
Abstract
In the central nervous system (CNS) complex endothelial tight junctions (TJs) form a restrictive paracellular diffusion barrier, the blood-brain barrier (BBB). During inflammation, BBB properties are frequently lost, resulting in brain edema. To investigate whether BBB leakiness correlates with molecular changes at BBB TJs, we performed immunofluorescence stainings for TJ molecules in a mouse model of experimental autoimmune encephalomyelitis (EAE) and in human tissue with glioblastoma multiforme (GBM). In TJs of healthy CNS vessels in both mouse and man we detected occludin, ZO-1, claudin-5 and claudin-3. In EAE brain and spinal cord sections we observed the selective loss of claudin-3 immunostaining from TJs of venules surrounded by inflammatory cuffs, whereas the localization of the other TJ proteins remained unchanged. In addition, selective loss of claudin-3 immunostaining was also observed in altered cerebral microvessels of human GBM. Our data demonstrate the selective loss of claudin-3 from BBB TJs under pathological conditions such as EAE or GBM when the integrity of the BBB is compromised, and therefore suggest that claudin-3 is a central component determining the integrity of BBB TJs in vivo.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Homeostasis of the central nervous system (CNS) microenvironment is essential for its normal function and is maintained by the blood-brain barrier (BBB). The BBB is formed by highly specialized endothelial cells, which inhibit transcellular passage of molecules across the BBB by an extremely low pinocytotic activity, and restrict the paracellular diffusion of hydrophilic molecules by an elaborate network of complex tight junctions (TJs) between the endothelial cells (reviewed by [14, 28]). The morphology of BBB TJs resembles those of epithelial TJs, which in ultrathin section electron micrographs appear as a chain of fusion points of the outer plasma membrane leaflet of adjacent cells [6]. Despite the structural similarities between BBB and epithelial TJs, BBB endothelial TJs are unique in their sensitivity to ambient factors. Cultured epithelial cells establish a high electrical resistance and low permeability in concert with a stable TJ morphology, mimicking the situation in vivo [13]. In contrast, cultured BBB endothelium loses many BBB characteristics including formation of proper TJs and a permeability barrier, indicating that integrity of BBB TJ strictly depends on signals provided by the CNS microenvironment [29]. During pathological conditions of the CNS such as tumors or during inflammation, when homeostasis within the CNS is disturbed, barrier properties of cerebral endothelium are frequently lost, leading to edema formation [15, 19]. The characterization of molecular changes at the BBB TJs responsible for the development of BBB leakiness and brain edema has been hampered by a lack of knowledge on the molecular architecture of BBB TJs.
In recent years, several proteins have been identified that are associated with epithelial and endothelial TJs. This group of TJ-associated proteins includes cytoplasmic peripheral membrane proteins of the MAGUK family, such as ZO-1 and ZO-2 (reviewed by [27]). Integral membrane proteins exclusively localized at TJs are occludin [1, 8] and the claudins, which comprise a novel gene family of four transmembrane TJ proteins with no sequence homology to occludin [9, 22]. Mice carrying a null mutation in the occludin gene develop morphologically normal TJs in most tissues including the brain [26], proving that occludin is not essential for proper TJ formation. In contrast, transfection of claudins into fibroblasts induced TJs in the absence of occludin, demonstrating that claudins are essential for TJ induction. To date, 20 members of the claudin family with different tissue distribution have been described [20]. In the CNS, claudin-1 and claudin-5 have been detected in BBB endothelium at the protein level [16, 21]. Furuse et al. [10] described in a Northern blot analysis the expression of claudin-1 and the absence of claudin-2 in whole brain preparations. As presence of claudin-1 and claudin-2 protein has been detected in the choroid plexus epithelium [18], the brain preparation used for this study was apparently devoid of the choroid plexus. In the absence of choroid plexus epithelial cells, endothelial cells are—besides oligodendrocytes—the only cells in the brain carrying TJs. In the morphologically unique TJs of oligodendrocytes only claudin-11 has been detected to date [23]. Thus, expression of claudin-1 in the brain tissue specimens free of the choroid plexus suggests that claudin-1 can only be expressed in the endothelial cells. This has been supported by our previous findings using an anti-claudin-1 antibody recognizing endothelial cell TJs in vitro [16] and in vivo [17, 18].
