
Abstract
We report the preparation and rheological characterization of interpenetrating polymer network (IPN) hydrogels made from alginate and hydrophobically modified ethyl hydroxyl ethyl cellulose (HMEHEC). To our knowledge, there have been no studies of the gelation behavior of IPNs. We found that the rheology of these systems can be easily tuned, with the elastic modulus of the IPN strongly dependent on the relative ratio of HMEHEC to alginate. The sol–gel transition of these systems was found to satisfy the Winter–Chambon criterion for gelation at various crosslinker densities. From the power law relationship of the dynamic moduli (G ′ ~G ″ ~ω n), the exponent n appears to be dependent on both the crosslinker density and relative amount of two polymers. The value of n was found to be ~0.5 for all samples for stoichiometric amounts of crosslinker. The effect of molecular weight of HMEHEC on the gel point and viscoelastic exponent has also been reported. Alginate seems to dominate the kinetics of the process but the effect of high molecular weight HMEHEC on the gel point, especially at lower proportion was also evident.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrogels, which typically contain more than 80% water, are highly popular for constructing three-dimensional scaffolding in tissue engineering, drug delivery systems, and cell encapsulation devices (Augst et al. 2006; Lee and Mooney 2001). The porous structure of hydrogels provide safe haven for cells to survive, proliferate, and sustain their metabolic activities while minimizing transport issues. Manipulating the bulk mechanical properties of the hydrogels is touted as one of the most critical criteria for successful implementation of such applications (Bhatia et al. 2005; Bhattarai et al. 2006; Discher et al. 2005). In soft materials like hydrogels, controlling the mechanical properties remains a great challenge, because they are relatively weak and tend to become unstable and disintegrate over a period of time. For drug delivery applications, it is also desirable to improve the solubility and stability of hydrophobic drugs in an aqueous media.
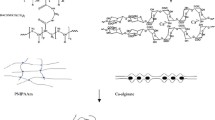
IPNs are defined as combinations of two or more interlocked polymer networks in which at least one such polymer is polymerized/crosslinked in the immediate presence of others (McNaught and Wilkinson 1997). They are a much cheaper option to fine tune the mechanical and physical properties of the system, in comparison to developing a new material from scratch. Materials having distinct characteristics can be synergistically combined to obtain novel systems. In this report, we prepared IPNs of alginate and hydrophobically modified ethyl hydroxyl ethyl cellulose (HMEHEC) (Fig. 1).

Chemical structures of alginate and hydrophobically modified ethyl hydroxyethyl cellulose (HMEHEC)
Alginate is a naturally occurring copolymer of (1–4) linked \(\upbeta\)-d-mannuronic (M) units and \(\upalpha\)–l-guluronic (G) units usually derived from brown algae (Rowley et al. 1999). Commercially, it is used extensively in various industries as a thickening agent and also as a detoxifier in removing heavy metals from water (Aamer et al. 2004; Klock et al. 1997; Moresi et al. 2004; Park and Chae 2004). G-blocks of adjacent alginate chain forms stable crosslinked junctions with divalent cations (e.g., Ca2 + , Ba2 + , Sr2 + ) leading to a three-dimensional network. The resultant mechanical strength of the gel depends strongly on the G/M ratio and the type of cations used. The ratio is also known to affect the biocompatibility characteristics (De Vos et al. 1997; Klock et al. 1997). Encapsulation of live cells with alginate hydrogels is widely popular because of the relatively mild conditions and simple chemistry with which biologically active entities can be immobilized (Rowley et al. 1999). However, the gelation rate is hard to control using calcium chloride solution as a source of calcium ions, and the resulting devices often have irregular shapes and non-homogeneity in the structure (Kuo and Ma 2001; Rowley et al. 1999). In order to slow down the rate of gelation and obtain uniform structure, we employed the method used by Liu et al. (2003). The calcium ions are released in situ from calcium–ethylene diaminetetraacetic acid complex (Ca–EDTA) by adding slowly hydrolysable d-glucono-\(\updelta\)-lactone (GDL) (Draget et al. 1993; Liu et al. 2003).
The second polymer of our choice is HMEHEC, derived from natural occurring cellulose, synthetically grafted with hydrophobic moieties typically less than 5 mol%. It is often used as a rheological modifier and stabilizer for disperse systems (Karlberg et al. 2004). HMEHEC is a type of amphiphilic polymer, which forms a physical gel by self-association in aqueous solution. The degree of substitution (DS), length, and chemical nature of the hydrophobic moiety determines the final properties of the gels. Often, there is a tradeoff between two opposing effects; a higher DS will theoretically yield stronger bonds but might lead to phase separation due to increasing insolubility in water (Rowley et al. 1999).
Previous investigators who worked on IPN-hydrogels of alginate and poly(N-isopropylacrylamide) had reported its utilities for drug delivery and other biomedical applications (de Moura et al. 2006; Ju et al. 2001; Lee et al. 2006; Park and Choi 1998). But these materials involve relatively lengthy steps for polymerization of one or more network that is initiated by “harmful” irradiation, which limit its applications where living cells or sensitive biologically active agents need to be encapsulated during the pre-gel stage. In that respect, the technique we used is gentle and simpler. Recently, hydrogel stiffness has been used as a controlling parameter to influence stem cell differentiation (Banerjee et al. 2009; Saha et al. 2008). Saha et al. (2008) utilized IPNs of synthetic materials as a substrate, whereas Banerjee et al. (2009) used alginate as a three-dimensional scaffold to control mechanical properties of the microenvironment.
