
Abstract
The linear and nonlinear viscoelastic behaviors of poly(ethylene oxide) (PEO) in aqueous media have been investigated as a function of concentration and molecular weight. A particular interest has been paid to study the effect of turbulent flow under stirring, inducing both shear and elongational stresses, on the rheological behavior of the polymer solutions. The comparison of intrinsic viscosity and viscoelastic properties between shaken and stirred PEO solutions is discussed at the molecular scale in terms of chain scission and aggregation. Results point out that the effect of the mechanical history on the rheological response of PEO solutions depends also on the concentration regime and molecular weight. Indeed, the influence of the dispersion procedure vanishes by decreasing both the concentration and the molecular weight.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Poly(ethylene oxide) (PEO) has been extensively studied due to its unique behavior in aqueous media and its important industrial application. Its biocompatibility and protein adsorption inhibitor capacity make PEO a good candidate for the development of a new drug delivery medicine (Lee et al. 1995; Allen et al. 1999), and it can be used to substitute some biopolymers as implant for tissue replacement or augmentation (Villain et al. 1996). The specific chemical structure of PEO, \(\mbox{HO}-[{({\mbox{CH}_{2}})_n -\mbox{O}}]_x -\mbox{H}\) with n = 2, confers to this polymer very unusual interactions with water. Indeed, while poly(methylene oxide) with n = 1 and poly(butylenes oxide) with n = 3 are both hydrophobic and insoluble in water, PEO is known as the simplest hydrosoluble hydrocarbon polymer, regarding its chemical structure. Its solubility in water originates from the competition between PEO–water and water–water hydrogen bonding (Dormidontova 2002, 2004), delicately balanced by hydrophobic interactions induced by the ethylene components. The rupture of hydrogen bonds with increasing temperature is responsible for the decrease of its solubility upon heating, a lower critical solution temperature behavior. Near room temperature, the water solubility of PEO is found to depend also on the polymer concentration. Indeed, water is a good solvent at low concentration and high temperature while it becomes a bad solvent at intermediate concentration, close to the critical concentration (Daoust and St Cyr 1984).
PEO is used for applications requiring high cation solvation and good electrochemical stability such as solid electrolyte used in lithium polymer cells (Gray and Armand 2000; Wright 1998), but surprisingly, it appears to be fragile and very sensitive to thermal (Vijayalakshmi et al. 2005), photochemical (Morlat and Gardette 2003; Hassouna et al. 2007), and ultrasound (Pritchard et al. 1966; Kanwal and Pethrick 2004) degradations in the bulk and in solution. For polymer solutions, thermal and photochemical degradations have been observed respectively for temperatures higher than 50°C and for samples exposed to irradiation corresponding to natural outdoor aging (λ > 300 nm). Both degradation processes induce the formation of formate and ester groups. The release of formic acid ions (HCOO − ) is responsible for a drastic decrease of the pH, leading to a random chain scission. In the case of ultrasound degradation, chain scission is due to intense shear stresses arising from the collapse of transient cavitation bubbles. Contrary to thermal and photochemical degradations, the resulting rupture of covalent bonds occurs preferentially in the middle of polymer chains (Madras and McCoy 2001) up to a lower limit of molecular weight of about 2 ×104 g/mol below which the polymer will not undergo scission. It has to be noticed that sonication produces heat, leading to some local increases of temperature and consequently associated thermodegradation, even if the temperature of the sample is externally controlled. Consequently, PEO in water requires controlled and precise conditions when handling, which make it a delicate polymer to work with. These difficulties are enhanced concerning high molecular weight PEO due to a more complex structural organization of the macromolecule. Indeed, contrary to low molecular weight PEO obtained from controlled polymerization techniques, high molecular weight PEO are obtained from condensation of low molecular weight PEO through multifunctional agent, leading to form both hydrophobic regions and branched structures.
The apparent sensitivity of PEO aqueous solutions to degradation could be at the origin of some uncertainties that remains concerning the presence of aggregates and the stability of polymer chains as regard to shear.
Let us focus first on the ability of PEO solution to form aggregates. Most works refer to the presence of molecular clusters (or aggregates) in aqueous PEO solutions, and various interpretations concerning their origin have been proposed. The aggregation has been attributed to the presence of impurities in water (Devanand and Selser 1990), but aggregates have been shown to spontaneously reformed within 1 day after filtration of the solution (Polverari and van de Ven 1996; Ho et al. 2003). The role of hydrogen bonds has also been raised to explain the aggregation phenomena (Dormidontova 2002), but aggregates seem to be stable upon heating (Khan 2006; Duval and Sarazin 2003), a situation for which hydrogen bonds are gradually broken. Aggregation can be seen as a phase separation at a microscopic scale. The de Gennes (1991) model, based on static light scattering data from Polik and Burchard (1983), proposes that PEO solutions below 70°C are phase-separated systems in which aggregates form a concentrated phase that coexists with a dilute phase of swollen coils. Such phase separation has been ascribed to the upper critical solution temperature behavior of PEO solutions. However, a recent small-angle neutron scattering investigation of PEO aqueous solutions has contradicted this hypothesis (Hammouda et al. 2004). This study pointed out chain ends effects on the clustering in PEO solutions with a molecular weight of 4 ×104 g/mol. Despite the fact that end groups represent only one unit per 1,000 units, Hammouda et al. (2004) have shown that the ability of the PEO to aggregate is enhanced in the presence of nonpolar CH3 groups at both ends of the polymer chain while it is strongly reduced in the case of chains end-capped by polar OH groups. The contribution of hydrophobic interactions, initially proposed by Polik and Burchard (1983) and Duval (2000) seems to be determinant when PEO chains are end-capped by nonpolar groups and would lead to polymer aggregation through –CH2–CH2– groups belonging to the chain and end groups. However, this interpretation cannot explain the ability of PEO chain end-capped with OH groups to form clusters. Several works refer to shear-induced aggregation in PEO aqueous solutions. According to Makogon et al. (1988), stirring of high molecular weight PEO solutions (\(M_{\rm w} = 2.4 \times 10^{6}\) g/mol) would favor steric screening of some ether oxygen atoms, reducing the affinity of PEO with water and leading to the dehydration and aggregation of polymer chains without decrease in molecular weight. Rheo-optical measurements have pointed out the trend of micrometric structures formed under shear to align along the flow direction (Liberator and McHugh 2005). As stressed by Hammouda et al. (2004), the aggregation mechanism depends on experimental conditions and could result from the contribution of various effects, such as hydrogen bonding, hydrophobic interaction depending on temperature, and concentration...
