
Abstract
A kind of electroactive nanoparticles (NPs) was prepared in solution by self-assembling amphiphilic 3-thiophenecarboxylic acid-modified starch (TPCA-St), which was synthesized through esterification reaction. By casting TPCA-St NPs on the surface of gold electrode and subsequent electropolymerization of the thiophene moieties in NP film, the electroconductive TPCA-St NP-based film was formed. After horseradish peroxidase and Nafion were sequentially cast on the film surface, a biosensor was successfully prepared. The prepared biosensor showed high sensitivity for H2O2 detection. The linear range from 1 × 10−10 to 1 × 10−5 mol L−1 with a detection limit of 3 × 10−11 mol L−1 was obtained for H2O2 sensing. The biosensor also showed good repeatability and stability, and it has been successfully used to sense H2O2 in commercial disinfector with satisfactory results.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Starch is a natural, biocompatible, biodegradable, non-toxic, inexpensive, and renewable biopolymer produced by many plants as a source of stored energy. With the development of nanomaterials, many kinds of starch-based NPs have been prepared from modified starch via various methods, such as precipitation of amorphous modified starch, combining complex formation and enzymatic hydrolysis, and microfluidization [1]. However, the prepared biocompatible starch-based NPs have only been used in drug delivery and gene carrier [2,3,4]. So, it is necessary to extend the application of starch-based NPs in some other fields.
Biosensor is a three-element system consisting of a bioreceptor, a transducer, and a signal processing unit. When the bioreceptor interacts with the analyte, a quantifiable signal is generated. Biosensor can be used in the fields of environmental sensing, medicine, and food testing [5]. Because of expediency, high sensitivity, and good selectivity, the enzyme-based biosensor has received significant attention [6, 7]. However, it is still a great challenge that effectively immobilize enzyme on solid electrode surface for preparing biosensor [8]. Many materials have been used as a platform to immobilize enzyme on the electrode surface, such as quantum dots [9], polymers [10, 11], mesoporous materials [12], and nanomaterials [13,14,15]. Among these materials, biocompatible nanomaterials are competitive candidates because of their unique advantages, such as large high surface reaction activity, strong adsorption ability, surface-to-volume ratio, and high-catalytic activity for enzyme immobilization [16]. Furthermore, biocompatible nanomaterial-based enzyme biosensor provides the microenvironment for keeping biological activity and enhancing the direct electron transfer between the enzyme active sites and the underlying electrode [17, 18].
Self-assembly of amphiphilic macromolecule is a kind of micro-phase separation behavior due to the weak interactions (such as hydrophilic and hydrophobic interaction, electrostatic interaction, hydrogen bond interaction, and others) between the polymer chains [19]. It is a common method to prepare polymer-based nanoparticles (NPs). The self-assembled NPs can be used in many fields, such as sensors, drug delivery, nanoreactors, catalysts, and others [19, 20]. Miao et al. [16] firstly reported using biocompatible botanical inositol hexakisphosphoric NPs as a platform to immobilize horseradish peroxidease for preparing enzyme-based biosensor. The resultant biosensor exhibited low detection limit and long-term stability for H2O2 sensing. In our previous work, a photosensitive amphiphilic poly(γ-glutamic acid)-graft-7-amino-4-methylcoumarin NPs were used as a matrix to entrap and immobilize hemoglobin on electrode surface for preparing biosensor, which showed good biocatalytic activity for H2O2 [21]. The biocompatible self-assembled NP-modified electrode has the advantages of high uniformity and organization, providing good microenvironment for enzyme immobilization and facilitating direct electron transfer from enzyme to the underlying electrode. In order to improve the electroconductivity of insulting self-assembled polymer NPs, we prepared an electroactive 3-aminothiophene-modified biocompatible poly(γ-glutamic acid) NPs, which were then used to prepare ultrasensitive biosensor. The prepared biosensor was ultrasensitive, stable, repeatable, and was successfully applied to the determination of H2O2 in a commercial disinfecting solution [22].
