
Abstract
Upon brain reperfusion following ischemia, there is widespread inhibition of neuronal protein synthesis that is due to phosphorylation of eukaryotic initiation factor 2α (eIF2α), which persists in selectively vulnerable neurons (SVNs) destined to die. Other investigators have shown that expression of mutant eIF2α (S51D) mimicking phosphorylated eIF2α induces apoptosis, and expression of non-phosphorylatable eIF2α (S51A) blocks induction of apoptosis. An early event in initiating apoptosis is the release of cytochrome c from mitochondria, and cytochrome c release corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. At present the signaling pathways leading to this are not well defined. We hypothesized that persistent eIF2α(P) reflects injury mechanisms that are causally upstream of release of cytochrome c and induction of apoptosis. At 4 h of reperfusion following 10-min cardiac arrest, vulnerable neurons in the striatum, hippocampal hilus and CA1 showed colocalized intense immunostaining for both persistent eIF2α(P) and cytoplasmic cytochrome c, while resistant neurons in the dentate gyrus and elsewhere did not immunostain for either. A lower intensity of persistent eIF2α(P) immunostaining was present in cortical layer V pyramidal neurons without cytoplasmic cytochrome c, possibly reflecting the lesser vulnerability of this area to ischemia. We did not observe cytoplasmic cytochrome c in any neurons that did not also display persistent eIF2α(P) immunostaining. Because phosphorylation of eIF2α during early brain reperfusion is carried out by PERK, these findings suggest that there is prolonged activation of the unfolded protein response in the reperfused brain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A transient interruption of the blood supply to the brain triggers a cascade of events culminating in selective neuronal death in vulnerable regions, such as hippocampal CA1 pyramidal cells, Purkinje cells of the cerebellum, striatal medium spiny neurons, and neurons in the third to fifth layers of the cerebral cortex [26, 46]. Morphological studies have shown that these neurons die, in varying proportions, from necrosis and/or apoptosis. The relative contribution of each pathway is dependent on the duration of ischemia and other as yet unidentified factors (reviewed in [34]). Moreover, both pathways may be active simultaneously. The precise events leading to neuronal death are under intense investigation [66].
Upon brain reperfusion following ischemia, there is widespread inhibition of neuronal protein synthesis that persists in selectively vulnerable neurons (SVNs) destined to die (reviewed in [15]). The immediate inhibition of ribosomal translation initiation is due to inhibited delivery of the initiator methionine caused by a greater than 20-fold increase in phosphorylation of serine 51 on eukaryotic initiation factor 2α [eIF2α, phosphorylated form eIF2α(P)] [15]. Several investigators have shown a link between apoptosis and phosphorylation of eIF2α [1, 9, 17, 56]. In vitro studies have shown that expression of mutant eIF2α (S51D) mimicking eIF2α(P) induces apoptosis, and expression of non-phosphorylatable eIF2α (S51A) blocks induction of apoptosis [17, 56]. The nature of the relationship of eIF2α(P) to apoptosis is unknown.
One early event in initiating apoptosis is the release of cytochrome c from the intramembranous space of mitochondria. This allows formation of the apoptosome complex, consisting of cytochrome c, apoptosis-activating factor 1 (Apaf-1), and caspase-9, which proteolytically activates caspase-3, a key apoptosis executioner (reviewed in [63]). Indeed, after 3–4 h of reperfusion, many SVNs begin to show release of cytochrome c and caspase-9 from mitochondria into the cytoplasm [7, 58]. In this study, we explored the potential relationship between eIF2α(P) and cytochrome c release utilizing immunohistochemistry. We found virtually complete co-mapping between eIF2α(P) and cytochrome c in CA1 pyramidal neurons at 4 h of reperfusion following 10-minute cardiac arrest. In addition, eIF2α(P) appears before cytochrome c release, suggesting that eIF2α(P) is upstream of cytochrome c release.