Here we report that claudin-3 is present in endothelial TJs in the CNS of mice and man, and is lost in inflamed microvessels of EAE brains and spinal cords and human glioblastoma multiforme (GBM) tissue. In contrast, when applying a novel anti-claudin-1 antibody we cannot confirm our previous observations of localization of claudin-1 in endothelial TJs in vivo. Rather, this novel antibody stained the entire endothelial cells of the CNS not allowing subcellular localization of claudin-1 at the protein level. Our data suggest that claudin-3 but not claudin-5 is a central component regulating the permeability of BBB TJs in vivo.
Material and methods
Antibodies
The following specific primary antibodies were used: M1/9 (rat anti-mouse CD45; PharMingen, Hamburg, Germany). In previous experiments [17] we used a rabbit polyclonal anti-human claudin-1 antibody (Zymed Laboratories, San Francisco, Calif., lot 01162834, cat. no. 71-7800), which we found to be cross-reactive with claudin-3 by immunofluorescence staining and Western blotting of mouse L cell transfectants expressing either murine claudin-1 or murine claudin-3 (data not shown; the transfectants were kindly provided by M. Furuse, Kyoto, Japan). This antibody was used again in this study and the staining described as "claudin-1/3" (Figs. 1g, h1, h2, and 2g, h). Rabbit polyclonal anti-human claudin-1 (Zymed, lot 10464713, cat. no. 51-9000), rabbit polyclonal anti-mouse claudin-3 (Zymed), rabbit polyclonal anti-human occludin, (Zymed), and as isotype control mouse IgG1 (clone MOPC-21; Sigma, Deisenhofen, Germany) were used. The rabbit polyclonal antiserum directed against mouse claudin-5/TMVCF was kindly provided by Dr. H. Kalbacher (Tübingen, Germany) [16]. Polyclonal rabbit anti-human fibronectin antiserum (Dako, Carpinteria, Calif.), FITC-conjugated polyclonal sheep anti-human fibronectin antiserum (Bio Trend Chemikalien, Köln, Germany). Secondary antibodies were goat anti-rat or goat anti-rabbit IgG (Jackson Immunoresearch Laboratories, West Grove, Pa.) conjugated with the cyanin-derivative dye CY3 (Dianova, Hamburg, Germany) or with Alexa Fluor (MoBiTec, Göttingen, Germany).
Induction of EAE
Female SJL/N mice were obtained from Bomholdgård Breeding (Ry, Denmark) and used for experiments at the age of 10 weeks. Active EAE (aEAE) was induced by immunization with syngeneic spinal cord homogenate as described in detail before [4, 5]. Animals were checked daily and clinical severity was documented as follows: +/−, limp tail; +, weak hindlimbs; ++, paraplegic; +++, paraplegic, incontinent and weakness. SJL mice with the clinical severity of EAE of ++ are presented in this study, as this is the clinical stage when BBB breakdown is definitely established in cerebral vessels surrounded by inflammatory cuffs, which are visible at the light microscopical level. Animal experiments were performed in accordance with the German legislation on the protection of animals and the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, revised 1985).
Immunofluorescence on frozen tissue sections
Mice afflicted with EAE or healthy littermates were killed, and their brains were removed, embedded in Tissue-Tek (OCT; Miles, Vogel, Giessen, Germany) and snap-frozen in a 2-methylbutane (Merck, Darmstadt, Germany) bath at -80°C. Some mice were perfused with 1% formaldehyde in PBS prior to removing the tissue.
Small fragments of seven GBM specimens were isolated from patients at the Department of Neurosurgery of Tübingen University Clinics. In addition, as control tissue, a small fragment of cerebral cortex was isolated from a patient with traumatic injury. After surgical removal, the tissue was embedded unfixed in Tissue Tec O.C.T. compound and frozen in liquid nitrogen. The time between surgery and freezing ranged from 20 min (GBM) to 2 h (traumatic injury).
Cryostat sections (8–12 µm thick) were cut using a Reichert-Jung Frigocut 2800 E cryotome (Reichert, Vienna, Austria), mounted on glass slides coated with poly-l-lysine (Sigma), and air dried. Sections from perfused mice were postfixed in 4% formaldehyde/HMSS on ice and washed in TRIS-buffered saline (pH 7.6) containing 1 mM CaCl2 (TBS). Brain-sections from non-perfused mice and from GBM tissue were fixed in ethanol (10 min; 4°C) followed by acetone (1 min at room temperature) and washed in TBS. Sections were blocked with 5% (w/v) skimmed milk, 0.3% (v/v) Triton X-100 (Serva, Heidelberg, Germany), 0.04% (w/v) NaN3 in TBS for 20 min. Primary antibodies were diluted in the same buffer and incubated overnight at 4°C. Following washes in TBS, sections were incubated with secondary antibodies for 1 h at room temperature. Sections were mounted in 90% glycerol/10% TBS. Fluorescence was visualized with a confocal laser-scanning microscope (Zeiss, Oberkochen, Germany). Images were computer processed using Adobe Photoshop (Adobe, Mountain View, Calif.). For controls, the primary antibodies were omitted. In double-labeling studies, controls included crossover incubation to exclude cross-reaction.