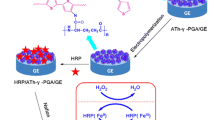
In this work, a novel kind of electroactive 3-thiophenecarboxylic acid-modified starch (TPCA-St) NPs was prepared and then employed to fabricate an electrochemical biosensor for H2O2 detection. The whole strategy is shown in Fig. 1. Firstly, the electroactive amphiphilic TPCA-St was synthesized via esterification reaction. Through solvent exchange method, TPCA-St can assemble into electroactive NPs, which were then cast onto the surface of gold electrode (GE) to form a NP-based film. The subsequent electropolymerization of the electroactive thiophene moieties in NP film induced the formation of electroconductive polymer network, which can provide a platform to accelerate the electron transfer and facilitate the signal transduction from the NP film to the GE, leading to high sensitivity and low detection limit of the electrochemical biosensor. After horseradish peroxidase and Nafion were sequentially cast on the NP film, a biosensor was successfully prepared.

Schematic illustration of the biosensor fabrication using the self-assembled electroactive TPCA-St NPs
Experimental
Reagents and materials
3-Thiophenecarboxylic acid (TPCA), 4-dimethylaminopyridine (DMAP), starch (St), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl), Nafion, phosphate, dimethyl sulfoxide (DMSO), acetonitrile, LiClO4, horse radish peroxidase (HRP), K4[Fe(CN)6], K3[Fe(CN)6], H2O2, and KCl were supplied by Aladdin Chemistry Co. Ltd. (Shanghai, China).
Synthesis of TPCA-St
TPCA-St was synthesized via esterification reaction between the hydroxyl group of starch and carboxyl group of TPCA in the presence of EDC·HCl and DMAP. Briefly, 2 mmol starch (repeat unit) was dissolved in 40 mL DMSO under stirring at 95 °C. After starch dissolved, the temperature of solution was gradually decreased to room temperature. DMAP of 12 mol, 6 mol TPCA, and 6 mol EDC·HCl were sequentially added into the starch solution. The reaction was carried out for 24 h at room temperature. The mixture was placed into a dialysis membrane (MC: 14000) and dialyzed in distilled water. Purification was considered to be finished as no free TPCA was detectable in the wash solution by UV. Finally, the TPCA-St solution was frozen and then lyophilized. The synthetic route of TPCA-St is shown in Fig. S1.
Preparation and characterization of TPCA-St NPs
With the hydrophobic TPCA moieties and hydrophilic starch chain, TPCA-St can self-assemble into NPs in DMSO/H2O solution through the solvent exchange method [23]. The self-assembly behavior of TPCA-St was monitored through turbidity experiment [24]. The turbidity was calculated from the equation: turbidity = 1 − 10−A, where A is the absorbance of the TPCA-St solution at 550 nm measured by UV–Vis spectrophotometry [25]. The solution was continued to be stirred for 3 h and then quenched into 10 mL distilled water. The resulted solution was then dialyzed to remove DMSO, obtaining TPCA-St NP solution. The size distribution and morphology of TPCA-St NPs were characterized by Zeta PALS instrument and transmission electron microscope (TEM) measurement.
Preparation and characterization of the biosensor
A bare GE was polished on polishing cloth with alumina particles and washed by ethanol and distilled water under ultrasonication. The cleaned GE was then subjected to cyclic sweeping between − 0.40 and 1.60 V in 1.0 mol L−1 H2SO4 until a stable cyclic voltammogram was obtained. Five microliters of TPCA-St NP solution was cast evenly onto the surface of GE and then dried in the air, obtaining the GE covered with TPCA-St NPs (TPCA-St/GE), which was subjected to electrochemical polymerization to induce the cross-linking of the thiophene moieties. The electrochemical polymerization was conducted via cyclic voltammetry (CV) for 20 cycles in a solution of LiClO4 dissolved in acetonitrile (0.1 mol L−1) [26]. The scan range and the scan rate are 0 ~ 1.5 V and 100 mV s−1, respectively. Five microliters of the HRP solution (5 mg mL−1, pH 7.4 PBS) was then cast on the surface of TPCA-St/GE and dried in 4 °C about 12 h, obtaining HRP/TPCA-St/GE, which was successively coated with 5 μL of the Nafion solution (five times diluted by pH 7.0 PBS). After Nafion/HRP/TPCA-St/GE was thoroughly rinsed three times with distilled water to remove the physically adsorbed HRP, the Nafion/HRP/TPCA-St/GE biosensor was fabricated. It was storied at 4 °C while not used.