Materials and methods
Animal model
All animal experiments were approved by our institutional review board and were conducted following the Guide for the Care and Use of Laboratory Animals (National Research Council, revised 1996). We utilized our well-characterized rat model of normothermic 10-min cardiac arrest followed by resuscitation, which causes: (1) neurological functional deficits in long-term survivors [55], (2) substantial loss of hippocampal SVNs [49], (3) inhibited translation initiation [11] associated with eIF2α phosphorylation [12] and μ-calpain-mediated degradation of eIF4G [12, 38], and (4) lipid peroxidation in SVNs [65] with associated ultrastructural alterations of the endoplasmic reticulum (ER) and Golgi apparatus [47]. Male Long Evans rats weighing 300–400 g were anesthetized with ketamine (70 mg/kg intraperitoneally) and xylazine (5 mg/kg intraperitoneally). Rats were orotracheally intubated and mechanically ventilated with room air at 80 breaths per minute, tidal volume of 10 ml/kg, and a positive end expiratory pressure of 3 cm H20. Femoral artery and femoral vein catheters were inserted and arterial pressure was continuously monitored. Core body temperature was maintained with a servo-controlled heating pad at 37.0±0.5°C for the entire experimental period. The electrocardiogram was continuously monitored by limb leads. After instrumentation, circulatory arrest was induced by thoracic compression [16, 64] and was confirmed by the complete loss of arterial pressure, which occurred within 5 s. After a 10-min cardiac arrest period, resuscitation was initiated by chest compressions (200–300/min) and mechanical ventilation (tidal volume 1 ml/100 g, 80 breaths per minute, 3 cm positive-end-expiratory pressure) with supplemental O2. The rate and depth of chest compressions was adjusted to maximize the arterial diastolic pressure. Epinephrine (10 µg/kg IV) was given during resuscitation. ECG and arterial pressure tracings were evaluated for return of spontaneous circulation, defined as a systolic blood pressure greater than 60 mmHg. If return of spontaneous circulation (ROSC) did not occur after 3.5 min of resuscitation efforts, the resuscitation was abandoned and the animal was not used in any studies. The time from onset of ischemia to ROSC was 700±100 s. In the early post-resuscitation phase, systolic blood pressure was maintained above 80 mmHg using 0.9% saline and dopamine infusion at 10–20 μg/kg/min as necessary. Artificial ventilation was continued until the end of the experiment.
Western blot analysis
The brain was homogenized and postmitochondrial supernatants were prepared as previously described [12, 13, 30, 38]. Proteins (50 μg) were separated by electrophoresis in SDS-polyacrylamide gel and electrophoretically transferred to nitrocellulose membranes. Cytochrome c and eIF2α(P) were detected with a mouse primary monoclonal antibody (BD PharMingen, San Diego, CA) and a rabbit primary polyclonal antibody (BioSource International, Camarillo, CA), respectively, and visualized by the enhanced chemiluminescence procedure (Amersham Pharmacia Biotech).
Fluorescence immunohistochemistry
Transaortic perfusion fixation with 50% methanol:10% acetic acid was performed and 40-μm vibratome sections prepared as previously described [13]. This fixative was used in preference to aldehydes because it allows much better anti-eIF2α(P) antibody penetration without resorting to antigen recruitment procedures [18]. Sections were quenched in 3% hydrogen peroxide (H2O2) for 10 min, washed three times with TRIS-buffered saline (TBS), solubilized in 0.1% Triton X-100 in TBS for 10 min, washed three times, and blocked in 10% normal goat serum in TBS for 1 h. Sections were then incubated in primary antibody (1:100 in TBS) at room temperature overnight, and unbound antibody then washed off with four TBS rinses. Fluorescent secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) anti-rabbit-FC rhodamine red-X (RRX) and anti-mouse-FC Cy2 were used to visualize eIF2α(P) and cytochrome c, respectively. Sections were incubated in the appropriate secondary antibody (1:100 in TBS) at room temperature overnight. Following four 15-min TBS rinses, the sections were mounted and coverslipped. Fluorescence photomicrographs were obtained utilizing a Nikon digital camera on a Leica DML fluorescence microscope with appropriate incident wavelengths (492 nm for Cy2, and 570 nm for RRX). For double immunofluorescence, sections were processed as above except that single sections were incubated first with the anti-cytochrome c primary and secondary antibodies and subsequently with the anti-eIF2α(P) primary and secondary antibodies. The resulting images were overlaid utilizing Adobe Photoshop.