All chemicals were purchased from Merck (Darmstadt, Germany), unless specifically mentioned.
Results
In this study, we investigated the molecular changes involved in BBB injury under EAE and glioma conditions focusing on claudins, which have been recognized as being both sufficient and essential for TJ reconstitution in vitro.
Selective loss of claudin-3 but not of claudin-5 from BBB TJs during inflammation in the CNS
During EAE, the recruitment of inflammatory cells into the CNS parenchyma is accompanied by the breakdown of the BBB. To investigate whether BBB breakdown in vivo is also accompanied by the loss of TJ-associated molecules from the BBB TJs, we performed immunohistochemistry on frozen sections of brains derived from both SJL/N mice afflicted with aEAE and healthy littermates. Localization of the TJ-associated proteins claudin-1, claudin-3, claudin-5, and occludin was investigated. In brain sections from healthy mice junctional localization of claudin-3, claudin-5, and occludin was demonstrated in cerebral vessels (Fig. 1a, c, e, g). Immunostaining for claudin-3, claudin-5, and occludin was exclusively localized at the endothelial cell junctions. In contrast, claudin-1 immunostaining using the novel claudin-1-specific antibody, which does not cross-react with claudin-3, revealed a diffuse endothelial cell-specific immunoreactivity not restricted to the junctional areas (Fig. 1i).

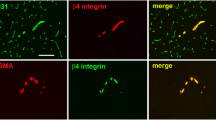
Comparative localization of TJ associated proteins in healthy (a, c, e, g, i) and inflamed (b, d, f, h, k) brains of SJL/N mice. Double immunofluorescence staining of TJ proteins of murine cerebral vessels (green) and CD45 as pan-leukocyte marker (red) is shown. In control brain (left panel), a strong staining for occludin (a) claudin-3 (c), claudin-5 (e) is visible. The staining using the old anti-claudin-1 antibody recognizing also claudin-3 (claudin-1/3 in g) is comparable with the staining for claudin-3 (c). The anti-claudin-1 staining (i) is diffuse and not restricted to the junctions. In EAE brain (right panel), typical CD45-positive inflammatory cell infiltrates (red) surrounding cerebral venules showing normal immunoreactivity of the TJ proteins (green) occludin (b) and claudin-5 (f). Claudin-3 is lost during EAE: In d1/d2, the green and red fluorescence channel are shown separately to clearly demonstrate that loss of claudin-3 immunostaining is not due to masking by the bright CD45 staining. Identical results are shown for the old anti-claudin-1 antibody (claudin-1/3 in h1/h2). The immunostaining for claudin-1 is diffuse and reduced (k1); again, the green and red fluorescence channel are shown separately to demonstrate that the reduction of claudin-1 is not due to masking by CD45 staining (k1/k2) (TJ tight junction, EAE experimental autoimmune encephalomyelitis)
Immunostaining of brain sections from mice afflicted with aEAE for CD45 as a pan-leukocyte marker showed massive perivascular infiltrates in the CNS white matter (Fig. 1b, d1, d2, f, h1, h2). Cerebral vessels lacking a surrounding inflammatory cell infiltrate showed the characteristic junctional staining pattern for claudin-3, claudin-5, and occludin indistinguishable from the staining pattern observed in vessels of healthy brains (data not shown). In vessels surrounded by inflammatory cuffs immunostaining for claudin-5 and occludin was also conserved (Fig. 1b, f), although due to the altered vessel morphology it was not possible to clearly define whether staining was solely localized at the endothelial cell junctions. In contrast, immunostaining for claudin-3 on brain sections from mice afflicted with EAE was completely lost from cerebral endothelial cells surrounded by inflammatory cuffs (Fig. 1d). Interestingly, immunoreactivity of cerebral vessels for claudin-1 was also strongly reduced (Fig. 1k). Thus, during EAE breakdown of the BBB was accompanied by the selective loss of claudin-3 but not of claudin-5 and occludin from BBB endothelial TJs.