Characterization and measurement
Infrared (IR) experiment was run by attenuate total reflexion (ATR) mode on a Nicolet FI-IR Spectrometer instrument. 1H NMR spectrum was recorded using a Bruker (400 MHz) instrument. Ultraviolet visible (UV–Vis) spectra and UV absorbance were recorded with a TU-1901 spectrophotometer (Beijing Purkinje General instrument Co., Ltd.). The particle size and size distribution were measured using a Zeta PALS instrument (Brookhaven Instruments Corporation, Holtsville, USA). TEM images were recorded in a JEOL JEM-2100 at 200 kV. The morphology of coated electrodes was observed by a Hitachi S-4800 field emission scanning electron microscope (FESEM) operating at 2.0 kV. Solution pH value was monitored using a pHS-3C precision digital pH meter (Shanghai Precision & Scientific Instrument Co. Ltd.). All electrochemical experiments were performed using a CHI 660E electrochemical workstation with a three-electrode cell. The three-electrode system included a platinum wire electrode as counter electrode, a saturated calomel electrode as reference electrode, and the bare or modified GE as the working electrode.
Results and discussion
Characterization of TPCA-St
The structure of TPCA-St was characterized by 1H NMR spectrum. The signals corresponding to the characteristic protons from both thiophene moieties and starch were assigned by numbers as shown in Fig. 2. The 1H-chemical shifts of H2–5 were possible assigned peaks in the range of 3.15–3.85 ppm. The chemical shifts of H1 and OH were possible to assign peaks between 4.58 and 5.50 ppm [27]. The signals at 6.9–7.5 ppm (H6) were assigned to the aromatic protons of thiophene groups, which indicated that TPCA had been successfully introduced into starch side chain. The degree of substitution (DS) of TPCA-St was determined by 1H NMR using integrals of the aromatic protons of thiophene groups and the H2–5 of starch skeleton. The effect of reaction conditions on the DS of TPCA-St is shown in Table S1. The structure of TPCA-St was further confirmed through FT-IR spectrum analysis. The FT-IR spectra of starch and TPCA-St are shown in Fig. S2. Compared with starch, a new peak at 1728 cm−1 corresponding to the C=O stretching mode of ester group appeared in the FT-IR spectra of the TPCA-St, confirming successful synthesis of TPCA-St.

1H NMR spectra of TPCA-St
Preparation and characterization of TPCA-St NPs
With the highest DS, TPCA-St0.56 was used in the following experiments. The turbidities of starch and TPCA-St0.56 in DMSO solutions (1 mL, 10.0 mg mL−1) at 550 nm as a function of water volumes are shown in Fig. S3. It can be seen that the turbidities of starch solution showed negligible change within the tested range, indicating that no NPs were formed because of the hydrophilicity of starch. Comparatively, the turbidities of TPCA-St0.56 were dramatically increased after the critical water content (CWC). The reason may be that at the point of CWC, the solvent DMSO/H2O was worse enough for the hydrophobic TPCA moieties of the TPCA-St0.56, resulting in the aggregation of TPCA-St0.56 by the hydrophobic interaction and the formation of TPCA-St0.56 NPs [28]. The effect of TPCA-St0.56 concentration on assembly process is shown in Fig. 3. It can be seen that with the decrease of TPCA-St0.56 concentrations from 10 to 0.2 mg mL−1, the turbidities of TPCA-St0.56 solutions were initially low and dramatically increased after the CWC. Additionally, the CWC values of TPCA-St0.56 solutions increased with the decrease of TPCA-St0.56 concentration (Fig. 3b). The reason can be ascribed to that with the increase of TPCA-St0.56 concentrations, less water content can induce TPCA-St0.56 chains to aggregate and self-assemble into NPs [29]. When TPCA-St0.56 concentrations further decreased to 0.1 mg mL−1, the turbidities showed negligible change in the tested range, indicating that no NPs were formed. It can be attributed to that the TPCA-St0.56 chain was difficult to aggregate each other via weak hydrophobic interactions at very low concentration.

a Turbidities of TPCA-St0.56 solutions with different concentrations as a function of water volume at 550 nm. b Effect of concentration of TPCA-St0.56 solution on the CWC value
The TPCA-St0.56 NPs in DMSO/H2O solution were stirred for 3 h and then quenched into 10 mL distilled water. The resulted solution was purified by dialysis to remove DMSO, obtaining TPCA-St0.56 NP aqueous solution. After 3 days’ standing, macroscopic precipitate can be observed in the TPCA-St0.56 NP solution except the TPCA-St0.56 NP solution prepared from the concentration of 0.2 mg mL−1. The size distribution and morphology of TPCA-St0.56 NPs are shown in Fig. 4, which revealed a spherical morphology with an average diameter of 100 nm.