The stability of PEO chain as regard to flow has also been the focus of attention in the past decades, but different interpretations were proposed. It is generally admitted that the decay of the drag reduction capacity in high molecular weight (\(M_{\rm w} > 10^{6}\) g/mol) PEO aqueous solutions under elongational flow is due to chain scission (Minoura et al. 1967; Hunston and Zakin 1978; Matthys 1991; Sung et al. 2004). A similar effect has also been reported in high-speed stirring. The number of bonds broken per chain seems to be independent of the polymer concentration but increases with increasing stirring speed (Odell and Keller 1986; D’Almeida and Dias 1997). The scission rate could be described properly by Jelinek’s or Ovenall’s rate equations (Jellinek and White 1951; Ovenall et al. 1958) used to characterize ultrasound degradation of polymer solutions. The scission rate depends also on the nature of the solvent and increases sharply with decreasing the solvent quality. As observed after ultrasound treatment, chain scission induced by high-speed stirring is likely to occur near the center of the macromolecule, leading to the decrease of the polydispersity index (Buchholz et al. 2004).
According to recent works of Duval et al. (Duval and Sarazin 2003; Duval and Boué 2007), aggregation and chain scission are probably combined phenomena. Indeed, stirring aqueous PEO solutions would lead first to chain scission and then to the formation of soluble aggregates. These entities would result from the intermolecular associations of monomers units previously aligned in the flow direction and stabilized by dipolar interactions. Aggregates can be dissolved by the addition of sodium chloride that breaks these interactions.
The work presented in this paper has been motivated by the important disparities reported in the literature in terms of viscosity, intrinsic viscosity, and hydrodynamic radius, respectively, obtained by rheological or light scattering measurements. For instance, zero-shear viscosities η 0 of 4.5 mPa s (Du et al. 2007) and 2 mPa s (Tam and Tiu 1989) are reported for a similar molecular weight of 4 ×106 g/mol at the concentration of 8.5 wt.% where polymer coil starts to contact. In this concentration range, the zero-shear viscosity is nearly proportional to the concentration, making this difference in η 0 significant. In this work, we present a thorough investigation of this polymer as a function of concentration and molecular weight, using rheological techniques. A peculiar attention has been paid to study the influence of the mechanical history induced during the dispersion procedure by comparing the rheological properties of polymer solutions prepared by shaking and by stirring. Dispersion procedures that govern the mechanical history of samples are generally summarily treated in the literature. This paper points out significant changes in the rheological behavior of polymer solutions according to the mechanical history of the solutions induced by the dispersion procedures. Differences are discussed at the molecular scale in terms of chain and aggregate scission and flow-induced aggregation.
Experimental section
Materials
Three poly(ethylene oxide) polymers (Sigma-Aldrich) have been used in this study, each characterized by an average molecular weight \(M_{\rm w} = 5 \times 10^{6}\) g/mol, 106 g/mol, and 3 ×105 g/mol announced by the manufacturer and a polydispersity index of 3.33. Referring to their expected molecular weight, these polymers are labeled 5, 1, and 0.3 M, respectively, in the text for more convenience.
Polymer solutions were obtained by adding the proper amount of polymer in distilled water and then by dispersing the polymer at room temperature using two conventional procedures:
-
The softer procedure consists of placing samples on an SM 30 Edmund Bühler GmbH to-and-fro motion shaker. A motion of 30 mm amplitude was applied at the frequency of 175 rpm during a minimum shaking time of 4 days needed to insure a total dispersion of low concentrated solutions, which can extend to 1 month for the most concentrated solutions. The dispersion time of 1 month is the upper limit beyond which natural degradation arises, leading to the decrease of rheological properties of PEO–water solutions. The homogeneous state of polymer solutions has been appreciated visually by checking the uniform natural light scattering of each sample, and it was confirmed by the reproducibility of rheological measurements for various samples from the same batch.
-
The second procedure, broadly used to disperse polymers, is based on magnetic stirring. Low viscous polymer solutions have been obtained using a Telesystem 06.40 Thermo Scientific Variomag magnetic stirrer while highly viscous solutions required an MR Hei-Standard Heidolph Instruments GmbH and Co. magnetic stirrer. The rotation speed of the stirring bar, initially immersed in the solvent, was fixed at 500 rpm, and the volume of the stirred sample did not exceed 50 ml to insure an efficient stirring of the whole sample. Polymer solutions have been continually stirred for 4 days. Samples have been prepared and stored in darkness to prevent photooxidation of polymer solutions.
Rheometry
The linear viscoelastic measurements and steady shear measurements were carried out using two rotational rheometers: a controlled strain TA Instruments ARES rheometer, equipped with a cone and plate geometry (diameter = 50 mm, cone angle = 1°, truncation = 46 \(\upmu\)m), and a controlled stress TA Instruments ARG2 rheometer, equipped with a cone and plate geometry (diameter = 60 mm, cone angle = 0.6°, truncation = 29 \(\upmu\)m). The viscosity versus time was measured at each shear stress or shear rate, and the steady-state viscosity values were determined as the limit, on long-time scales, of the transient viscosity. To prevent solvent evaporation during measurements, geometries were enclosed in a solvent trap which saturates the atmosphere. The lower plate was equipped with a Peltier thermoelectric device that insures a controlled temperature, fixed at 21 ±0.1°C for this study.