Results
Because we were interested in the potential relationship between persistence of eIF2α(P) and neuronal selective vulnerability, we first utilized Western blotting to examine relative levels of eIF2α(P) by region in brain homogenates obtained from three rats in each of four experimental groups: (1) non-ischemic controls, (2) 10-min ischemia followed by no reperfusion, (3) 10-min reperfusion, and (4) 60-min reperfusion. There was no increase of eIF2α(P) during ischemia (Fig. 1), but after 10 min of reperfusion a whole-brain homogenate showed a 24-fold increase over controls. Similarly, brain homogenates from microdissected regions of the brain stem, cerebellum, cortex, hippocampus, and thalamus all showed large increases. By 1 h of reperfusion, there was a consistent trend toward reduction of the eIF2α(P) levels in all regions except the cortex and hippocampus, although this decrease was statistically significant only in the ischemia-resistant brain stem.

Regional changes in eIF2α(P) Western blot density during the first hour of reperfusion with the data normalized against the NIC mean. Groups are: (1) non-ischemic controls (NIC) and (2) 10 min. ischemia followed by no reperfusion (10I), (3) 10 min. reperfusion (10R), and (4) 60 min. reperfusion (60R). ANOVA followed by Fisher's LSD post hoc tests showed that all reperfused samples had significantly greater eIF2α (P) levels (*P<0.05) than seen in NIC or 10I; NIC and 10I were not different for any group. The decrease between 10 and 60 min reperfusion in the brain stem was also significant [eIF2α(P) phosphorylated eukaryotic initiation factor 2α, BS brain stem, CER cerebellum, CTX cortex, HIP hippocampus, THAL thalamus, WB whole-brain homogenate]
We chose the 4-h reperfusion time point to investigate potential colocalization of eIF2α(P) and cytoplasmic cytochrome c because of the evidence from Cao et al. [7] that mitochondrial release of cytochrome c had begun by that time. By Western blot we observed that levels of eIF2α(P) remained approximately sixfold elevated after 4-h reperfusion in forebrain (rostral to the thalamus) homogenates (Fig. 2A). Immunoblot examination of the anti-eIF2α(P) (Fig. 2B) and anti-cytochrome c (Fig. 2C) antibodies showed that both were substantially monospecific.

A eIF2α (P) after 10-min and 4-h reperfusion is increased ~24- and 6-fold, respectively, over controls in forebrain homogenates. B The rabbit polyclonal anti-eIF2α (P) is monospecific for eIF2α(P) produced by 30-min thapsigargin (tg) treatment of NB104 cells, and generates little signal in homogenates from normal control brains; it is monospecific for eIF2α(P) in brain homogenates obtained after 10-min cardiac arrest and 10-min reperfusion. C The mouse monoclonal anti-cytochrome c is monospecific and recognizes much more cytochrome c in the mitochondrial pellet than in post-mitochondrial supernatant (PMS) obtained from normal rat brains
Immunofluorescence examination of eIF2α(P) and cytochrome c was performed on three animals each in two groups: (1) control and (2) 10-min cardiac arrest with 4-h reperfusion. Representative photomicrographs are shown. Omission of primary antibody ablated the strong immunofluorescence signal seen in reperfused brains (not shown). Control brains revealed only background fluorescence for eIF2α(P) and scant punctate cytoplasmic immunofluorescence for cytochrome c (shown for hippocampus CA1 in Fig. 3). Although cytochrome c is constitutively present in the mitochondrial intramembranous space, other investigators have also noted scant immunostaining before cytochrome c is released into the cytoplasm [32, 40].

Representative photomicrographs of immunofluorescence staining for eIF2α(P) or cytochrome c in the CA1 region (arrowheads indicate CA1 border) from normal controls show little signal above background, although sparse punctate cytoplasmic immunofluorescence for cytochrome c is noted. Results were confirmed by at least three independent studies for each time point
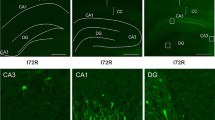
In marked contrast to controls, there was a strong immunofluorescence signal in corresponding populations of SVNs in the striatum and hippocampal CA1 and hilus after 4-h reperfusion when serial consecutive sections were stained for either eIF2α(P) or cytochrome c (Fig. 4). There was a well-demarcated reduction of fluorescence for both species in the injury-resistant CA2 (left of arrow in Fig. 4A, B) as compared to CA1. The injury-resistant hippocampal dentate granule cells also showed a paucity of fluorescence for either species, although a few cells in the polymorphic layer nearest the hilar cells demonstrated fluorescence (Fig. 4C, D). In the striatum, the radial bundles did not fluoresce, but the vulnerable medium spiny neurons showed fluorescence for both eIF2α(P) and cytochrome c (Fig. 4E, F).