Selective loss of claudin-3 but not of claudin-5 from BBB TJs in human GBM
In seven cases of human GBM, the expression pattern of four different junctional markers, occludin, claudin-1, claudin-3 and claudin-5 was investigated and compared by means of double-labeling immunofluorescence (together with fibronectin) and confocal laser scanning microscopy. The majority of tumor microvessels showed an immunoreactivity of occludin, claudin-3 and claudin-5, which was identical to that in mature rat brain microvessels (Fig. 2a, c, e). Other vessel profiles revealed less occludin, which was no longer restricted to the junctions, or no occludin (Fig. 2b). In some microvessels, claudin-3 and even claudin-5 were completely lacking (Fig. 2d, f). As described above in EAE vessels, the immunoreactivity for claudin-1 when using the new antibody revealed a dispersed, vesicular and non-junctional staining of many but not all endothelial cells (Fig. 2i, k). When using the old anti-claudin-1 antibody, which also recognizes claudin-3, the staining was identical to that of the anti-claudin-3 antibody (compare Fig. 2c, d with Fig. 2g, h).

Comparative localization of TJ-associated proteins in microvessels of human glioblastoma multiforme. Double immunofluorescence staining of TJ proteins of human cerebral vessels (red) and fibronectin (green) is shown. In the left panel, vessels are shown which look relatively normal. In the right panel, vessels are shown which lack tight junctional proteins. In most vessels, occludin (a), claudin-3 (c) and claudin-5 (e) is present, but a considerable portion shows a loss of these proteins (b, d, f). The staining using the old anti-claudin-1 antibody recognizing also claudin-3 (claudin-1/3 in g and h) confirm the results for claudin-3 (c, d). The anti-claudin-1 staining is punctuate or vesicular and not restricted to the junctions (i) or not detectable (k)
Discussion
Breakdown of the endothelial BBB is a hallmark of pathological alterations of the CNS, as in brain tumors or inflammatory diseases such as multiple sclerosis or its animal model EAE [3, 15, 19]. The molecular changes involved in dysfunction of BBB endothelial TJs remain to be characterized.
In two foregoing studies we investigated the molecular alterations of BBB TJs in human gliomas and showed a specific down-regulation of claudin-1, a stable expression of claudin-5 in the microcapillary endothelial cells, and a dependency of both molecules on the integrity of the extracellular matrix composition around the vessels [16, 25]. As demonstrated here, junctional localization of claudin-1 might have been a misinterpreted finding due to the cross-reactivity of the previously available anti claudin-1 antibody with claudin-3. Hitherto, many investigators used the commercially available anti-claudin-1 antibody and described the expression of claudin-1 in different tissues including the brain endothelial cells.
We were able to prove the cross-reactivity of the first commercially available anti-claudin-1 antibody with claudin-3 by immunofluorescence staining and Western blotting of murine fibroblasts transfected with either murine claudin-3 or murine claudin-1 (data not shown). The old anti-claudin-1 antibody recognized claudin-1 and claudin-3, whereas the anti-claudin-3 antibody did not detect claudin-1. In the meantime, a new anti-claudin-1 antibody has become available, which recognizes only claudin-1 and not claudin-3 in both immunofluorescence staining and Western blotting of transfected murine fibroblasts. Immunocytochemically, the new anti-claudin-1 antibody stained capillaries in the brain, but the staining was diffuse and not restricted to the junction. Therefore, whereas claudin-3 is a regular component of BBB TJs, presence of claudin-1 in BBB TJs cannot be shown with the reagents available to date.
To date, presence of claudin-3 has only been described in epithelial cells of organs of the gastrointestinal system including liver, pancreas, stomach and intestine [24]. The presence of claudin-3 in BBB TJs supports our notion that the stoichiometry of different claudins in a given TJ reflects the ratio of tight junctional particles associated with either the P- or the E-face of the endothelial cell in freeze fracture replicas [14, 17, 18, 28]. L-fibroblasts transfected with claudin-3 form TJs associated exclusively with the P-face as is the case for claudin-1 [11].