In this paper, we have engineered IPN hydrogels by mixing two water-soluble naturally occurring polymers having different gelling characteristics. Simply, the presence of hydrophobic domain in the aqueous HMEHEC system improves the solubility of lipophilic drugs but because of the inherent weakness of the physical gels, often results in burst release. Strength and integrity of a matrix plays a critical role in the release characteristics. Hence, we surmised that including ionically crosslinked alginate chain should provide the necessary mechanical rigidity and help in controlling release of drug molecules. Thus the mechanical properties of the resultant materials depend on two distinct gelation mechanisms; one ionic crosslinking and other hydrophobically driven self-assembly. Our technique also has potential for cell immobilization/transplantation with a minimally disruptive procedure. We obtained a wide variety of materials with different mechanical properties simply by varying the ratio of two polymers. Using polymers with different molecular weights but the same DS of hydrophobic moiety of HMEHEC gives us an extra handle to tweak the mechanical properties of the hydrogels.
We also investigated the gelation kinetics of the aqueous IPNs with rheological techniques. Gelation studies are important in understanding the static and dynamic characteristic features of the chemically crosslinked polymers (Daoud 2000; Mu et al. 2010; Muthukumar 1989; Norisuye et al. 1998). The intricacy or complexity in our system arrives due to the presence of hydrophobic interaction or physical gelation which we expect would play a role in the chemical gelation of the system. From these experiments, we can understand how hydrophobic modification affects the rheological signature of the system during the gelation process. It will be shown that the addition of hydrophobic groups has a significant effect on the mechanical properties and gel point. In future, it is our intention to use these materials as a model system to simulate extra cellular matrices, whose mechanical properties can be adjusted for different cell-type in line with the works of Banerjee et al. (2009) and Saha et al. (2008).
Materials and methods
Materials
Medium viscosity sodium alginate, EDTA and GDL were obtained from Sigma–Aldrich. The molecular weight and G/M of the alginate, as per the supplier, was 120,000 and 60–70, respectively. Hydrophobically modified ethyl hydroxyl ethyl cellulose was a gift from Akzo-Nobel Inc. Table 1 summarizes the different grades of HMEHEC used for the study along with important characteristics. The degree of alkyl (C10–C18) substituted hydrophobic modification for each grade was approximately 1 mol%. Calcium chloride was purchased from Fischer Scientific. All the materials were of ultra-pure grade and used as obtained. All the solutions were prepared in nanopure water (resistivity 18 MΩ cm). The NIH-3T3 fibroblast cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA). Dubelcco’s Modified Eagle’s Medium (DMEM), 0.05% Trypsin/0.02% EDTA, penicillin, and streptomycin were obtained from Mediatech, INC. (Manassas, VA), and heat-inactivated fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Atlanta, GA). Sodium pyruvate, sodium bicarbonate, and l-glutamine were obtained from Sigma–Aldrich (St. Louis, MO).
Methods
We employed the method of in situ release of calcium ions to obtain the homogenous alginate gels (Draget et al. 1990). Ca2 + was released from a Ca–EDTA complex by slowly hydrolyzing GDL. Ca–EDTA was prepared by mixing 0.3 M CaCl2 solution with 0.3 M EDTA in equal proportion. The pH of the resultant complex was adjusted to 7 by addition of 3 M NaOH solution, and volume adjusted by adding water to make 0.3 M Ca–EDTA.
Gel preparation
Gelling of alginate by Ca–EDTA and GDL were obtained as described by Draget et al. (Martin et al. 1988). In a typical method of sample preparation, 0.3 M Ca–EDTA was mixed thoroughly with 2 wt.% alginate solution. The stoichiometric amount of Ca–EDTA added was calculated according to what was actually needed to crosslinked all the G units in the polymer backbone. Freshly prepared GDL solution was then added to the mixture and stirred for another 5 min. The final concentration of the sample was adjusted to desired concentration (1 wt.%) by adding water. The mixture was then poured either directly into the rheometer or a teflon dish to gel for at least 48 h. For preparing higher concentration samples (10 wt.%), the gelled sample was allowed to dry first at ambient conditions and then held at 40°C until the weight became constant. The required amount of water was added, and sample was air sealed and left to equilibrate for another 24 h before taking any measurements.
IPN preparation
Dilute solution of alginate and HMEHEC (2 wt.% for alginate and HMEHEC-200 and 1 wt.% for HMEHEC-500) were mixed thoroughly along with required amount of Ca–EDTA solution for at least 12 h before adding GDL. The samples were allowed to gel in the similar manner as explained before. To compare the gel with its precursor, a certain amount of above mixture was set aside before adding GDL. Different concentration of IPNs was prepared similar to the ones described in gel preparation. The legend, for e.g. A-xC5-y refers to A—alginate, C5—HMEHEC-500 (C2 in case of HMEHEC-200), x and y are wt.% of alginate and HMEHEC, respectively, in the mixture. The ratio r, which is equal to the weight ratio of HMEHEC to alginate, can be determined by dividing y with x. The total polymer concentration in the aqueous mixture is equal to x+y wt.%.
Dynamic mechanical analysis
Rheological measurements were carried out using TA AR-2000 or TA AR-G2 stress-controlled rheometer. All measurements were taken under ambient condition of 25°C unless otherwise specified. Viscoelastic measurements of 1 wt.% gel and all sols were performed using a double Couette geometry with a sample volume of 6.43 mL. To study the dynamic mechanical behavior of the IPNs and pure polymers, oscillatory shear experiments in the linear viscoelastic region was performed. Stress sweep was performed to determine the linear viscoelastic region (LVR) at frequency of 1 Hz. The value of controlling stress or strain was chosen from the mid-region of linear regime to ascertain that strain fall under LVR throughout the entire frequency range. The final mixture (after the addition of GDL) containing 1 wt.% of total polymer was immediately transferred to rheometer, followed by shorter equilibration time of 5 min. A time sweep was performed for 24–30 h at multi-frequency mode. The time reported in this article starts from the instant GDL is added to the mixture. After the time sweep, frequency sweeps were performed in the linear viscoelastic regime to determine elastic (G ′) and viscous (G ″) moduli. The value of G ′ reported in this article is either plateau modulus or measured at 10 rad/s frequency.