After 10-min cardiac arrest and 4-h reperfusion, representative immunofluorescence photomicrographs for eIF2α(P) (red) or cytochrome c (green) in serial consecutive sections are shown for: the junction (large arrows) of the hippocampus CA1 and CA2 (A, B); the dorsal and ventral blades of the DG and the interposed hilus (C, D); and the striatum (E, F). A striatal radial bundle is outlined in E and F. Results were confirmed by at least three independent studies for each time point (DG dentate granule cells)
In the cortex after 4 h of reperfusion, we observed a somewhat attenuated eIF2α(P) immunofluorescence that was mainly present in layer III pyramidal neurons without any consistent cytoplasmic cytochrome c immunofluorescence in these cells (Fig. 5).

After 10-min cardiac arrest and 4-h reperfusion, representative immunofluorescence photomicrographs for eIF2α (P) (red) or cytochrome c (green) in serial consecutive sections are shown for the cerebral cortex. Many layer III neurons show some degree of cytoplasmic immunofluorescence for eIF2α (P) (left); however, the lower magnification image at the right (small arrow points to the pial surface) shows the widespread paucity of immunofluorescence for cytoplasmic cytochrome c in this region. Results were confirmed by at least three independent studies for each time point
Double immunofluorescence studies in the three areas shown in Fig. 4 revealed strong immunofluorescence signals for both eIF2α(P) and cytoplasmic cytochrome c with nearly complete cellular congruity (CA1 shown in Fig. 6). Higher magnification shows that cytochrome c staining in normal controls (Fig. 7, left) is mainly punctate as would be expected with mitochondrial localization. However, after 4-h reperfusion following 10-min cardiac arrest, there is diffuse cytoplasmic cytochrome c staining consistent with its release into the cytosol (Fig. 7, right).

Representative double immunofluorescence photomicrographs for eIF2α(P) and cytochrome c in the hippocampus CA1 at 4-h reperfusion after 10-min cardiac arrest. Results were confirmed by at least three independent studies for each time point

Representative immunofluorescence photomicrographs for cytochrome c in the hippocampus CA1 from brain perfusion fixed with 4% paraformaldehyde. In normal controls (left), there is mainly punctate staining. After 4-h reperfusion following 10-min cardiac arrest (right), there is diffuse cytoplasmic staining consistent with cytochrome c release into the cytosol. Bar 10 μm
Discussion
Mitochondria are thought to play a pivotal role in the early phase of apoptosis in mammalian cells [54, 63]. Following certain apoptotic stimuli, cytochrome c is released from the mitochondria and binds to Apaf-1, which leads to activation of caspase-9 and other caspases, including caspase-3. Caspase-3, believed be the final committed step in neuronal apoptosis, cleaves several cellular targets producing the hallmark morphological features of apoptosis. Sugawara et al. [58] have shown that mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. At present, the apoptotic signaling pathways leading to mitochondrial permeability are not well defined. There are several pathways by which eIF2α(P) and apoptosis might be linked. Further work is needed to discern which pathway is correct, and it is certainly possible that multiple pathways may be simultaneously active [29].
A direct mechanism from persistent eIF2α phosphorylation to apoptosis induction could involve an eIF2α(P)-induced bypass-scanning mechanism of translation initiation for ATF4 [20] (the transcription factor for CHOP) and CHOP [51]. CHOP (C/EBP homology protein) production is required for the loss of mitochondrial membrane integrity and apoptosis caused by depletion of ER Ca2+ following nitric oxide-induced nitrosylation and inactivation of the ER Ca2+ pump [42], and its synthesis precedes release of cytochrome c to the cytoplasm [61]. ER stress-induced CHOP transcription is blocked in cells containing a non-phosphorylatable mutant of eIF2α (S51A) [51]. Although Paschen et al. [45] have shown that CHOP transcription is induced during brain reperfusion, CHOP protein levels in the reperfused brain have yet to be determined.