Results and discussion
Influence of the dispersion procedure at various concentrations for the 5 M PEO
Visual observation and flow curves
The first part of this work is dedicated to the rheological investigation of the 5 M PEO solutions. In order to illustrate the significant influence of the dispersion procedure on the rheological properties of the solutions, two polymer solutions at the concentration of 1.5 wt.% have been prepared, one by stirring and the other by shaking. Due to its intermediate level of viscosity, the 1.5 wt.% polymer solution is specially appropriated to perform both steady shear flow and linear viscoelastic investigations, and its rheological behavior is representative of the rheological response of PEO solutions. As a consequence, the 1.5 wt.% polymer solution has been chosen as a reference for the 5 M PEO. A droplet of each sample has been placed into the gap of a parallel plate geometry, and the upper plate has been pulled up at a constant speed of 5 mm/s, producing an extensional flow. As shown in Fig. 1, the shaken solution forms a long filament that does not break, even for the maximum displacement of the upper geometry, while the filament for stirred solution is broken for a low displacement of the geometry. This basic observation clearly shows that the dispersion procedures used to disperse high molecular weight PEO in water lead to the formation of two different final products: a shaken PEO solution with high elongational properties and a stirred solution with nearly no elongational property. To go further in the investigation of the 1.5 wt.% 5-M PEO solutions, flow curves of the two systems are plotted in Fig. 2. Both solutions exhibit a low-shear Newtonian viscosity, followed by a shear thinning behavior. From a quantitative point of view, the way the polymer has been dispersed within distilled water has two major influences on viscous parameters: the zero-shear Newtonian viscosity of the stirred solution of about 35 Pa s is one decade lower than that of the shaken solution (η 0 ~360 Pa s), while the linear regime marking the limit of Newtonian behavior is ten times broader.

Basic characterization of elongational properties of shaken and stirred 5 M PEO solutions at 1.5 wt.%

Flow curves of a 1.5 wt.% 5 M PEO solution for which the polymer has been dispersed by shaking (open symbol) or stirring (full symbol)
This study has been extended to polymer concentrations lying from 0.008 to 5 wt.%. The zero-shear viscosities of both shaken and stirred polymer solutions have been reported in Fig. 3 as a function of polymer concentration. For the most concentrated solutions for which the Newtonian behavior is difficult to access, experimental data have been fitted using the Bird-Carreau model (Eq. 1) to extrapolate zero-shear viscosities.
Three concentration regimes are clearly noticed, each regime being characterized by power law functions of η 0 with distinct exponents
-
For stirring solutions at concentration c ∗ < 0.1 wt.%, the power law exponent is found to be equal to 0.7. In this dilute regime, macromolecules are supposed to be isolated. The concentration c ∗ is known as the overlap concentration which marks the transition from a dilute to a semidilute solution (Doi and Edwards 1986). In the semidilute regime, 0.1 wt.% < c < 0.5 wt.%, η 0 increases sharply with a power law exponent equal to 2.7, corresponding to the rise of molecular overlaps. The power law exponent reaches 4.8 in the upper concentrated regime, for which an entangled network is formed.
-
For stirred polymer solutions, the power law dependence of η 0 is not modified in the diluted and concentrated regime but the exponent of the power law of the intermediate regime is 3.3 instead of 2.7.

Zero-shear viscosity of 5 M PEO solutions as a function of concentration dispersed by shaking (open symbol) and stirring (full symbol)
In the whole concentration range explored, the zero-shear viscosity of stirred solutions is lower than that of shaken solutions. Moreover, the two critical concentrations marking the boundaries between concentration regimes are significantly increased, passing from 0.1 to 0.25 and 0.5 to 0.8 wt.%, respectively, and these new values are in good agreement with the literature (Powell and Scharz 1975; Tam and Tiu 1993). From a phenomenological point of view, the shift of the concentration regimes towards higher concentrations associated with a lower value of the viscosity for stirred solutions can be ascribed to the presence of smaller molecular entities, i.e., individual polymer chains or aggregates. From a molecular point of view, the decrease in the size of molecular entities after stirring results from the mechanical stress of the stirring bar, inducing either a molecular scission for PEO chains and/or the breakup of aggregates.
Intrinsic viscosity and Huggins coefficient
To attempt to identify the nature of the elementary objects that contribute to the viscous behavior of PEO solutions, the reduced viscosity \(\eta _{\rm red} ={({\eta _0 -\eta _\emph{w}})} /{\eta _\emph{w} c}\) of both shaken and stirred solutions is plotted in Fig. 4 as a function of concentration c. The reduced viscosity is defined with η 0, the zero-shear viscosity of polymer solutions, and \(\eta_{\emph{w}}= 0.97\) mPa s, the Newtonian viscosity of distilled water at 21°C. In the dilute regime, the reduced viscosity is a linear function of the concentration and could be described by the Huggins (1942) equation:
where [η] is the intrinsic viscosity and k H the Huggins coefficient. The intrinsic viscosity, obtained from Fig. 4 by extrapolation of the reduced viscosity at zero concentration, is a unique function of the molecular weight for a given polymer–solvent pair. Alternatively, [η] can be obtained by fitting the so-called inherent viscosity, \(\eta _{\rm inh} ={({\ln \eta _{\mbox{rel}}})} / c\) with the Kraemer equation
where η rel is the relative viscosity; \(\eta _{\rm rel} =\eta _0 /{\eta _{\emph{w}}}\), and k K is the Kraemer coefficient. For shaken solutions, the intrinsic viscosity obtained from both Huggins and Kraemer representations is about 4,000 ± 800 cm3 g − 1 and decreases to 900 ±150 cm3 g − 1 in the case of stirred solutions. The intrinsic viscosity is related to the molecular weight M by the Houwink–Mark–Sakurada (HMS) equation:
where K and α are constants (Flory 1953). For PEO solutions, HMS constants obtained at T = 25°C in a molecular weight range from 6 ×105 to 106 g/mol are K = 6.103 ×10 − 3 cm3 g − 1 and α = 0.83 (Khan 2006).