For concentrated gels (10 wt.%), 40 mm aluminum-coated parallel-plate geometry was used. Samples were gently pressed with tissue paper to remove surface water before loading, and exposed surfaces were coated with low-viscosity silicone oil to prevent evaporation during the course of experiments. Samples were allowed to equilibrate for 15–30 min before starting the run. A solvent trap was used in all rheological experiments to reduce solvent evaporation.
Drug release protocol
Preliminary studies were performed to test our system for drug delivery applications. Thirty milligrams of a model drug, sulindac, was dissolved in 20 ml of 1 wt.% total polymer solution followed by gelation with calcium. The system was concentrated to 10 wt.% total polymer as described earlier in gel preparation. The details of the drug release protocol can be found elsewhere (Agrawal et al. 2006). In brief, samples weighing about 1 g were transferred to the dialysis membrane (molecular weight cutoff = 1000), ends were sealed and immersed in a bottle containing 900 ml of phosphate-buffered saline (PBS) maintained at 37°C. A small amount of the aliquot was withdrawn at regular interval and analyzed with UV spectrometer at a wavelength of 283 nm. The amount of drug released was determined by comparing the UV readings with a calibration curve. All release experiments were performed in triplicate.
Viability of encapsulated cells
Preliminary studies were performed to assess the utility of our materials for cell encapsulation applications. The NIH-3T3 cell line was maintained in DMEM supplemented with 110 mg/L sodium pyruvate, 3.7 g/L sodium bicarbonate, 584 mg/L l-glutamine, 0.06% penicillin, 0.1% streptomycin, 10% heat-inactivated FBS prepared in nanopure water using Beckman Dickinson vented-cap cell culture flasks, and are passaged every 5 to 6 days. Cells were detached using 0.05% Trypsin/0.02% EDTA collected by centrifugation (800 × g for 7.5 min) and resuspended in fresh medium. Cells were incubated in a 37°C/5% CO2 incubator (Thermo Electron, Waltham, MA). To encapsulate cells in IPN materials, cells were detached using the method described above and resuspended in alginate/cellulose mixtures at 1 × 106 cells/mL of 1% \(\emph{w}/v\) IPN solution (A-0.66C2-0.33 and A-0.66C5-0.33) with 1% \(\emph{w}/v\) alginate as the control). The IPN solution and the control solution were supplemented with 0.00235 g/ml solution HEPES buffer and 0.001 g/mL solution glucose. The mixture was pumped through a Masterflex peristaltic pump equipped with size 13 Masterflex tubing at 0.6 ml/min. The mixture flowed through a custom-made coaxial air flow glass encapsulation piece, fitted with a 25-G needle. Air was flowed past the tip of the needle as the cell mixture was slowly extruded out to decrease the droplet size that collects on the needle tip. The solution then dripped into a bath of 50 mM barium chloride and allowed to cross-link for 30 min forming spherical capsules. The capsules were then rinsed in fresh cell culture medium and stored in 6-well non-tissue culture plates in an incubator at 37°C/5% CO2 on a gentle rocker to keep the capsules moving and maintain their structure. Media was changed every two days. Capsules were soaked in media containing 10 \(\upmu\)l of 1 mg/ml propidium iodide and 20 \(\upmu\)l of 1 mg/ml acridine orange per milliliter of cell culture medium for 10 min. Capsules were then rinsed in fresh media for 10 min to reduce background fluorescence. Images were taken using an Olympus IX71 microscope with a ×4 objective at 1 and 5 days post-encapsulation.
Results and discussion
Rheological characterization
The critical point (c*) at which HMEHEC forms a gel was determined by a tube inversion test, and ascertained more rigorously by rheological measurements. Frequency independence of G ′ indicates solid-like characteristics of the material at gel state (Fig. 2a). Storage modulus were found to be varying between ~4,000 to 0.5 Pa (measured at 10 rad/s) depending upon the grade and polymer strength. As expected, the mechanical strength of the HMEHEC-500 was found to be always higher than the lower grade at a given concentration. HMEHEC-200 even at a high concentration of 5 or even 10 wt.% was not frequency-independent over the entire range (Fig. 2a), in that scenario ability to resist flow for more than 30 s during tube inversion test was chosen as criteria for gelation. The c* for high molecular weight HMEHEC-500 and low molecular weight grade HMEHEC-200 was found to be 3 and 5 wt.%, respectively, at 25°C. Since the degree of substitution of HMEHEC in both cases was the same, the difference can be attributed to entanglement of polymer chains, which tends to increase with molecular weight. HMEHEC are a class of polymers known to exhibit lower critical solution temperature behavior, i.e. they tend to phase separate upon heating (Karlberg et al. 2004; Tsianou et al. 2001). The phase separation temperature of 1 wt.% HMEHEC-200 and HMEHEC-500, as determined by visual observation, was found to be 45°C and 50°C, respectively. For the scope of our experiments, which was between ambient (25°C) and physiological temperature (37°C), we were not concerned with the issues due to phase separation.

Frequency sweep of pure polymer at different concentration in water; a HMEHEC (only G ′), b alginate gels. Legend in (a) for e.g. 500-1 wt.% refers to HMEHEC-500 at 1 wt.% concentration, and so forth
The rheological data of pure alginate gel is shown in Fig. 2b. The plateau storage modulus (G ′) of alginate gels for 1 and 2 wt.% sample were found to be 400 and 850 Pa, respectively. Figure 3 shows frequency dependence of modulus for IPN of alginate with high molecular weight HMEHEC (HMEHEC-500) at two different total concentrations, 1 and 10 wt.%. At low concentration, addition of HMEHEC had little effect on the mechanical strength, as can be seen from the elastic moduli of 1 wt.% alginate gel which is higher than any of the IPNs at the same concentration. The effect is probably due to the fact that the concentration of HMEHEC was below the c*; hence, it just behaves as a non-contributing component having little effect on overall elasticity. “Liquid like” behavior or slight frequency dependence of A-0.5C5-0.5 (G ′ ~ω 0.19 and G ′ ~ω 0.21) compared to other samples in Fig. 3a indicate gels are not strong enough to maintain its structural integrity.