A second pathway by which eIF2α(P) and apoptosis might be linked is through caspase-8. Caspase-8 can cleave the eIF2α kinase PKR (RNA-dependent protein kinase), which separates the PKR regulatory domain from its kinase domain; the released kinase domain can then efficiently phosphorylate eIF2α [50]. Caspase-8 also cleaves Bid, and the resulting Bid fragment interacts with the mitochondrial membrane to release cytochrome c [33, 35] by a process that does not involve permeability transition [52]. Such a mechanism for cytochrome c release is attractive because Fiskum's group has developed compelling evidence that brain ischemia and reperfusion do not cause mitochondrial permeability transition [3]. Yin et al. [68] have recently shown activated Bid and early release of cytochrome c in the brain following transient cerebral ischemia. However, previous work in our laboratory has shown that mice bearing a homozygous PKR knockout do not have reduced levels of eIF2α(P) during early reperfusion, which argues against this pathway [14].
A third potential pathway involves the role of reactive oxygen species in the phosphorylation of eIF2α and release of cytochrome c. Neurons produce large amounts of H2O2 following ischemia/reperfusion [24], and H2O2 inhibits protein translation in cortical neurons by a process that involves the phosphorylation of both eIF2α and eEF2 [2]; the relative contribution of these two events depends on the duration of H2O2 treatment. H2O2 treatment is known to induce release of cytochrome c from cultured cells [57, 60, 67].
Finally, ischemia/reperfusion-induced ER stress could trigger a cytochrome c-independent apoptosis pathway involving IRE1α and caspase-12. The reperfusion-induced phosphorylation of eIF2α is likely caused by activation of the eIF2α kinase PERK (PKR-like ER kinase); knockouts of the other three eIF2α kinases (PKR, HRI, GCN2) do not reduce levels of eIF2α(P) during reperfusion [30]. The only known mechanism by which PERK is activated is as a component of the ER unfolded protein response (UPR) [19, 25]. Originally described as a response to an overload of unfolded proteins in the ER [28], several types of ER stress trigger a compensatory response, the UPR, whose most proximal sensors are the ER membrane-bound proteins ATF6, IRE1α, and PERK. Thus, induction of the UPR is also expected to cause activation of IRE1α, which is both a kinase and a RNase [53].
In mammalian cells active IRE1α carries out cytoplasmic endoribonucleolytic processing of XBP1 mRNA [6]. Translation of the processed XBP1 participates in the resolution of the UPR [31] by generating a transcription factor that strongly enhances gene transcription, such as that of the ER chaperone GRP78. Resolution of the ER stress is associated with GRP78 binding of the ER luminal domains of both IRE1α and PERK, which attenuates their activation [5]. In the case of cultured hippocampal neurons, antisense-mediated depletion of GRP78 substantially enhances cell death induced by glutamate or Fe2+-mediated radical damage [70]. Paschen's group has preliminary evidence of XBP1 processing consistent with IRE1α activation during post-ischemic reperfusion in a stroke model (personal communication), and induction of GRP78 transcription by brain ischemia and reperfusion is well established [15].
Activated IRE1 recruits tumor necrosis factor receptor-associated factor-2 (TRAF2) [62]. Normally TRAF2 forms a stable complex with procaspase-12 [69], but ER stress [37] dissociates TRAF2 from procaspase-12, which then oligomerizes, is activated [69] and can cleave caspase-9 [36, 48], which is presumably followed by activation of caspase-3 [8]. In addition, activation of IRE1α also leads to activation of the apoptosis-associated stress-induced protein Ser/Thr kinases known as SAPKs (stress activated protein kinases) or JNKs (Jun N-terminal kinases) via interaction of the IRE1α cytoplasmic domain with TRAF2 [62] and JIK (Jun inhibitor kinase) [69]. During early reperfusion there is rapid activation of JNK1 in SVNs [59] associated with enhanced N-terminal Jun phosphorylation [21].
The initial roles of both PERK and IRE1α in the UPR appear protective: PERK by inhibiting protein synthesis to reduce the load of new peptides requiring ER luminal processing; and IRE1α by up-regulation of genes encoding chaperones that facilitate the protein-folding process in the ER. The ischemia-reperfusion-induced activation of PERK [30] suggests that ER stress has caused activation of the UPR, consistent with evidence developed by Paschen [43, 44].