Reduced viscosity η red (open symbols) and inherent viscosity ln (η rel)/c (full symbols) of shaken and stirred 5 M PEO solutions as a function of concentration. Intrinsic viscosities are the extrapolated values to zero concentration of the reduced viscosity and the inherent viscosity using linear fits
Assuming that HMS constants are not significantly modified between 25°C and 21°C and are available for a broader range of molecular weight, the intrinsic viscosity of about 4,000 ± 800 cm3 g − 1 obtained for shaken solutions corresponds to elementary objects having an average molecular weight of about \(({1.02\pm 0.15})\times 10^7\mbox{g} / {\mbox{mol}}\). Such big objects, characterized by an average molecular weight significantly higher than that given by the provider, can be reasonably ascribed to the presence of aggregates. An elementary observation confirms this interpretation: during the incorporation of the polymer in powder form in distilled water, PEO remains at the water surface, forming a hydrated layer of concentrated and viscous polymer solution. With time, this layer forms a macroscopic aggregate that vanishes by diffusion mechanisms. This observation stresses the spontaneous tendency of PEO chains to aggregate in distilled water at room temperature (T ~18°C). The dispersion of polymer under shaking is mainly due to the diffusion of water molecules within the hydrated polymer layer, and the mechanical energy brought during shaking is significantly lower compared to the stirring process. As a consequence, the presence of PEO clusters is highly expected under shaking.
On the contrary, for stirred solutions, the intrinsic viscosity of about 900 ±150 cm3 g − 1 corresponds to elementary objects having an average molecular weight of about \(({1.69\pm 0.2})\times 10^6\mbox{g} /{\mbox{mol}}\), which is well below the theoretical value and could be due to chain scission after stirring. This interpretation is consistent with the spectacular decrease of the elongational behavior pointed out for stirred solutions in Fig. 1. Indeed, the elongation at break is known to significantly decrease with decreasing molecular weight. In the case of polymer solutions, elongational properties are governed by dispersed chains, and the contribution of possible aggregates is negligible. Consequently, differences in the elongational properties between shaken and stirred polymer solution can be clearly understood with a decrease of PEO molecular weight, confirming our interpretation of polymer scission during stirring.
In order to have physicochemical insight into the microstructure of this complex polymeric system, we have considered the Huggins coefficient k H obtained from the slopes of η red in Fig. 4. The value of k H that depends on the solvent quality is linked to the second virial coefficient A 2 (Yamakawa 1961) and is generally considered as a parameter that quantifies the interactions between two molecules also called pair interactions. For shaken solutions, k H, shaken = 0.32±0.12 while k H, stirred = 0.61±0.09 in the case of stirred solutions. The lower value of k H, shaken suggests that aggregates interact weakly and can be considered as isolated entities in dilute solutions. On the contrary, the higher value of k H, stirred implies that PEO chains interact strongly with neighboring chains. The more associative character of stirred polymer solutions could be assigned to chain scissions. Indeed, polymer scission could break C–C or C–O bonds with nearly the same probability since their bond energy of 83 and 81 kcal mol − 1, respectively, are very close (Cottrell 1958). The specific interactions of the oxygen lone pair with the hydrogen of water, which bears a partial positive charge, may however influence the predominant bond breaking. If the breaking occurs at the C–C bonds, it will result in the formation of two \(\mbox{R}-\mbox{O}-\mbox{CH}_{2}^\bullet\) where R designates the polymer chains. If the breaking occurs at the C–O bonds, it will result in the formation of two radicals \(\mbox{R}-\mbox{O}-\mbox{CH}_2 -\mbox{CH}_{2}^\bullet\) and R–O ∙ . The stability of carbon radicals decreases as follows: \(\mbox{R}-\mbox{O}^\bullet >\mbox{R}-\mbox{O}-\mbox{CH}_{2}^\bullet >\mbox{R}-\mbox{O}-\mbox{CH}_2 -\mbox{CH}_{2}^\bullet\). As a C–O breaking provides both the more stable and less stable radicals while the C–C bond breaking provides two radicals of intermediary stability, both types of breakings are realistic. Chains end-capped by free radicals can recombine, giving a variety of polymer chains but involve no modification of the end-group nature.
Broken bonds giving rise to \(-\mbox{CH}_{2}^\bullet\) and –O ∙ radicals could lead to the formation of –CH3 and –OH end groups, respectively, through H ∙ transfer mechanism. This H ∙ transfer may proceed from a transfer to polymer; the resulting radical may recombine with the other macromolecular chain fragments, leading to a branched PEO. When the H ∙ transfer is a transfer solvent, it generates a very aggressive hydrophilic OH ∙ radical that can attack the polymer chain.
The formation of –OH end groups through polymer scission would not modify interactions with the solvent. On the contrary, the presence of hydrophobic –CH3, –CH=CH2, or –CH2–CH3, depending on where the polymer scission takes place in the ethylene groups, favors associative interactions between broken polymer chains. Such hydrophobic interactions may contribute to the high pair interactions measured for stirred PEO solutions. Despite their small fraction, end groups are found to play a dominant role in PEO solutions (Dormidontova 2004; Hammouda et al. 2004). The density of end groups, i.e., their contribution in the rheological response of polymer solutions, increases when decreasing the molecular weight. Hammouda et al. 2004 have suggested that hydrophobic interactions between end groups and ethylene groups of the chains are at the origin of clustering in PEO solutions. We cannot exclude the formation of small aggregates in dilute solutions dispersed by stirring, but their rheological signature is not noticeable. So, the schematic insight into PEO chain organization in dilute solutions is that shaking dispersion procedure could favor the formation of aggregates of nondegraded chains while stirring dispersion prevents their formation and induces chain scission.