Frequency sweep of IPN of alginate and HMEHEC-500 at 25°C a 1 wt.%, b 10 wt.% polymer content in water. The legend, A-xC5-y refers to A alginate, \(C_{\emph{5}}\) HMEHEC-500, x and y are wt.% of alginate and HMEHEC, respectively, in the mixture. Closed symbol refer to G ′, open to G ″
Significant improvement in the storage modulus was observed when the total polymer concentration was raised to 10 wt.% (G ′ ~ 20,000 Pa for A-3.33C5-6.66 compared to 1,700 and 4,000 Pa for 10 wt.% alginate and HMEHEC-500, respectively). Overall, we found that G ′ of IPNs were greater than the pure polymers (compare 10 wt.% sample from Figs. 2 with 3b) for all the formulation reported in this article. One of the plausible reasons is the higher crosslinking density of the matrix with the increase in concentration or simply higher polymer content which tends to restrict the dynamics of the system. The ratio, r, is found to be a critical parameter in determining the final strength of the gel. It was observed that at 10 wt.%, samples with greater proportion of either polymer (r = 0.5, 2) yields better G ′ compared to r equal to 1. Among the unequal proportion (\(r \ne 1\)) samples, A-3.33C5-6.66 exhibited best properties, which suggests that hydrophobically modified cellulose plays a more prominent role in determining the elastic modulus for IPNs at higher concentration. This can be attributed to the fact that the concentration of HMEHEC in the mixture is above the c*, which enables intermolecular bond between the hydrophobic domains, resulting in stronger networks. The G ′ of A-6.66C5-3.33 is lower than A-3.33C5-6.66 at least by a factor of 2, despite having a higher proportion of ionically-crosslinked alginate.
For the 1 wt.% IPN of alginate with HMEHEC-200, we saw the similar trend as in the case of A-xC5-y samples (Fig. 4a). The G ′ and G ″ of sample for an r of 2, which are proportional to ω 0.21 and ω 0.3, respectively, were found to be lower than for A-0.33C5-0.66. The effect of molecular weight on the mechanical properties is clearly visible; dynamics of high molecular weight are much slower as expected. The frequency dependent modulus of 1 wt.% A-xC2-y samples indicates that the gels are weak and “not solid-like.” Similar to 500 series IPNs, 10 wt.% samples had much higher G ′ to that of individual polymer (see Figs. 2 and 4b). However, the trend was opposite to those of A-xC5-y samples; r = 0.5 show the higher storage moduli compared to r = 2. Why the system behaves differently at r = 1 at both the grades of HMEHEC is not clearly understood.

Frequency sweep of IPN of alginate and HMEHEC-200 in water at 25°C a 1 wt.%, A-0.5C2-0.5 data has been shifted vertically up by 160 Pa for clarity. b 10 wt.% total polymer content. The legend, A-xC2-y refers to A alginate, \(C_{\emph{2}}\) HMEHEC-200, x and y are wt.% of alginate and HMEHEC, respectively, in the mixture. Closed symbol refer to G ′, open to G ″
Finally, we note that for the 2 wt.% alginate gel, as well as for the IPNs with the highest proportion of alginate, a minima is observed in the frequency dependence of G ″. Such behavior has been experimentally observed in hard sphere colloidal glasses and dense emulsions and has been predicted in other soft glassy systems (Mason et al. 1997; Mason and Weitz 1995; Sollich 1998). Because this behavior occurs in the pure alginate at 2 wt.% as well as in the IPNs, it may be related to some slow relaxation process associated with the alginate network.
We were able to develop a suite of materials having elastic moduli in the range of ~20 to 20,000 Pa. These biomaterials can be used to develop tissue engineering scaffolds for soft tissues whose moduli falls in this range (Erkamp et al. 1998; Yu et al. 1993).
Gel kinetics
Classically, the gel point is defined as the instance when the viscosity diverges or when a non-zero equilibrium modulus is detected during the gelation reaction. This is the time when the polymer network first appears to span the containing vessel during a crosslinking reaction. The complex viscosity in the linear viscoelastic region for a model single-relaxation-time Maxwell fluids is given by η ∗ = G ∗ /iω, the absolute value can be determined by the dynamic moduli obtained from frequency sweep data as
Also, from scaling relations, complex modulus can be described as η ∗ (ω) ~ω n − 1 (Muthukumar 1989) where n is the viscoelastic exponent, defined below. The frequency response of the complex viscosity during the course of gelation is shown in Fig. 5a. Weaker dependence of η ∗ on frequency was observed during the sol state, when the system is more liquid-like. The exponent tends to approach −1 value as the reaction progresses to completion. At the gel point, the exponent (Table 3) falls in between the two limits. A log–log plot of complex modulus versus time is shown in Fig. 5b. The viscosity of the system in the pre-gel stage was much lower as compared to the gel stage; an increase in excess of order 2 was observed. At or near the critical gel condition a sudden surge in the |η ∗ (ω)| value was observed. The viscosity seems to reach a plateau in the later stage (t > > t c) of the gelation (complete data not shown) indicating that the crosslinking was complete. Determining the exact gel point from the viscosity data, however, is not straightforward. It requires an intrusive measurement and involves extensive extrapolation which is prone to error.