Importantly, if the ER stress is not resolved, both PERK and IRE1α can also drive apoptotic mechanisms [39], and there is evidence of prolonged ER stress in the reperfused brain. Brain ischemia and reperfusion induce abnormal protein aggregates in the ER and other neuronal compartments [22, 23]; these aggregates clear from injury-resistant neurons but persist in vulnerable neurons that progress to cell death. Moreover, Kohno et al. [27] have shown prolonged loss of ER luminal Ca2+ in reperfused SVNs, and low ER Ca2+ concentrations leads to inhibition of the ER luminal protein disulfide isomerases (PDI) and thus causes misfolding of newly synthesized peptides in the ER lumen [4, 10, 41].
In this study, we have shown that there are large increases in eIF2α(P) in both vulnerable and non-vulnerable brain regions at 10 min of reperfusion following 10-min cardiac arrest. By 1 h of reperfusion, there was a consistent trend toward reduction of the eIF2α(P) levels in all regions except the cortex and hippocampus. We also demonstrated by double immunofluorescence that eIF2α(P) and cytochrome c show substantial congruity at 4-h reperfusion; we did not observe cytoplasmic cytochrome c in any neurons that did not also display persistent eIF2α(P) immunostaining. These findings suggest that there is prolonged activation of the UPR in the reperfused brain. These results are also consistent with a hypothesis that persistent eIF2α(P) reflects injury mechanisms that are causally upstream of release of cytochrome c to the cytoplasm, an event documented as proapoptotic [66].
Interestingly, at 4-h reperfusion, persistent eIF2α(P) is seen in layer III cortical neurons without cytoplasmic release of cytochrome c, while neurons in the other selectively vulnerable areas demonstrate both persistent eIF2α(P) and cytoplasmic release of cytochrome c. Because cortical pyramidal neurons are more tolerant of ischemia than hippocampal CA1 pyramidal neurons, this suggests that mechanisms other than simply a transient elevation in eIF2α(P) levels play an important role in determining the ultimate fate of the post-ischemic neuron.
References
Alcazar A, Bazan E, Rivera J, Salinas M (1995) Phosphorylation of initiation factor 2 alpha subunit and apoptosis in Ca2+ ionophore-treated cultured neuronal cells. Neurosci Lett 201:215–218
Alirezaei M, Marin P, Nairn AC, Glowinski J, Premont J (2001) Inhibition of protein synthesis in cortical neurons during exposure to hydrogen peroxide. J Neurochem 76:1080–1088
Andreyev AY, Fahy B, Fiskum G (1998) Cytochrome-c release from brain mitochondria is independent of the mitochondrial permeability transition. FEBS Lett 439:373–376
Baksh S, Burns K, Andrin C, Michalak M (1995) Interaction of calreticulin with protein disulfide isomerase. J Biol Chem 270:31338–31344
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of GRP78 and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2:326–332
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92–96
Cao G, Minami M, Pei W, Yan C, Chen D, O'Horo C, Graham SH, Chen J (2001) Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab 21:321–333
Cao G, Luo Y, Nagayama T, Pei W, Stetler RA, Graham SH, Chen J (2002) Cloning and characterization of rat caspase-9: implications for a role in mediating caspase-3 activation and hippocampal cell death after transient cerebral ischemia. J Cereb Blood Flow Metab 22:534–546
Clemens MJ (2001) Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog Mol Subcell Biol 27:57–89
Corbett EF, Oikawa K, Francois P, Tessier DC, Kay C, Bergeron JJM, Thomas DY, Krause KH, Michalak M (1999) Ca2+ regulation of interactions between endoplasmic reticulum chaperones. J Biol Chem 274:6203–6211
DeGracia DJ, O'Neil BJ, Krause GS, Skjaerlund JM, White BC, Grossman LI (1993) Studies of the protein synthesis system in the brain cortex during global ischemia and reperfusion. Resuscitation 25:161–170
DeGracia DJ, Neumar RW, White BC, Krause GS (1996) Global brain ischemia and reperfusion: modifications in eukaryotic initiation factors are associated with inhibition of translation initiation. J Neurochem 67:2005–2012
DeGracia DJ, Sullivan JM, Neumar RW, Alousi SS, Hikade KR, Pittman JE, White BC, Rafols JA, Krause GS (1997) Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2α. J Cereb Blood Flow Metab 17:1291–1302