Linear viscoelastic measurements and relaxation time dynamics
In this dilute regime, where polymer chains are not entangled, the elongational stress that induces chain scission during stirring is most likely due to hydrodynamic forces transmitted by the suspending medium through friction. What happens in the concentrated regimes for which polymer chains are entangled? To answer this question, the rheological investigation has been completed by linear viscoelastic measurements for polymer solutions at a concentration higher than 0.5 wt.%, for which a viscoelastic response can be measured. Figure 5 shows the frequency dependence of the storage modulus G′ and loss modulus G′′ for the reference solutions at 1.5 wt.%. For stirred solutions, G′ and G′′ moduli are, respectively, proportional to ω 2 and ω 1 at low frequencies, corresponding to the terminal portion of the curves, while both moduli increase slowly at higher explored frequencies. Such a linear viscoelastic behavior is observed in dense macromolecular systems having a broad relaxation time distribution, generally ascribed to the molecular polydispersity, and for which long-time dynamics, characterized by the terminal zone, is governed by reptation (de Gennes 1979).

Storage modulus G′ and loss modulus G′′ versus frequency of 1.5 wt.% 5 M PEO solutions dispersed by shaking (open symbols) and stirring (full symbols). Continuous lines through data represent the fit using the generalized Maxwell model. Dot lines are added to account for the power law dependence of shaken solutions at low frequencies
For shaken PEO solution, it is first worth noting that the viscoelastic moduli are higher than those reported for stirred solutions. This observation is valid in the whole explored frequency range. A significant departure of G′ and G′′ moduli from the terminal zone is observed at lower frequencies. Indeed, the pulsation dependence of viscoelastic moduli follows a power law of ω 1 and ω 0.6, respectively, in this zone. Such a weaker power law dependence of viscoelastic moduli in the terminal zone has already been noticed for soft microgel suspensions of commercial associative guar gum (Aubry et al. 2002) and confirms the presence of aggregates in shaken solutions as suggested previously in this work.
The linear viscoelastic behavior can be expressed as a function of a discrete spectrum using the generalized Maxwell model:
where G i is the elastic modulus corresponding to a relaxation time λ i . The fit of G′ and G′′ data using Eqs. 5 and 6 has been performed in order to monitor the concentration dependence of the longest relaxation time, ascribed to the slow dynamics of the longest molecular entities that are individual chains or aggregates. These slower relaxation time dynamics, noticed λ s , are reported as a function of concentration in Fig. 6. Both G′ and G′′ moduli have been fitted separately and give very close values of λ s for each concentration. Error bars reported in Fig. 6 correspond to the uncertainty on relaxation times obtained from the fitting.

Concentration dependence of the longest relaxation time, λ s , for shaken (open symbols) and stirred (full symbols) 5 M PEO solutions obtained by fitting linear viscoelastic moduli with the generalized Maxwell model
Let us consider first the shaken solutions. According to the previous discussion, the high values of λ s for shaken solutions are probably associated with the relaxation dynamics of aggregates while relaxation dynamics of long chains dispersed in the suspending medium are represented by the distribution of faster relaxation times. In the concentration regime ranging from 0.5 wt.% to a critical concentration of about 1.25 wt.%, marking roughly the beginning of the entangled regime, the relaxation time increases with the concentration according to a power law with an exponent of about 3.8. It is worthwhile to note that the concentration dependence of λ s is much more important than that expected, with a power law of 0.5 generally observed for well-dispersed chains in the semidilute nonentangled regime. The discrepancy between exponents could be explained by the presence of aggregates for shaken solutions: for polymer solutions of individualized chains, the increase of the relaxation dynamics with increasing polymer concentration is due to an increase of constraints induced by neighboring chains. For aggregates, their relaxation dynamics increases with increasing the aggregate size. By adding polymer, the relaxation dynamics of aggregates is more efficiently hindered than that of single molecules.
In addition, a direct relation between zero-shear viscosity and relaxation time can be obtained by comparing their concentration dependence. Below the critical concentration regime, the zero-shear viscosity for shaken solutions is found to be proportional to the ratio λ i /c. In the entangled regime c > 1.25 wt.%, λ s still increases with concentration but seems to level off in the highest concentration regime. It is worth mentioning that λ s values in the highest concentration regime are more difficult to obtain, and this result will not be commented.
Shaken solutions can be reasonably considered as suspensions of polymeric aggregates dispersed in a water solution of long polymer chains. Such coexistence of a concentrated phase dispersed in a more dilute phase has been proposed by de Gennes (1991) for PEO solutions in water and can be evidenced by the turbid character of polymer solution for concentrations higher than ~1 wt.%.
What about the slow relaxation time dynamics for stirred solutions? With increasing concentration in the lowest concentration regime 0.5 wt.% < c < 1.25 wt.%, λ s increases with an exponent of the power law of about 2.5, and the zero-shear viscosity is found to be proportional to the product λ s ·c. Then, λ s reaches a maximum around the critical concentration and finally tends to a nearly constant value at high polymer concentrations. The exponent of the power law of λ s is lower than that obtained for shaken solution, but it is still higher than the expected value. This point will be discussed further in the article.