a Frequency sweep of complex viscosity at different stages of gelation, the number denotes the slope in log–log plot. b Evolution of complex viscosity of 1 wt.% alginate gel at different frequency
Gel point during the crosslinking of a polymer solution is more commonly determined by the Winter–Chambon criterion (Winter and Chambon 1986). The frequency-independence of loss tangent (\(\mbox{tan}\;\delta ={G}^{\prime\prime}(\omega)/G'(\omega)\)) near the gel point, obtained by plotting tan δ over gelation period at different angular frequencies, indicates the formation of incipient gels. The same information can be obtained by plotting the G ′ and G ″ at various frequencies, which at gel point can be represented by a simple power law; the Kramers–Kroenig relationship (Scanlan and Winter 1991b).
where, phase angle δ = nπ/2, or tan δ (=tan (nπ/2)) is independent of frequencies, and S is the gel strength (Pa sn). Although the Winter–Chambon approach was developed for chemically crosslinked systems, it has since been applied to a number of different systems, including physically associated gels and colloidal glasses. To our knowledge, this type of analysis has not been previously applied to IPNs. Doubtless there are differences in the physics of gelation between chemically crosslinked gels and IPNs. However, as seen in Figs. 6, 7, 8, and 9, the data at the gel point appear to be well-described by this analysis for our IPNs. We do not have sufficient data to comment on how well the Winter–Chambon approach may describe gelation of other IPNs nor to account for these differences in the theory.

Time evolution of loss tangent during gelation of 1 wt.% alginate at different frequencies. Similar profile of different samples was obtained to determine the gel point

Frequency sweep of 1 wt.% alginate gel at different stages (time, t) of gelation process. Closed symbol refer to G ′, open to G ″

Frequency sweep of 1 wt.% IPN of alginate and HMEHEC-500 at gel point a r = 0.5, b r = 1, c r = 2. Data are shifted vertically on some plots for clarity. Closed symbol refer to G ′, open to G ″

Frequency sweep of 1 wt.% IPN of alginate and HMEHEC-200 at gel point a r = 0.5, b r = 1, c r = 2. Data are shifted vertically on some plots for clarity. Closed symbol refer to G ′, open to G ″
Figure 6 illustrates the typical method to determine the gel point (t c) shown for 1 wt.% alginate. Similar analyses were performed for all gel samples and are not shown due to space considerations; the results are summarized in Table 3. The crossover point, which represents the same value of tan δ at all frequencies, gives the gel point (t c). The tan δ away from the gelation point was found to be frequency-dependent starting with values more than 1 during the initial stages, falling sharply close to the gelation time, and finally reaching a plateau at time t > > t c . This indicates that the system becomes mechanically stronger and stabilizes as it reaches the final stages of gelation. Equation 2 can be further simplified as
Thus, frequency sweep of dynamic moduli, G ′ and G ″, at the gel point should be parallel to each other and their slope equal to the viscoelastic component. The power law behavior as predicted by Eqs. 2 and 3 are illustrated in Fig. 7. The figure depicts the dynamic response of alginate gels at three different stages of gelation: initial or sol, at gel point, and final stage or gel state. As can be seen clearly as the system evolves towards gels, the viscoelastic exponent n decreases and approaches to zero, indicating frequency independence of dynamic moduli. At some point in between sol and gel, we observe that G ′ and G ″ became parallel; this is the indication of formation of incipient gel. Similar behavior is exhibited by all samples at the frequency range reported in this article.
It can be seen clearly from the values of viscoelastic component during the initial stage of gelation or “sol” state, shown in Table 2, that the n ′ is always higher than the n ″ which gives an evidence of fragility of gel network. The values of n much greater than zero also indicate that the network is far from its fully developed stable state. At the frequency range studied for this work, it was found that G ′ and G ″ are proportional to ω 0.53 − 0.81 and ω 0.49 − 0.73, respectively for HMEHEC-500 IPNs. Deviation from values as predicted by Rouse theory (Rubinstein and Colby 2006) may be because of entanglement of polymer chains or frequencies at which these were performed, not exactly probing the terminal region. Corresponding values of n ′ and n ″ for low molecular HMEHEC-200 IPNs are 1.0 ±0.03 and 0.82, respectively. These values were always found to be lower than pure alginate which indicates that the addition of HMEHEC improves the strength of the materials.
Figure 8 shows the dynamic mechanical behavior of high molecular weight cellulose IPN near the gel point. At the gel point, the dynamic moduli yielded a parallel profile in a log–log plot similar to the one observed in the case of pure alginate (Fig. 7). The viscoelastic exponent was found to be 0.59, 0.55, and 0.49 for r equal to 0.5, 1, and 2, respectively. Although the decrease in the value of n with the cellulose content was not substantial, it may indicate that the system is becoming more mechanically stable with the addition of HMEHEC. For IPNs based on HMEHEC-200 the variation in the value of n is comparatively larger (Fig. 9). However, for all samples with HMEHEC-200, the value of n was found to be lower than those of HMEHEC-500. At first thought, this was not expected from low molecular weight grade of modified cellulose. However, it must be noted that the gel point of IPNs of HMEHEC-200 (see Table 3) was much higher than that of HMEHEC-500. Hence, since these values correspond to much later stage of gelation compared to HMEHEC-500 samples for similar r, the crosslinking reaction must have proceeded to a larger extent, leading to a denser network, and hence a lower n than expected. The trends in the case of HMEHEC-200 IPNs were not as straightforward as in the HMEHEC-500 ones. The n (= 0.55) for r = 1 was highest among other proportion, indicating that equimolar content of two polymers perhaps is not the optimum ratio as suggested earlier.
Gelation is generally described by the bond percolation model (De Gennes 1979; Larson 1999; Rubinstein and Colby 2006) which defines “incipient gel” as the system near gel point, which is still mostly a polydisperse cluster of branched polymers but contains at least one such structure constituting a space filling network. Using the Rouse–Zimm limits for the viscosity exponent, \(\mbox{k}=6\;(\eta \sim | {\mbox{p}-\mbox{pc}}|^{\rm k})\), the experimental value of 2/3 ≤ n ≤ 1 was established (Martin et al. 1988). Other rheological studies in different polymeric systems also give a value of n close to 0.7 (Chambon and Winter 1987; Martin et al. 1988). Experimentally, value of n has been found to be dependent on the percentage of crosslinker (Chambon and Winter 1987), and for a stoichiometric amount of crosslinker a value closer to 1/2 was obtained. Higher values in the range of 1/2 < n < 1 were seen when the system has a crosslinker content below stoichiometric. Even smaller n were reported for sets of different systems and % crosslinker (Nystrom et al. 1996; Silioc et al. 2007). Still, more studies (Martin et al. 1988; Nystrom et al. 1996; Scanlan and Winter 1991a) have reported that the exponent near gel point can have values in the entire range from 0 to 1. Thus, there appears to be no “universal” value for n; it depends on molecular weight, concentration, stoichiometry, and material composition among other variables.