DeGracia DJ, Adamczyk S, Folbe AJ, Konkoly LL, Pittman JE, Neumar RW, Sullivan JM, Scheuner D, Kaufman RJ, White BC, Krause GS (1999) Eukaryotic initiation factor 2α kinase and phosphatase activity during post-ischemic brain reperfusion. Exp Neurol 155:221–227
DeGracia DJ, Kumar R, Owen CR, Krause GS, White BC (2002) Molecular pathways of inhibited translation during brain reperfusion: implications for neuronal survival or death. J Cereb Blood Flow Metab 22:127–141
Edmonds HL Jr, Raque GM Jr, Zhang PY, Jenkins SA, Sheilds CB (1989) Cerebroprotective effects of a parenteral flunarizine formulation. In: Hartmann A., Kuchinsky W (eds) Cerebral ischemia and calcium, Springer, Berlin, pp 494–500
Gil J, Alcami J, Esteban M (1999) Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-κB. Mol Cell Biol 19:4653–4663
Goldstein E, Owen C, White BC, Rafols JA (1999) Ultrastructural localization of phosphorylated eIF2α [eIF2α(P)] in rat dorsal hippocampus during reperfusion. Acta Neuropathol 98:493–505
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274
Harding HP, Novoa II, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6:1099–1108
Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin MJ (1998) Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. Neuroscience 18:5124–5135
Hu BR, Martone ME, Jones YZ, Liu CL (2000) Protein aggregation after transient cerebral ischemia. J Neurosci 20:3191–3199
Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjo BK, Liu CL (2001) Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab 21:865–875
Hyslop PA, Zhang Z, Pearson DV, Phebus LA (1995) Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res 671:181–186
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational control. Genes Dev 13:1211–1233
Kirino T (1982) Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239:57–69
Kohno K, Higuchi T, Ohta S, Kohno K, Kumon Y, Sakaki S (1997) Neuroprotective nitric oxide synthase inhibitor reduces intracellular calcium accumulation following transient global ischemia in gerbil. Neurosci Lett 224:17–20
Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332:462–464
Kulms D, Schwarz T (2002) Independent contribution of three different pathways to ultraviolet-B-induced apoptosis. Biochem Pharmacol 64:837–841
Kumar R, Azam S, Sullivan JM, Owen CR, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, White BC, Krause GS, DeGracia DJ (2001) Brain ischemia and reperfusion activates the eukaryotic initiation factor 2α kinase, PERK. J Neurochem 77:1418–1421
Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16:452–466
Lewén A, Fujimura M, Sugawara T, Matz P, Copin JC, Chan PH (2001) Oxidative stress-dependent release of mitochondrial cytochrome c after traumatic brain injury. J Cereb Blood Flow Metab 21:914–920
Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–501
Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79:1431–1568
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481–490
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y (2002) An ER stress-specific caspase cascade in apoptosis: cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277:34287–34294
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yanker BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403:98–103
Neumar RW, DeGracia DJ, Konkoly LL, White BC, Krause GS (1998) Calpain I mediates eukaryotic initiation factor 4G degradation during global brain ischemia. J Cereb Blood Flow Metab 18:876–881
Niwa M, Walter P (2000) Pausing to decide. Proc Natl Acad Sci USA 97:12396–12397
Noshita N, Sugawara T, Fujimura M, Morita-Fujimura Y, Chan, PH (2001) Manganese superoxide dismutase affects cytochrome c release and caspase-9 activation after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab 21:557–567
Oliver JD, Roderick HL, Llewellyn DH, High S (1999) Erp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell 10:2573–2582
Oyadomari S, Takeda K, Takiguchi M, Gotoh, T, Matsumoto M, Wada I, Akira S, Araki E, Mori M (2001) Nitric oxide-induced apoptosis in pancreatic β cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA 98:10845–10850
Paschen W (1996) Disturbances of calcium homeostasis within the endoplasmic reticulum may contribute to the development of ischemic-cell damage. Med Hypotheses 47:283–288
Paschen W (2000) Role of calcium in neuronal cell injury: which subcellular compartment is involved? Brain Res Bull 53:409–413