Discussion of results at the molecular scale is more delicate for concentrated solutions prepared by stirring. For this purpose, two stirred solutions at 1.5 wt.% corresponding to the highest value of λ s and 3.6 wt.% for which λ s is low have been diluted in the concentration range c < 0.01 wt.% in order to estimate their intrinsic viscosity and the size of individual objects formed in the solution. Figure 7 shows the reduced viscosity of stirred solutions obtained by the dilution of two mother stirred solutions at 1.5 and 3.6 wt.% as a function of concentration. The intrinsic viscosity [η]1.5 ~3,620 cm3 g − 1 of solutions obtained by dilutions of the 1.5 wt.% is significantly higher than [η]3.6 ~1,800 cm3 g − 1 obtained by dilutions of the 3.6 wt.%. According to Eq. 4, stirred solutions at 1.5 wt.% would contain polymeric objects with an average molecular weight of about 9.0 ×106 g/mol, while at 3.6 wt.%, the average molecular weight is about 3.9 ×106 g/mol.

Reduced viscosity of stirred 5 M PEO solutions obtained from the dilution of a 1.5 wt.% mother solution and a 3.6 wt.% mother solution as a function of concentration. Intrinsic viscosities are the extrapolated values to zero concentration of the reduced viscosity using linear fits
Stirring has been shown to reduce the average molecular weight of PEO. Moreover, in the semidilute and concentrated regime, PEO chains are likely to form aggregates with increasing concentration as chain contact increases considerably. This aggregation mechanism is macroscopically evidenced by a more turbid sample, also observed for shaken solutions. Consequently, both molecular weights have to be considered as the signature of an aggregation state of short polymer chains for which the molecular weight determination is difficult. It has to be stressed that [η]1.5 > [η]3.6, and both are significantly higher than that obtained from dilute solutions in Fig. 4, [η] ~900 cm3 g − 1. This observation shows that aggregates formed in the semidilute regime are bigger that those obtained in the concentrated regime, themselves bigger than probably individualized and degraded PEO chains. Moreover, this result suggests that big aggregates formed under stirring as concentration increases are stable in water after dilution and appear consequently as insoluble entities, strengthening the interpretation of the hydrophobic character of PEO aggregates. Indeed, the hydrophile/lipophile balance that quantifies the hydrophilic character of PEO chains decreases with both increasing the concentration (Kim and Cao 1993) and decreasing the molecular weight (Cao and Kim 1994) due to hydrophobic end effects.
At this point of the study, we can propose a schematic insight into PEO chain organization in semidilute and concentrated solutions dispersed by stirring. In these concentration regimes, stirred solutions contain aggregates for which the effect of stirring is twofold:
-
It favors the formation of shorter chains having an enhanced hydrophobic character that contributes to aggregation.
-
As pointed out by Pang and Englezos (2002) for phase-separated PEO, aggregates are very sensitive to shear and can breakup under flow.
The molecular dynamics seems to be governed by the competition between the hindering effects induced by the growth rate of aggregate size and the speed up effect induced by chain and aggregate scissions.
An increase of the polymer concentration below the critical concentration of 1.25 wt.% seems to favor the hindering effect with a gradual growth of aggregates. The lower exponent of the power law of λ s between shaken and stirred solutions may be ascribed to the weaker increase of the growth rate of aggregate size induced by shorter polymer molecules compared to nondegraded chains for shaken solutions.
As polymer concentration increases above the critical concentration of 1.25 wt.%:
-
Entanglements induces additional stresses that favors the formation of shorter chains with a more randomized scission, probably between knots of the entangled network that leads to increase of the polydispersity index (Odell et al. 1992).
-
Frictions between aggregates and hydrodynamic interactions with the more viscous suspending medium increase, favoring the breakup of aggregates.
In this latter case, the breakup of aggregates is probably more important than the growth rate of aggregates, leading to the formation of smaller aggregates at high concentrations.
Influence of the molecular weight
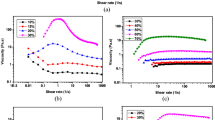
In order to complete the physical insight into the microstructure of PEO solutions, rheological measurements have been extended to two lower molecular weight polymers with an expected value of 106 and 3 ×105 g/mol, labeled 1 and 0.3 M. Figure 8 shows the zero-shear viscosity curves for both molecular weight polymer solutions prepared by shaking and stirring as a function of polymer concentration.

Zero-shear viscosity of 1-M (square symbols) and 0.3 M (triangle symbols) PEO solutions as a function of concentration and dispersed by shaking (open symbol) and stirring (full symbol)
For 1 M polymer solutions in the dilute regime c < 0.5 wt.%, η 0 does not depend on the dispersion procedure. This result suggests that hydrodynamic forces transmitted by the suspending medium are not strong enough to produce chain scission of PEO solutions with molecular weight lower than 1 M. Indeed, the critical elongational strain rate \(\dot{\varepsilon}_f\) required for chain scission of high molecular weight PEO in dilute regime is found to be proportional to \(M_{\emph{w}}^{-2.25}\) (Islam et al. 2004). By decreasing \(M_{\emph{w}}\) below 1 M, \(\dot{\varepsilon}_f\) cannot be achieved under stirring at 500 rpm, and PEO chains are stretched but do not break.
In the semidilute regime, the zero-shear viscosity of stirred solutions is slightly lower than that of shaken solutions, and this difference is more pronounced in the concentrated regime. However, the gap between zero-shear viscosities of shaken and stirred solutions is lower than that observed previously for the 5 M molecular weight polymer. As the concentration increases, polymer chains undergo additional stress due to entanglements that could be responsible for either chain and/or aggregate scission. This additional stress enhances as molecular weight increases.