Size distribution (a) (inset: photograph of TPCA-St0.56 NP solution) and TEM image (b) of TPCA-St0.56 NPs. The concentration of TPCA-St0.56 solution was 0.2 mg mL−1
Fabrication and characterization of the biosensor
The prepared TPCA-St0.56 NPs were then cast on the surface of GE to form a NP film. The electroactive TPCA moieties can form a large conjugated electroconductive structure (Fig. S4) throughout the NP film via electropolymerization [30], which can enhance the electroconductivity of NP film and then accelerate the electron transfer from the enzyme to the underlying electrode. Figure S5 shows typical cyclic voltammograms (CVs) obtained by scanning electropolymerization from 0.0 to 1.5 V vs. SCE in a fresh LiClO4 in acetonitrile solution. It can be seen that after 15 circles, the current of oxidation peak of thiophene units was stable, indicating the completion of electropolymerization. So, the electropolymerization circle was set as 15. HRP solution of 5 μL was then cast onto the surface of elctroconductive TPCA-St0.56 NP film. The interfacial properties and morphologies of different modified electrodes were characterized by electrochemical impedance spectroscopy (EIS), CV, and SEM.
EIS is an effective measurement to investigate the interfacial property of the modified electrode [31]. To get an equilibrated situation at the electrode surface, the impedance spectra were recorded at an open-circuit voltage and an AC amplitude of 5 mV over a frequency range from 105 to 10−2 Hz. All the EIS experiments were carried out in 0.01 mol L−1 PBS (pH 7.4) containing 5 mmol L−1 K3[Fe(CN)6]/K4[Fe(CN)6] (1:1) and 0.1 mol L−1 KCl. Figure 5a shows the impedance spectra of bare GE, TPCA-St NPs/GE, TPCA-St NPs/GE after electropolymerization, and HRP/TPCA-St NPs/GE. Obviously, the bare GE exhibited a small semicircle domain with the value of 90 Ω (Rct). After being modified by the TPCA-St0.56 NPs, the Rct increased to about 105 Ω. The increased Rct value indicated that NP film-modified GE had a large electron transfer resistance. However, the Rct of NP film-modified GE after electropolymerization was reduced to 30 Ω, demonstrating the improved electroconductivity of the NP film. After immobilization of HRP, the Rct of modified electrode increased to about 880 Ω, suggesting a decrease in electroconductivity due to the adsorption of insulating HRP.

Nyquist plots (a) and CVs (b) corresponding to various modified electrodes. The impedance spectra were recorded at an open-circuit voltage and an AC amplitude of 5 mV over a frequency range from 105 to 10−2 Hz. All the EIS experiments were carried out in 0.01 mol L−1 PBS (pH 7.4) containing 5 mmol L−1 K3[Fe(CN)6]/K4[Fe(CN)6] (1:1) and 0.1 mol L−1 KCl. All the CV experiments were carried out in 0.1 mol L−1 KCl containing 0.01 mol L−1 [Fe(CN)6]4−/3−. The scan rate was 100 mV s−1
CVs also demonstrated the interfacial property of the modified electrode. All the CV experiments were carried out in 0.1 mol L−1 KCl containing 0.01 mol L−1 [Fe(CN)6]4−/3− [31]. The scan rate was 100 mV s−1. As shown in Fig. 5b, a pair of excellently reversible redox peaks of [Fe(CN)6]4−/3− was observed on the bare GE. When TPCA-St0.56 NPs were cast on the GE surface, the redox peaks of [Fe(CN)6]4−/3− decreased, indicating the formation of an insulating film on the GE. After electropolymerization, an almost 2-fold increase in the peak current response was observed, confirming the improved electrical property of the film. After immobilization of HRP, the redox peaks of [Fe(CN)6]4−/3− decreased, implying the adsorption of insulating HRP on the film. After Nafion was cast on the surface of HRP/TPCA-St NPs/GE, the electrochemical biosensor Nafion/HRP/TPCA-St NPs/GE was fabricated.