Paschen W, Gissel C, Linden T, Althausen S, Doutheil J (1998) Activation of gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Brain Res Mol Brain Res 60:115–122
Pulsinelli WA, Brierley JB, Plum F (1982) Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol 11:491–498
Rafols JA, O'Neil BJ, Krause GS, Neumar RW, White BC (1995) Global brain ischemia and reperfusion: Golgi apparatus ultrastructure in neurons selectively vulnerable to death. Acta Neuropathol (Berl) 30:17–30
Rao RV, Hermel E, Castro-Obregon S, Rio G del, Ellerby LM, Ellerby HM, Bredesen DE (2001) Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem 276:33869–33874
Sadowski M, Wisniewski HM, Jakubowska-Sadowska K, Tarnawski M, Lazarewicz JW, Mossakowski MJ (1999) Pattern of neuronal loss in the rat hippocampus following experimental cardiac arrest-induced ischemia. J Neurol Sci 168:13–20
Saelens X, Kalai M, Vandenabeele P (2001) Translation inhibition in apoptosis. Caspase-dependent PKR activation and eIF2-α phosphorylation. J Biol Chem 276:41620–41628
Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7:1165–1176
Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y (2001) Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol 152:237–250
Sidrauski C, Walter P (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90:1031–1039
Sims NR, Anderson MF (2002) Mitochondrial contributions to tissue damage in stroke. Neurochem Int 40:511–526
Skjaerlund JM, Krause GS, Feldman DM, White BC (1991) The effect of ethyl-methyl-hydroxy-pyrid-4-one on post-cardiac arrest survival of rats. Resuscitation 22:139–149
Srivastava SP, Kumar KU, Kaufman RJ (1998) Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J Biol Chem 273:2416–2423
Stridh H, Kimland M, Jones DP, Orrenius S, Hampton MB (1998) Cytochrome c release and caspase activation in hydrogen peroxide- and tributyltin-induced apoptosis. FEBS Lett 429:351–355
Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH (1999) Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 19:RC39
Takagi Y, Nozaki K, Sugino T, Hattori I, Hashimoto N (2000) Phosphorylation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase after transient forebrain ischemia in mice. Neurosci Lett 294:117–120
Takeyama N, Miki S, Hirakawa A, Tanaka T (2002) Role of the mitochondrial permeability transition and cytochrome c release in hydrogen peroxide-induced apoptosis. Exp Cell Res 274:16–24
Totoh T, Oyadomari S, Mori K, Mori M (2002) Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J Biol Chem 277:12343–12350
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–666
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15:2922–2933
Wauquier A, Melis W, Janssen PAJ (1989) Long-term neurological assessment of the post-resuscitation effects of flunarizine, verapamil and nimodipine in a new model of global complete ischaemia. Neuropharmacology 28:837–846
White BC, Daya A, DeGracia DJ, O'Neil BJ, Skjaerlund JM, Krause GS, Rafols JA (1993) Fluorescent histochemical localization of lipid peroxidation during brain reperfusion following cardiac arrest. Acta Neuropathol 86:1–9
White BC, Sullivan JM, DeGracia DJ, O'Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS (2000) Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 179:1–33
Yang JC, Cortopassi GA (1998) Induction of the mitochondrial permeability transition causes release of the apoptogenic factor cytochrome c. Free Radic Biol Med 24:624–631
Yin XM, Luo Y, Cao G, Bai L, Pei W, Kuharsky DK, Chen J (2002) Bid-mediated mitochondrial pathway is critical to ischemic neuronal apoptosis and focal cerebral ischemia. J Biol Chem 277:42074–42081
Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 276:13935–13940
Yu ZF, Luo H, Fu W, Mattson MP (1999) The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol 155:302–314.
Acknowledgement
Supported by NIH-NINDS grant NS33196 (G.S.K., D.J.D., C.R.O.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Page, A.B., Owen, C.R., Kumar, R. et al. Persistent eIF2α(P) is colocalized with cytoplasmic cytochrome c in vulnerable hippocampal neurons after 4 hours of reperfusion following 10-minute complete brain ischemia. Acta Neuropathol 106, 8–16 (2003). https://doi.org/10.1007/s00401-003-0693-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0693-2