The value of n for nearly all our stoichiometric samples was found to be similar, close to 0.5 (Table 3) and falls within a range of ± 0.15. It is clear that the percolation model alone is insufficient to explain the values of n obtained in our case. Screening of excluded volume effect of polymeric chains combined with percolation was proposed to describe the system (Muthukumar 1989). The value of n = 0.5 has been attributed to stoichiometrically balanced crosslinking networks (Chambon and Winter 1987; Scanlan and Winter 1991a), which is true for some of our systems. In another approach, a ladder model (Schiessel and Blumen 1995) consisting of a mechanical spring and dashpot that simulate the elastic and viscous component of the viscoelastic response predicts a valute of n equal to 0.5. It has also been suggested that entanglement effects can also lower n, which can never be avoided for the high molecular weight precursor used in our system. Moreover, hydrophobically induced intermolecular attraction, as also observed by Silioc et al. (2007), might also influence the viscoelastic signature at the critical gel conditions. The hydrophobic moiety attached to the HMEHEC backbone interwoven with alginate contributes to denser junctions as we see that the n drops slightly with the increase in modified cellulose content. Although the effect of addition of HMEHEC is minimal in the value of n, interplay of two gelation techniques is evident in the evolution of critical gel.
The effect of hydrophobically modified ethyl hydroxyl ethyl cellulose on the gelation time is much higher compared to final gel modulus. As summarized in Table 3, an increase in the gel point t c was observed as the amount of HMEHEC was increased in the IPNs, with a dramatic inflection at r = 2. For high molecular weight (HMEHEC-500) IPNs, we observed a drop in t c at r = 0.5 and 1 compared to pure alginate. The apparent decrease in the gel point is perhaps due to association of hydrophobic groups attached to cellulose, which owing to intimate contact with alginate pull the alginate chains into closer contact. As the alginate chains, facilitated by intermolecular hydrophobic attractions, come closer, the chances of crosslinking of G units via calcium ions also increases. However, a very high gel time of 215 min was observed at the highest r. It should be noted that since the amount of crosslinker to alginate ratio is kept uniform in all the cases, as the IPNs gets progressively richer in cellulose, the amount and probability of forming ionically crosslinked junctions also decreases. The net result is that more time is needed to form the incipient gel. Indeed, as we have found the gel time increases from 56, 66, to 215 min for corresponding r values of 0.5, 1, and 2.
In the case of IPNs of alginate with HMEHEC-200, t c was found to be higher than those of pure alginate in all cases. The effect of addition of HMEHEC on the gel point was much less prominent. Relatively low molecular weight of HMEHEC-200 offer less entanglement that may not influence the rate of gelation as much as 500 series cellulose. So, expectedly 200 series IPNs exhibited monotonic increase in the gel point.
When the crosslinker density was increased to twice (2×) that of the stoichiometric amount (1×), we saw a decrease in the gel-point. This was expected; as higher crosslinker density should increase the rate of gelation. More importantly, the trend in both the dataset was found to be similar, validating our earlier reasoning. In general, viscoelastic exponent for 2× data were found to be higher, indicating that although the arrival of first sample-spanning macromolecule was comparatively faster, the system remains predominantly liquid-like at gel point.
From the data, it seems as though the concentration of alginate had far more influence on the rate of gelation. At a relatively low concentration of <1 wt.% (both the HMEHEC are below the critical concentration), the influence of HMEHEC is low. Modified cellulose has a secondary role, influencing the system through entanglements and/or hydrophobic interaction. That said, addition of HMEHEC does have a significant impact on the mechanical properties at higher concentration, as we have seen earlier.
Drug release test
The model drug, sulindac, used in this study is a highly hydrophobic drug (log P o/w = 3.29 (Caron et al. 1997)). Its maximum solubility in water/PBS/pure alginate was found to be approximately 0.15 wt.% at 37°C. Dissolving and stabilizing sufficient amount of drug in a formulation is critical for maintaining drug bioavailability. We were able to dissolve the drug as high as 0.5 wt.% in our system (A-xC5-y). Figure 10 shows the total amount of drug released over time for different systems. The total amount of drug loaded in each IPN is also plotted for comparison. While all three IPNs show similar release kinetics, it is interesting to see that the IPNs with the highest amount of alginate do not release all of the drugs initially loaded, even after several days.

Drug release profile of sulindac from IPNs of alginate and HMEHEC-500 in PBS at 37°C. Data points are the average of three repeats, and error bars represent the standard deviation. Data points on right-hand y-axis indicate total amount of drug loaded in each IPN
Presumably, the higher solubility of sulindac in IPNs as compared to pure alginate is due to migration of drug into hydrophobic domains provided by the HMEHEC chains. However, our release data indicate that sulindac does not become “trapped” in these domains; IPNs with the highest amount of HMEHEC are still able to release all of the sulindac initially loaded into the IPN. Interestingly, the IPNs with higher amounts of alginate do appear to trap some sulindac. The mechanism behind this is currently under study. However, these initial results are encouraging, as they indicate that these IPNs may be used to improve drug loading into alginate-based gels.