Performance of the electrochemical biosensor
In order to confirm the concentration of TPCA-St0.56 NP solution for biosensor fabrication, 5 μL of TPCA-St0.56 NP solutions with different concentrations (0.05, 0.02, 0.01, 0.005 mg·mL−1) was cast on the surface of GE, respectively. The electrochemical currents of prepared Nafion/HRP/TPCA-St NPs/GE in pH 7.0 PBS were determined by differential normal pulse voltammetry (DNPV) technique. The scan rate was 20 mV s−1. As shown in Fig. S6, the electrochemical current of biosensor prepared from TPCA-St solution of 0.02 mg mL−1 was larger than that of biosensors prepared from other concentrations. So, the Nafion/HRP/TPCA-St NPs/GE prepared from 0.02 mg mL−1 was used in the following experiments.
The DNPV voltammograms of Nafion/HRP/TPCA-St NPs/GE in 0.1 mol L−1 PBS (pH 7.0) with different concentrations of H2O2 are shown in Fig. 6. As can be seen, the DNPV peak currents of the biosensor increased with increasing H2O2 concentration and showed linear relationships in the range of 1 × 10−10 ~ 1 × 10−5 mol L−1. The calibration curve in Fig. 6b revealed that the peak currents were proportional to the concentrations of H2O2. The linear regression equation was I(uA) = 9.142 + 0.271 × logC(H2O2) (R2 = 0.999).

a Differential normal pulse voltammograms of H2O2 with different concentrations at Nafion/HRP/TPCA-St NPs/GE in 0.1 mol L−1 PBS (pH 7.0). The scan rate is 20 mV s−1. b Calibration curves corresponding to the response recorded on the Nafion/HRP/TPCA-St NPs/GE biosensor versus the concentration of H2O2
Additionally, we did a comparison of the prepared Nafion/HRP/TPCA-St NPs/GE biosensor with other previously reported detection methods for H2O2. As can be seen in Table 1, the prepared Nafion/HRP/TPCA-St NPs/GE biosensor exhibited wider detection range with lower detection limit for sensing H2O2 than other detection methods in the previous reports. The wider detection range with lower detection limit can be attributed to the large specific surface area of the TPCA-St NP-based film, which could immobilize a large number of HRP. In addition, the electropolymerization of thiophene moieties formed the elcetroconductive network accelerated the electron transfer from the enzyme to the underlying electrode, leading to the increase of sensitivity of Nafion/HRP/TPCA-St NPs/GE biosensor.
To investigate the reproducibility of the Nafion/HRP/TPCA-St NPs/GE biosensor, four biosensors were prepared under identical experimental conditions. For detecting H2O2 of 1 × 10−8 mol L−1 in 0.1 mol L−1 PBS (pH 7.0), the peak current was obtained by using each of Nafion/HRP/TPCA-St NPs/GE biosensor. As shown in Fig. S7, the standard deviation of the obtained response did not exceed 3%, implying good reproducibility of the prepared biosensor.
The stability of the Nafion/HRP/TPCA-St NPs/GE biosensor was also investigated through sensing 1 × 10−8 mol L−1 H2O2 in 0.1 mol L−1 PBS (pH 7.0). The peak currents determined by Nafion/HRP/TPCA-St NPs/GE versus time are shown in Fig. S8. The calculated RSD was about 2.4% (n = 8). The prepared biosensor can retain its properties for 4 weeks as it was stored in air at room temperature, indicating that the Nafion/HRP/TPCA-St NPs/GE biosensor response was quite stable.
To investigate the applicability of prepared Nafion/HRP/TPCA-St NPs/GE, the biosensor was applied to sensing H2O2 in commercial disinfector. The sample was diluted 10,000,000-fold with 0.1 mol L−1 PBS (pH 7.0). The average concentration of H2O2 in sample was 9.75 mol L−1 determined by the Nafion/HRP/TPCA-St NPs/GE as shown in Table S2. The concentration of H2O2 in sample was 9.79 mol L−1 determined by the method of potassium permanganate titration. The relative standard deviation of results obtained by two methods was 2.8%, indicating that it was practicable using the Nafion/HRP/TPCA-St NPs/GE biosensor to determine H2O2 in real sample.
Conclusion
In this work, a novel kind of electroactive TPCA-St NPs was prepared from self-assembly via solvent exchange method. The electroactive NPs were then cast on the surface of GE. After subsequent electropolymerization, HRP and Nafion were cast on the electroconductive NP film to prepare Nafion/HRP/TPCA-St NPs/GE biosensor. The electrochemical response current of prepared biosensor for H2O2 increased with increasing H2O2 concentration and showed linear relationships in the range of 1 × 10−10 ~ 1 × 10−5 mol·L−1. The prepared biosensor also showed good stability, repeatability, and practical application.