For the 0.3 M PEO solutions, no difference in η 0 is noticeable between stirred and shaken solutions in the whole range of concentration explored. Indeed, it has been reported that the ability of PEO chains to form aggregates vanishes below the critical molecular weight of about 6 ×105 g/mol, and macromolecules behave as flexible chains in a good solvent (Rangelov and Brown 2000). For such low molecular weights in the concentrated regime, elongational stresses that are strengthened by local stresses of neighbor chains are not strong enough to induce chain scission. It has to be noticed that concentration dependence of η 0 seems to be independent on both the molecular weight and the dispersion procedure and characterized by a power law with an exponent of about 0.6 in the dilute regime and 5 in the concentrated regime.
The reduced viscosity η red for both molecular weight polymer solutions obtained by shaking and stirring is plotted in Fig. 9 as a function of concentration. Contrary to results obtained for 5 M PEO, η red for stirred and shaken solutions is very close for the 1 M polymer, especially at low concentrations, and superimpose for the 0.3 M PEO, forming a master curve. The intrinsic viscosities [η]1 M ~820 cm3 g − 1 and [η]0.3 M ~250 cm3 g − 1 extrapolated from Fig. 9 correspond, respectively, to objects with an average molecular weight of about 1.5 ×106 and 3.6 ×105 g/mol. Both values are close to the expected molecular weight, confirming that polymer scission induced by stirring dilute water PEO solutions at 500 rpm occurs only for molecular weight higher than about 106 g/mol.

Reduced viscosity of 1 M (square symbols) and 0.3-M (triangle symbols) PEO solutions as a function of concentration and dispersed by shaking (open symbol) and stirring (full symbol)
Conclusion
Rheological properties of PEO aqueous solutions have been characterized as a function of the polymer concentration and molecular weight for two dispersion processes: stirring and shaking. The whole set of data reported in this paper clearly shows that shaking favors aggregation and stirring favors chain scission, depending on concentration and molecular weight.
For \(M_{\emph{w}} \le 3\times 10^5~\mbox{g}/{\mbox{mol}}\), neither chain scission nor aggregation is noticed using rheological investigation techniques. As a consequence, the dispersion process does not modify the rheological properties of low molecular weight PEO solutions. However, chain scission and aggregation cannot be excluded.
For \(M_{\emph{w}} \ge 3\times 10^5~\mbox{g} / {\mbox{mol}}\), differences in composition appear in PEO solutions according to the concentration and the dispersion procedure. In the dilute regime, moderated stirring is able to break PEO long chains via hydrodynamic forces while shaking leads to the formation of aggregates. The ability of polymer scission induced by the dispersion procedure increases with increasing \(M_{\emph{w}}\). In the concentrated regime, aggregation of PEO chains is favored by increasing the concentration. Hydrodynamic forces combined to additional stresses due to entanglement enhance the breakup of polymer chains and aggregates.
These results are of potential practical importance as regards to industrial applications involving frequently turbulent flow processes. In particular, it stresses that the mechanical history of PEO solutions leads to the formation of polymer solutions having differences in their compositions (aggregates, chain scission) and in their rheological properties. The general rule that recommends the use of high molecular weight to provide high viscous and viscoelastic solutions is still available but should be balanced by considering the dispersion process.
References
Allen C, Maysinger D, Eisenberg A (1999) Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf B 16:3–27
Aubry T, Bossard F, Moan M (2002) Rheological study of compositional heterogeneity in an associative commercial polymer solution. Polymer 43:3375–3380
Buchholz BA, Zahn JM, Kenward M, Slater GW, Barron AE (2004) Flow-induced chain scission as a physical route to narrowly distributed, high molar mass polymers. Polymer 45:1223–1234
Cao BH, Kim MW (1994) Molecular weight dependence of the surface tension of aqueous poly(ethylene oxide) solutions. Faraday Discuss 98:245–252
Cottrell TL (1958) The strengths of chemical bonds, 2nd edn. Butterworths Scientific, London
D’Almeida AR, Dias ML (1997) Comparative study of shear degradation of carboxymethylcellulose and poly(ethylene oxide) in aqueous solution. Polym Degrad Stab 56:331–337
Daoust H, St Cyr D (1984) Microcalorimetric study of poly(ethylene oxide) in water and in water–ethanol mixed solvent. Macromolecules 17:596–601
Devanand K, Selser JC (1990) Polyethylene oxide does not necessarily aggregate in water. Nature 343:739–741
Doi M, Edwards SF (1986) The theory of polymer dynamics, Chapter 5. Oxford University Press, Oxford
Dormidontova EE (2002) Role of competitive PEO–water and water–water hydrogen bonding in aqueous solution PEO behaviour. Macromolecules 35:987–1001
Dormidontova EE (2004) Influence of end groups on phase behaviour and properties of PEO in aqueous solutions. Macromolecules 37:7747–7761
Du M, Maki Y, Tominaga T, Furukawa H, Gong JP, Osada Y, Zheng Q (2007) Friction of soft gel in dilute polymer solution. Macromolecules 40:4313–4321
Duval M (2000) Monitoring of cluster formation and elimination in PEO solutions. Macromolecules 33:7862–7867
Duval M, Boué F (2007) Dilute poly(ethylene oxide) aqueous solutions in a turbulent flow. Macromolecules 40:8384–8388
Duval M, Sarazin D (2003) Properties of PEO in dilute solution under stirring. Macromolecules 36:1318–1323