Cell encapsulation
In order to investigate the use of these IPNs for cell growth and viability, NIH-3T3 fibroblasts were encapsulated in various formulations and monitored daily. Preliminary results show that cells are viable in the A-0.66C2-0.33 sample capsules after 5 days in culture, as shown in Fig. 11. However, cells grown in capsules formed with A-0.66C5-0.33 were not viable 1 day post-encapsulation (images not shown). Additionally, it should be emphasized that these preliminary experiments were performed only on samples with a low total polymer concentration of 1.0 wt.%. The technique we have used to concentrate our IPNs (e.g., drying of dilute IPNs) is not compatible with cell encapsulation. To utilize IPNs of a higher polymer concentration for cell encapsulation would require development of an alternate encapsulation protocol. For example, techniques used to encapsulate cells in more highly concentrated polyelectrolyte gels via an initial encapsulation step with a dilute precursor (reviewed in Bhatia et al. 2005) could be modified for IPNs. Another possible route for encapsulation in higher concentration IPNs would be to freeze-dry the gels (Karlson et al. 2000, 2002) and then rehydrate with a cell suspension. Therefore, while these materials can likely be used for biomaterials applications, optimization is necessary to determine the appropriate overall polymer concentration, cell encapsulation protocol, ratio of the IPN components, and molecular weight of the HMEHEC that will promote viability and functionality of the desired cell type.

Viability of NIH-3T3 fibroblasts in IPNs. a–c the alginate control. d–f the A-0.66C2-0.33 sample. b, e a live stain (acridine orange); c, f a dead stain (propidium iodide) after 5 days post-encapsulation. Preliminary results show that fibroblasts are viable in the A-0.66C2-0.33 IPN
Conclusions
Biomaterials with adjustable mechanical strength were prepared from readily available naturally occurring polymers and their derivatives. IPNs of alginate and HMEHEC were found to be compatible and synergistic when blended with each other. The mechanical strength of the IPN is strongly dependent on the relative ratio of polymer. High molecular weight of HMEHEC in general showed better mechanical properties.
The addition of HMEHEC had a profound impact on the gel kinetics of the IPNs. At the gel point, the dynamic modulus of the IPN show a power law behavior; G ′(ω) ~G ″(ω) ~ω n. The value of the exponent n was found to be roughly 0.5 for all samples, consistent with the system having a stoichiometric amount of crosslinker. In general, for lower molecular weight HMEHEC-200, the time of gelation increases with increase in cellulose content, indicating that the concentration of alginate in the IPN is the governing parameter for the gel time. For HMEHEC-500, the gel point at first decreases, probably due to entanglement or inter-molecular hydrophobic attraction but increases as the proportion of HMEHEC gets higher. Hydrophobic modification at a low concentration has a minimal effect; the real advantage of it can be felt only at a concentration above a critical point. It is expected that the presence of hydrophobic domain will increase the solubility of hydrophobic drugs whereas surrounding crosslinked region should help in providing the mechanical stability and extended release of the encapsulated drugs.
References
Aamer KA, Sardinha H, Bhatia SR, Tew GN (2004) Rheological studies of PLLA-PEO-PLLA triblock copolymer hydrogels. Biomaterials 25:1087–1093
Agrawal SK, Sanabria-DeLong N, Coburn JM, Tew GN, Bhatia SR (2006) Novel drug release profiles from micellar solutions of PLA-PEO-PLA triblock copolymers. J Control Release 112:64–71
Augst AD, Kong HJ, Mooney DJ (2006) Alginate hydrogels as biomaterials. Macromol Biosci 6:623–633
Banerjee A, Arha M, Choudhary S, Ashton RS, Bhatia SR, Schaffer DV, Kane RS (2009) The influence of hydrogel modulus on the proliferation and differentiation of encapsulated neural stem cells. Biomaterials 30:4695–4699
Bhatia SR, Khattak SF, Roberts SC (2005) Polyelectrolytes for cell encapsulation. Curr Opin Colloid Interface Sci 10:45–51
Bhattarai N, Li ZS, Edmondson D, Zhang MQ (2006) Alginate-based nanofibrous scaffolds: structural, mechanical, and biological properties. Adv Mater 18:1463–1467
Caron G, Gaillard P, Carrupt PA, Testa B (1997) Lipophililicity behavior of model and medicinal compounds containing a sulfide, sulfoxide or sulfone moiety. Helv Chim Acta 80:449–462
Chambon F, Winter HH (1987) Linear viscoelasticity at the gel point of a cross-linking PDMS with imbalanced stoichiometry. J Rheol 31:683–697
Daoud M (2000) Viscoelasticity near the sol-gel transition. Macromolecules 33:3019–3022
De Gennes PG (1979) Scaling concepts in polymer physics. Cornell University Press, Ithaca
de Moura MR, Aouada FA, Guilherme MR, Radovanovic E, Rubira AF, Muniz EC (2006) Thermo-sensitive IPN hydrogels composed of PNIPAAm gels supported on alginate-Ca2+ with LCST tailored close to human body temperature. Polym Test 25:961–969
De Vos P, De Haan B, Van Schilfgaarde R (1997) Effect of the alginate composition on the biocompatibility of alginate-polylysine microcapsules. Biomaterials 18:273
Discher DE, Janmey P, Wang YL (2005) Tissue cells feel and respond to the stiffness of their substrate. Science 310:1139–1143
Draget KI, Ostgaard K, Smidsrod O (1990) Homogeneous alginate gels—a technical approach. Carbohydr Polym 14:159–178
Draget KI, Simensen MK, Onsoyen E, Smidsrod O (1993) Gel strength of ca-limited alginate gels made in situ. Hydrobiogia 260–261:563–569
Erkamp RQ, Wiggins P, Skovoroda AR, Emelianov SY, O’Donnell M (1998) Measuring the elastic modulus of small tissue samples. Ultrason Imag 20:17–28