References
Corre DL, Bras J, Dufresne A (2010) Starch nanoparticles: a review. Biomacromolecules 11:1139–1153
Thiele C, Auerbach D, Jung G, Qiong L, Schneider M, Wenz G (2011) Nanoparticles of anionic starch and cationic cyclodextrin derivatives for the targeted delivery of drugs. Polym Chem 2:209–215
Dandekar P, Jain R, Stauner T, Loretz B, Koch M, Wenz G, Lehr CM (2012) A hydrophobic starch polymer for nanoparticle-mediated delivery of docetaxel. Macromol Biosci 12:184–194
Simi CK, Abraham TE (2007) Hydrophobic grafted and cross-linked starch nanoparticles for drug delivery. Bioprocess Biosyst Eng 30:173–180
Goode JA, Rushworth JVH, Millner PA (2015) Biosensor regeneration: a review of common techniques and outcomes. Langmuir 31:6267–6276
Songa EA, Okonkwo JO (2016) Recent approaches to improving selectivity and sensitivity of enzyme-based biosensors for organophosphorus pesticides: a review. Talanta 155:289–304
Bhakta SA, Evans E, Benavidez TE, Garcia CD (2015) Protein adsorption onto nanomaterials for the development of biosensors and analytical devices: a review. Anal Chim Acta 872:7–25
Du D, Ding JW, Cai J, Zhang AD (2007) Determination of carbaryl pesticide using amperometric acetylcholinesterase sensor formed by electrochemically deposited chitosan. Colloids Surf B: Biointerfaces 58:145–150
Jahanbakhshi M, Habibi B (2016) A novel and facile synthesis of carbon quantum dots via salep hydrothermal treatment as the silver nanoparticles support: application to electroanalytical determination of H2O2 in fetal bovine serum. Biosens Bioelectron 81:143–150
Sun YX, Zhang JT, Huang SW, Wang SF (2007) Hydrogen peroxide biosensor based on the bioelectrocatalysis of horseradish peroxidase incorporated in a new hydrogel film. Sensors Actuators B Chem 124:494–500
Li J, Lin XQ (2007) Glucose biosensor based on immobilization of glucose oxidase in poly(o-aminophenol) film on polypyrrole-Pt nanocomposite modified glassy carbon electrode. Biosens Bioelectron 22:2898–2905
Evans SAG, Elliott JM, Andrews LM, Bartlett PN, Doyle PJ, Denuault G (2002) Detection of hydrogen peroxide at mesoporous platinum microelectrodes. Anal Chem 74:1322–1326
Delvaux M, Walcarius A, Demoustier-Champagne S (2004) Electrocatalytic H2O2 amperometric detection using gold nanotube electrode ensembles. Anal Chim Acta 525:221–230
Martín M, Salazar P, Villalonga R, Campuzano S, Pingarrónd JM, González-Mora JL (2014) Preparation of core–shell Fe3O4@poly(dopamine) magnetic nanoparticles for biosensor construction. J Mater Chem B 2:739–746
Araque E, Arenas CB, Gamella M, Reviejo J, Villalonga R, Pingarrón JM (2014) Graphene-polyamidoamine dendrimer–Pt nanoparticles hybrid nanomaterial for the preparation of mediatorless enzyme biosensor. J Electroanal Chem 717-718:96–102
Miao Y, Wen Y, Dong J, Zhou WX, Zhang ZR, Yang HF (2011) Botanical micelle and its application for direct electrochemical biosensor. Biosens Bioelectron 26:2994–2999
Gorton L, Lindgren A, Larsson T, Munteanu FD, Ruzgas T, Gazaryan I (1999) Direct electron transfer between heme-containing enzymes and electrodes as basis for third generation biosensors. Anal Chim Acta 400:91–108
Jia JB, Wang BQ, Wu AG, Cheng GJ, Li Z, Dong SJ (2002) A method to construct a third-generation horseradish peroxidase biosensor: self-assembling gold nanoparticles to three-dimensional sol-gel network. Anal Chem 74:2217–2223
Yang YQ, Luo J, Liu R, Liu JK, Jiang JQ, Liu XY (2011) Glucose sensors based on electrodeposition of molecularly imprinted polymeric micelles: a novel strategy for MIP sensors. Biosens Bioelectron 26:2607–2612
