of Ca2+-release channel in cardiac sarcoplasmic reticulum. Am J Physiol 276:H865–H872
Sakuma K, Nishikawa J, Nakao R, Watanabe K, Totsuka T, Nakano H, Sano M, Yasuhara M (2003) Calcineurin is a potent regulator for skeletal muscle regeneration by association with NFATc1 and GATA-2. Acta Neuropathol 105:271–280
Sanchez H, Zoll J, Bigard X, Veksler V, Metthauer B, Lampert E, Lonsdorfer J, Ventura-Clapier R (2001) Effect of cyclosporine A and its vehicle on cardiac and skeletal muscle mitochondria: relationship to efficacy of the respiratory chain. Br J Pharmacol 133:781–788
Sartorius CA, Lu BD, Acakpo-Satchivi L, Jacobsen RP, Byrnes WP, Leinwand LA (1998) Myosin heavy chains IIa and IId are functionally distinct in the mouse. J Cell Biol 141:943–953
Schiaffino S, Serrano AL (2002) Calcineurin signaling and neural control of skeletal muscle fiber type and size. Trends Pharmacol Sci 23:569–575
Seale P, Rudnicki MA (2000) A new look at the origin, function, and “stem cell” status of muscle satellite cells. Dev Biol 218:115–124
Semsarian C, Wu M.-J, Ju Y-K, Marciniec T, Yeoh T, Allen DG, Harvey RP, Graham RM (1999) Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signaling pathway. Nature 400:576–581
Serrano AL, Murgia M, Pallafacchina G, Calabria E, Coniglio P, Lømo T, Schiaffino S (2001) Calcineurin controls nerve activity-dependent specification of slow skeletal muscle fibers but not muscle growth. Proc Natl Acad Sci USA 98:13108–13113
Spangenburg EE, Williams JH, Roy RR, Talmadge RJ (2001) Skeletal muscle calcineurin: influence of phenotype adaptation and atrophy. Am J Physiol 280:R1256–R1260
Talmadge RJ, Roy RR (1993) Electrophoretic separation of rat skeletal muscle myosin heavy chain isoforms. J Appl Physiol 75:2337–2340
Watchko JF, O’Day TL, Hoffman EP (2002) Functional characteristics of dystrophic skeletal muscle: insights from animal models. J Appl Physiol 93:407–417
Weller B, Massa R, Karpati G, Carpenter S (1991) Glucocorticoids and immunosupressants do not change the prevalence of necrosis and regeneration in mdx skeletal muscle. Muscle Nerve 14:771–774
Xu Q, Yu L, Liu L, Cheung CF, Li X, Yee S-P, Yang X-J, Wu Z (2002) p38 Mitogen-activated protein kinase-, calcium-calmodulin-dependent protein kinase-, and calcineurin-mediated signaling pathways transcriptionally regulate myogenin expression. Mol Biol Cell 13:1940–1952
Acknowledgements
Supported by research grants from the Muscular Dystrophy Association (USA). We thank Dr. Eva Chin (Pfizer Global Research and Development, Groton, CT) for her critical reading of the manuscript and helpful comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stupka, N., Gregorevic, P., Plant, D.R. et al. The calcineurin signal transduction pathway is essential for successful muscle regeneration in mdx dystrophic mice. Acta Neuropathol 107, 299–310 (2004). https://doi.org/10.1007/s00401-003-0807-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0807-x