Ju HK, Kim SY, Lee YM (2001) pH/temperature-responsive behaviors of semi-IPN and comb-type graft hydrogels composed of alginate and poly (N-isopropylacrylamide). Polymer 42:6851–6857
Karlberg M, Thuresson K, Piculell L, Lindman B (2004) Mixed solutions of hydrophobically modified graft and block copolymers. Colloids Surf, A Physicochem Eng Asp 236:159–164
Karlson L, Joabsson F, Thuresson K (2000) Phase behavior and rheology in water and in model paint formulations thickened with HM-EHEC: influence of the chemical structure and the distribution of hydrophobic tails. Carbohydr Polym 41:25–35
Karlson L, Thuresson K, Lindman B (2002) A rheological investigation of the complex formation between hydrophobically modified ethyl (hydroxy ethyl) cellulose and cyclodextrin. Carbohydr Polym 50:219–226
Klock G, Pfeffermann A, Ryser C, Grohn P, Kuttler B, Hahn HJ, Zimmermann U (1997) Biocompatibility of mannuronic acid-rich alginates. Biomaterials 18:707–713
Kuo CK, Ma PX (2001) Ionically crosslinked alginate hydrogels as scaffolds for tissue engineering: part 1. Structure, gelation rate and mechanical properties. Biomaterials 22:511–521
Larson RG (1999) The structure and rheology of complex fluids. Oxford University Press, New York
Lee KY, Mooney DJ (2001) Hydrogels for tissue engineering. Chem Rev 101:1869–1879
Lee SB, Park EK, Lim YM, Cho SK, Kim SY, Lee YM, Nho YC (2006) Preparation of alginate/poly(N-isopropylacrylamide) semi-interpenetrating and fully interpenetrating polymer network hydrogels with gamma-ray irradiation and their swelling behaviors. J Appl Polym Sci 100:4439–4446
Liu XX, Qian LY, Shu T, Tong Z (2003) Rheology characterization of sol-gel transition in aqueous alginate solutions induced by calcium cations through in situ release. Polymer 44:407–412
Martin JE, Adolf D, Wilcoxon JP (1988) Viscoelasticity of near-Critical Gels. Phys Rev Lett 61:2620–2623
Mason TG, Lacasse MD, Grest GS, Levine D, Bibette J, Weitz DA (1997) Osmotic pressure and viscoelastic shear moduli of concentrated emulsions. Phys Rev E 56:3150–3166
Mason TG, Weitz DA (1995) Linear viscoelasticity of colloidal hard-sphere suspensions near the glass-transition. Phys Rev Lett 75:2770–2773
McNaught AD, Wilkinson A (1997) IUPAC: compendium of chemical technology, 2nd edn. Blackwell, Oxford
Moresi M, Bruno M, Parente E (2004) Viscoelastic properties of microbial alginate gels by oscillatory dynamic tests. J Food Eng 64:179–186
Mu C, Sakai S, Ijima H, Kawakami K (2010) Preparation of cell-enclosing microcapsules through photopolymerization of methacrylated alginate solution triggered by irradiation with visible light. J Biosci Bioeng 109:618–621
Muthukumar M (1989) Screening effect on viscoelasticity near the gel point. Macromolecules 22:4656–4658
Norisuye T, Shibayama M, Nomura S (1998) Time-resolved light scattering study on the gelation process of poly(N-isopropyl acrylamide). Polymer 39:2769–2775
Nystrom B, Kjoniksen AL, Lindman B (1996) Effects of temperature, surfactant, and salt on the rheological behavior in semidilute aqueous systems of a nonionic cellulose ether. Langmuir 12:3233–3240
Park HG, Chae MY (2004) Novel type of alginate gel-based adsorbents for heavy metal removal. J Chem Technol Biotechnol 79:1080–1083
Park TG, Choi HK (1998) Thermally induced core-shell type hydrogel beads having interpenetrating polymer network (IPN) structure. Macromol Rapid Commun 19:167–172
Rowley JA, Madlambayan G, Mooney DJ (1999) Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 20:45–53
Rubinstein M, Colby R (2006) Polymer physics. Oxford University Press, Oxford
Saha K, Keung AJ, Irwin EF, Li Y, Little L, Schaffer DV, Healy KE (2008) Substrate modulus directs neural stem cell behavior. Biophys J 95:4426–4438
Scanlan JC, Winter HH (1991a) Composition dependence of the viscoelasticity of end-linked Poly(Dimethylsiloxane) at the gel point. Macromolecules 24:47–54
Scanlan JC, Winter HH (1991b) The evolution of viscoelasticity near the gel point of end-linking Poly(Dimethylsiloxane)S. Makromol Chem Macromol Symp 45:11–21
Schiessel H, Blumen A (1995) Mesoscopic pictures of the sol–gel transition—ladder models and fractal networks. Macromolecules 28:4013–4019
Silioc C, Maleki A, Zhu KZ, Kjoniksen AL, Nystrom B (2007) Effect of hydrophobic modification on rheological and swelling features during chemical gelation of aqueous polysaccharides. Biomacromolecules 8:719–728
Sollich P (1998) Rheological constitutive equation for a model of soft glassy materials. Phys Rev E 58(1):738–759
Tsianou M, Thuresson K, Piculell L (2001) Phase separation in aqueous mixtures of hydrophobically modified cellulose derivatives with their nonmodified analogues. Colloid Polym Sci 279:340–347
Winter HH, Chambon F (1986) Analysis of linear viscoelasticity of a cross-linking polymer at the gel point. J Rheol 30:367–382
Yu QL, Zhou JB, Fung YC (1993) Neutral axis location in bending and youngs modulus of different layers of arterial-wall. Am J Physiol 265:H52–H60
Acknowledgements
We acknowledge support from the NSF-funded Center for Hierarchical Manufacturing (CMMI-0531171), use of central facilities of the NSF-funded MRSEC on Polymers (DMR-0213695), a NSF IGERT Fellowship for JCW (DGE-0654128) and an NIH Chemistry-Biology Interface Traineeship for WLS (National Research Service Award T32 GM08515).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Choudhary, S., White, J.C., Stoppel, W.L. et al. Gelation behavior of polysaccharide-based interpenetrating polymer network (IPN) hydrogels. Rheol Acta 50, 39–52 (2011). https://doi.org/10.1007/s00397-010-0499-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00397-010-0499-9