Flory PJ (1953) Principles of polymer chemistry, Chapter 7. Cornell University Press, Ithaca
de Gennes PG (1979) Scaling concepts in polymer physics. Cornell University Press, Ithaca
de Gennes PG (1991) A second type of phase separation in polymer solution. C R Acad Sci Ser II 313:1117–1122
Gray FM, Armand M (2000) In: Osaka T, Datta M (eds) New trends in electrochemical technology: energy storage systems for electronics. Gordon and Breach, New York, pp 351–406
Hammouda B, Ho DL, Kline S (2004) Insight into clustering in poly(ethylene oxide) solutions. Macromolecules 37:6932–6937
Hassouna F, Morlat-Thérias S, Mailhot G, Gardette JL (2007) Influence of water on the photodegradation of poly(ethylene oxide). Polym Degrad Stab 92:2042–2050
Ho DL, Hammouda B, Kline SR (2003) Clustering of poly(ethylene oxide) in water revisited. J Polym Sci Polym Phys 41:135–138
Huggins ML (1942) The viscosity of dilute solutions of long-chain molecules. IV. Dependence on concentration. J Am Chem Soc 64:2716–2718
Hunston DL, Zakin JL (1978) Effects of flow-assisted degradation on the drag reduction phenomenon. Polym Prepr Am Chem Soc Div Polym Chem 19:430–438
Islam MT, Vanapalli SA, Solomon MJ (2004) Inertial effect on polymer scission in planar elongational cross-slot flow. Macromolecules 37:1023–1030
Jellinek HHG, White G (1951) The degradation of long-chain molecules by ultrasonic waves. I. Theoretical. J Polym Sci 6:745–756
Kanwal F, Pethrick RA (2004) Ultrasonic degradation studies of poly(ethylene oxide), poly(ethylene adipate) and poly(dimethylsiloxane). Polym Degrad Stab 84:1–6
Khan MS (2006) Aggregate formation in poly(ethylene oxide) solutions. J Appl Polym Sci 102:2578–2583
Kim MW, Cao BH (1993) Additional reduction of surface tension of aqueous polyethylene oxide (PEO) solution at high polymer concentration. Europhys Lett 24:229–234
Lee JH, Lee HB, Andrade JD (1995) Blood compatibility of polyethylene oxide surfaces. Prog Polym Sci 20:1043–1079
Liberator MW, McHugh AJ (2005) Dynamics of shear-induced structure formation in high molecular weight aqueous solutions. J Non-Newton Fluid Mech 132:45–52
Madras G, McCoy BJ (2001) Molecular-weight distribution kinetics for ultrasonic reactions of polymers. AIChE J 47:2341–2348
Makogon BP, Bykova EN, Klenin SI, Povkh IL (1988) Instability in polyethylene oxide solutions in a hydrodynamic field. J Eng Phys Thermophys 54:161–164
Matthys EF (1991) Heat transfer, drag reduction, and fluid characterization for turbulent flow of polymer solutions: recent results and research needs. J Non-Newton Fluid Mech 38(2–3):313–342
Minoura Y, Kasuya T, Kawamura S, Nakano A (1967) Degradation of poly(ethylene oxide) by high-speed stirring. J Polym Sci Part A-2 5:125–142
Morlat S, Gardette JL (2003) Phototransformation of water-soluble polymers. Part II: photooxidation of poly(ethylene oxide) in aqueous solution. Polymer 44:7891–7897
Odell JA, Keller A (1986) Flow-induced chain fracture of isolated linear macromolecules in solution. J Polym Sci Part B 24:1889–1916
Odell JA, Keller A, Muller J (1992) Thermomechanical degradation of macromolecules. Colloid Polym Sci 270:307–324
Ovenall DW, Hastings GW, Allen PEM (1958) Degradation of polymer molecules in solution under influence of ultrasonic waves. I. Kinetic analysis. J Polym Sci 33:207–212
Pang P, Englezos P (2002) Kinetics of the aggregation of polyethylene oxide at temperatures above the polyethylene oxide–water cloud point temperature. Colloids Surf A Physicochem Eng Asp 204:23–30
Polik WF, Burchard W (1983) Static light scattering from aqueous poly(ethylene oxide) solutions in the temperature range 20–90°C. Macromolecules 16:978–982
Polverari M, van de Ven TGM (1996) Dilute aqueous poly(ethylene oxide) solutions: clusters and single molecules in thermodynamic equilibrium. J Chem Phys 100:13687–13695
Powell RL, Scharz WH (1975) Rheological properties of polyethylene oxide solutions. Rheol Acta 14:729–740
Pritchard NJ, Hughes DE, Peacocke AR (1966) The ultrasonic degradation of biological macromolecules under conditions of stable cavitation. I. Theory, methods, and application to deoxyribonucleic acid. Biopolymer 4:259–273
Rangelov S, Brown W (2000) Microgel formation in high molecular weight poly(ethylene oxide). Polymer 41:4825–4380
Sung JH, Lim ST, Kim CA, Chung H, Choi HJ (2004) Mechanical degradation kinetics of poly(ethylene oxide) in a turbulent flow. Korea Aust Rheol J 16:57–62
Tam KC, Tiu C (1989) Steady and dynamic shear properties of aqueous polymer solutions. J Rheol 33:257–280
Tam KC, Tiu C (1993) Improved correlation for shear-dependent viscosity of polyelectrolyte solutions. J Non-Newton Fluid Mech 46:275–288
Vijayalakshmi SP, Chakraborty J, Madras G (2005) Thermal and microwave-assisted oxidative degradation of poly(ethylene oxide). J Appl Polym Sci 96:2090–2096
Villain FL, Parel JM, Lee W, Simon G (1996) Injectable polyethylene oxide gel implant and method for production. WO Pat 006883
Wright PV (1998) Polymer electrolytes—the early days. Electrochim Acta 43(10–11):1137–1143
Yamakawa H (1961) Concentration dependence of polymer chain configurations in solution. J Chem Phys 34:1360–1372
Author information
Authors and Affiliations
Corresponding author
Additional information
Paper presented at the de Gennes Discussion Conference held February 2–5, 2009 in Chamonix, France.
Rights and permissions
About this article
Cite this article
Bossard, F., El Kissi, N., D’Aprea, A. et al. Influence of dispersion procedure on rheological properties of aqueous solutions of high molecular weight PEO. Rheol Acta 49, 529–540 (2010). https://doi.org/10.1007/s00397-009-0402-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00397-009-0402-8