Zhang RL, Xu S, Luo J, Liu XY (2015) Molecularly imprinted photo-sensitive polyglutamic acid nanoparticles for electrochemical sensing of hemoglobin. Microchim Acta 182:175–183
Zhang RL, Sun JD, Luo J, Xu S, Liu XY (2014) Preparation of photo-sensitive poly(γ-glutamic acid) nanoparticles and application for immobilizing hemoglobin on electrode. Colloid Polym Sci 292:2295–2302
Zhang RL, Jiang C, Fan XX, Yang RC, Sun YY, Zhang CG (2018) A gold electrode modified with a nanoparticulate film composed of a conducting copolymer for ultrasensitive voltammetric sensing of hydrogen peroxide. Microchim Acta 185:58–66
Zhang L, Eisenberg A (1995) Multiple morphologies of ‘crew-cut’ aggregates of polystyrene-b-poly (acrylic acid) block copolymers. Science 268:1728–1731
Zhang RL, Xu S, Luo J, Shi DJ, Liu C, Liu XY (2014) One-pot green synthesis of nanohybrid structures: gold nanoparticles in poly(γ-glutamic acid) copolymer nanoparticles. RSC Adv 4:25106–25113
Xu J, Bai HY, Yi CL, Luo J, Yang C, Xia WS, Liu XY (2011) Self-assembly behavior between native hyaluronan and styrylpyridinium in aqueous solution. Carbohydr Polym 86:678–683
Luo J, Ma Q, Wei W, Zhu Y, Liu R, Liu XY (2016) Synthesis of water-dispersible molecularly imprinted electroactive nanoparticles for the sensitive and selective paracetamol detection. ACS Appl Mater Interfaces 8:21028–21038
Namazi H, Fathi F, Dadkhah A (2011) Hydrophobically modified starch using long-chain fatty acids for preparation of nanosized starch particles. Scientia Iranica 18:439–445
Zhang L, Shen H, Eisenberg A (1997) Phase separation behavior and crew-cut micelle formation of polystyrene-b-poly (acrylic acid) copolymers in solutions. Macromolecules 30:1001–1011
Zhang CG, Yang SH, Zhu Y, Zhang RL, Liu XY (2015) Formation of bowl-shaped nanoparticles by self-assembly of cinnamic acid-modified dextran. Carbohydr Polym 133:637–643
Pang YH, Li XY, Ding HL, Shi GY, Jin LT (2007) Electropolymerization of high quality electrochromic poly(3-alkyl-thiophene)s via a room temperature ionic liquid. Electrochim Acta 52:6172–6177
Prodromidis MI (2010) Impedimetric immunosensors-a review. Electrochim Acta 55:4227–4233
Zhu Y, Lu S, Manohari AG, Dong XX, Chen F, Xu W, Shi ZL, Xu CX (2017) Polydopamine interconnected graphene quantum dots and gold nanoparticles for enzymeless H2O2 detection. J Electroanal Chem 796:75–81
Chen QG, Lin TR, Huang JL, Chen Y, Guo LQ, Fu FF (2018) Colorimetric detection of residual hydrogen peroxide in soaked food based on au@ag nanorods. Anal Methods 10:504–507
Song MR, Wang JL, Chen BY, Wang L (2017) A facile, nonreactive hydrogen peroxide (H2O2) detection method enabled by ion chromatography with UV detector. Anal Chem 89:11537–11544
Kitte SA, Gao WY, Zholudov YT, Qi LM, Nsabimana A, Liu ZY, Xu GB (2017) Stainless steel electrode for sensitive luminol electrochemiluminescent detection of H2O2, glucose, and glucose oxidase activity. Anal Chem 89:9864–9869
Funding
This work was supported by the National Natural Science Foundation of China (NSFC 21504001, 51703001, 51572004), the foundation of Key Laboratory of Food Colloids and Biotechnology, Ministry of Education, Jiangnan University (JDSJ2015-02).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(PDF 432 kb)
Rights and permissions
About this article

Cite this article
Zhang, R., Jin, Z., Zhang, C. et al. An ultrosensitive biosensor based on electroactive nanoparticles self-assembled from 3-thiophenecarboxylic acid-modified starch. Colloid Polym Sci 296, 1365–1372 (2018). https://doi.org/10.1007/s00396-018-4359-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-018-4359-y