The molecular changes observed previously in human brain tumor capillaries using the first anti-claudin-1 antibody cross-reacting with claudin-3 mainly consisted in a loss of claudin-1 immunostaining [16, 25]. Here, we see that it is claudin-3 that is mainly reduced in glioma as well as in EAE vessels. Breakdown of the BBB and edema formation are well-characterized hallmarks of inflammation in the CNS and have been demonstrated to be involved in the pathogenesis of EAE [3]. Using immunohistochemistry we detected a selective loss of the TJ molecule claudin-3 from vessels surrounded by inflammatory cuffs. In contrast to claudin-3, the presence of other TJ molecules like claudin-5, occludin or ZO-1 in cerebral vessels was not affected during EAE. However, it should be mentioned that in electron microscopic investigations (manuscript in preparation), the overall morphology of vessels characterized by the presence of inflammatory cells is altered such that the TJs are maintained and intact, but irregularly formed. Thus, the impression of an altered staining pattern for occludin and claudin-5 at the light microscopic level (Fig. 1b, f) might reflect an alteration of the arrangement of TJ proteins in inflamed vessels. In any case, occludin and claudin-5 remain well detectable in inflamed vessels at the protein level, whereas immunostaining for claudin-3 is completely lost. Our observations differ from a previous report in which loss of ZO-1 and occludin from cerebral vascular endothelium was observed during CNS inflammation caused by injection of LPS in juvenile rats [2]. In the previous study the cellular infiltrate was dominated by neutrophils, in contrast to EAE where the cellular infiltrate is mostly composed of mononuclear cells. Interestingly, only juvenile rats younger than 3 weeks developed LPS-induced neutrophil-mediated inflammation in the CNS, whereas injection of LPS into older rats did not lead to any CNS inflammation or alterations at the BBB, indicating that maturation of the BBB was not complete at that age. Thus, loss of occludin and ZO-1 from cerebral vessels upon neutrophil recruitment might be specific to the immature BBB of juvenile rats. Alternatively, recruitment of different leukocyte populations might cause different alterations of the TJs due to usage of different routes of transendothelial migration, i.e., transcellular versus paracellular through the TJs [7, 12]. In our electron microscopical investigation of EAE material, we found fibrin in the subendothelial space around inflammatory cells, indicating leakage through the inflamed vessel wall (manuscript in preparation). This is despite the fact that TJs remained morphologically intact, indicating a transendothelial passage not only of cells but of molecules as well. The clarification of the way by which this passage occurs—whether paracellularly through the TJs or transcellularly apart from the TJs—is beyond the scope of this study. What we see is that the presence of mononuclear cells (this study) and fibrin (unpublished) at the abluminal aspect of cerebral vessels is connected to an alteration of the molecular composition of the TJ. The findings in the GBM are characterized, in the absence of inflammation, by alterations of TJ composition identical to those seen during EAE, suggesting that in vivo the loss of claudin-3 from BBB TJs correlates with BBB breakdown at these sites and that this is not necessarily dependent on the presence of inflammatory cells.
Taken together, loss of claudin-3 from BBB TJs accompanies the breakdown of the diffusion barrier provided by the TJs. Our observation that both during inflammation in the CNS, in EAE, and in glioma, claudin-3 is selectively lost from TJs of cerebral vessels suggest that claudin-3 is the key component determining the permeability of BBB endothelial TJs in vivo. Future studies will have to demonstrate the signals determining the subcellular distribution of claudin-3. Defining these signals might allow to develop new therapeutic strategies to reduce BBB permeability and thus severity of brain edema in CNS diseases.
References
Ando-Akatsuka Y, Saitou M, Hirase T, Kishi M, Sakakibara A, Itoh M, Yonemura S, Furuse M, Tsukita S (1996) Interspecies diversity of the occludin sequence: cDNA cloning of human, mouse, dog, and rat-kangaroo homologues. J Cell Biol 133:43–47
Bolton SJ, Anthony DC, Perry VH (1998) Loss of tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil.induced blood-brain barrier breakdown in vivo. Neuroscience 86:1245–1257
Claudio L, Kress Y, Factor J, Brosnan CF (1990) Mechanisms of edema formation in experimental autoimmune encephalomyelitis. The contribution of inflammatory cells. Am J Pathol 137:1033–1045
Engelhardt B, Vestweber D, Hallmann R, Schulz M (1997) E- and P-selectin are not involved in the recruitment of inflammatory cells across the blood-brain barrier in experimental autoimmune encephalomyelitis. Blood 90:4459–4472
Engelhardt B, Laschinger M, Schulz M, Samulowitz U, Vestweber D, Hoch G (1998) The development of experimental autoimmune encephalomyelitis in the mouse requires alpha4-integrin but not alpha4beta7-integrin. J Clin Invest 102:2096–2105
Farqhuar MG, Palade. GE (1963) Junctional complexes in various epithelia. J Cell Biol 17:375–412
Faustmann PM, Dermietzel R (1985) Extravasation of polymorphonuclear leukocytes from the cerebral microvasculature. Cell Tissue Res 242:399–407
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S (1993) Occludin—a novel integral membrane-protein localizing at tight junctions. JCell Biol 123:1777–1788
Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S (1998) Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 141:1539–1550
Furuse M, Sasaki H, Fujimoto K, Tsukita S (1998) A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol 143:391–401
Furuse M, Sasaki H, Tsukita S (1999) Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol 147:891–903
Greenwood J, Howes R, Lightman S (1994) The blood-retinal barrier in experimental autoimmune uveoretinitis—leukocyte interactions and functional damage. Lab Invest 70:39–52
Gumbiner B, Simons K (1986) A functional assay for porteins involved in establishing an epithelial occluding barrier: idenification of an uvomorulin-like polypeptide. J Cell Biol 102:457–468
Kniesel U, Wolburg H (2000) Tight junctions of the blood-brain barrier. Cell Mol Neurobiol 20:57–76
Lassmann H (1983) Comparative neuropathology of chronic experimental allergic enecephalomyelitis and multiple sclerosis. Springer, Berlin Heidelberg New York
Liebner S, Kniesel. U, Kalbacher H, Wolburg H (2000) Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol 79:707–717
Liebner S, Fischmann A, Rascher G, Duffner F, Grote E-H, Kalbacher H, Wolburg H (2000) Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol 100:323–331
Lippoldt A, Liebner S, Andbjer B, Kalbacher H, Wolburg H, Haller H, Fuxe K (2000) Organization of choroid plexus epithelial and endothelial cell tight junctions and regulation of claudin-1, -2 and -5 expression by protein kinase C. NeuroReport 11:1427–1431
Long DM (1970) Capillary ultrasturcture and the blood-brain barrier om human malignant brain tumors. J Neurosurg 32:127–144
Mitic LL, Van Itallie CM, Anderson JM (2000) Molecular physiology and pathophysiology of tight junctions. I. Tight junction structure and function: lessons from mutant animals and proteins. Am J Physiol 279:G250–G254
Morita K, Sasaki H, Furuse M, Tsukita S (1999) Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol147:185–194
Morita K, Furuse M, Fujimoto K, Tsukita S (1999) Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci USA 96:511–516
Morita K, Sasaki H, Fujimoto K, Furuse M, Tsukita S (1999) Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J Cell Biol 145:579–588
Rahner C, Mitic LL, Anderson JM (2001) Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology 120:411–422
Rascher G, Fischmann A, Kröger S, Duffner F, Grote E-H, Wolburg H (2002) Extracellular matrix and the blood-brain barrier in glioblastoma multiforme: spatial segregation of tenascin and agrin. Acta Neuropathol 104:85–91
Saitou M, Furuse M, Sasaki H, Schulzke J-D, Fromm M, Takano H, Noda T, Tsukita S (2000) Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 22:4131–4142
Tsukita S, Furuse M, Itoh M (1999) Structural and signalling molecules come together at tight junctions. Curr Opin Cell Biol 11:628–633
Wolburg H, Lippoldt A (2002) Tight Junctions of the blood-brain barrier: development, composition, and regulation. Vasc Pharmacol 38:323–337
Wolburg H, Neuhaus J, Kniesel U, Krauss B, Schmid EM, Öcalan M, Farrell C, Risau W (1994) Modulation of tight junction structure in blood-brain-barrier endothelial-cells—effects of tissue-culture, 2nd messengers and cocultured astrocytes. J Cell Sci 107:1347–1357
Acknowledgements
We gratefully thank Dr. Mikio Furuse (Kyoto, Japan) for providing claudin-1 and -3 transfected fibroblasts. The diagnostic work of the Institute of Brain Research (University of Tübingen, Germany) is gratefully acknowledged. J.K. was supported by a postdoctoral fellowship of the DFG.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is dedicated to the memory of Werner Risau (†13.12.1998) who initiated this collaboration.
Rights and permissions
About this article
Cite this article
Wolburg, H., Wolburg-Buchholz, K., Kraus, J. et al. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol 105, 586–592 (2003). https://doi.org/10.1007/s00401-003-0688-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0688-z