
Abstract
In situ emulsion polymerization was employed for synthesizing carbon nanotube (CNT) composites in a colloidal system with poly(styrene) or PS to form nanostructured brush. CNTs were initially functionalized with oleic acid, followed by silanization with (3-aminopropyl) triethoxysilane to impart cross-linking properties. Styrene monomers were efficiently grafted to surface modified CNT via emulsion polymerization with variable CNT concentrations. FTIR analyses of the functionalized CNT and PS/CNT composites confirmed the bond formation and effectiveness of the developed experimental method. X-ray photoelectron spectroscopy confirmed the presence of the desired bonds and the composition of the composites. Structural properties of the composites characterized by TEM confirmed excellent deagglomeration and dispersion of CNTs in PS/CNT composite. Thermal characteristics from TGA and DSC data showed enhanced properties for the nanocomposites as a function of the CNT content. BET measurements indicated significant improvements in surface area and pore volume with enhancements in gas sorption for the polymer nanocomposites.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nanocomposites demonstrate significant improvements in mechanical strength, toughness, and electrical and thermal conductivity [1]. Incorporating surface chemical and functional moieties on nanomaterials offers a way to tailor nanoscale and microscale interactions [2]. Polymer brushes, that is, assemblies of polymer chains tethered at one end to a substrate [3] allow attenuation of sequestration capacity, wettability, colloidal stability, and biocompatibility of composites which are suitable for applications ranging from electronics, separation to biology [4, 5]. Polymer brushes can be fabricated by “grafting to” via covalent tethering of preformed or end-functionalized polymer chains to a functionalized surface, and “grafting from” via polymerization on a substrate previously modified with initiators to provide polymerizable sites [6].
Techniques reported for synthesizing polymer brushes include ring-opening metathesis polymerization (ROMP) [7], nitroxide-mediated polymerization (NMP) [8], atom transfer radical polymerization (ATRP) [9], single electron-transfer living radical polymerization (SET-LRP) [10], and reversible addition fragmentation chain transfer (RAFT) [11, 15]. Most techniques have disadvantages such as risk of overheating, removal of solvents via costly separation, and challenges with large-scale production. These can be overcome by in situ emulsion polymerization (IEP), a method though environmentally friendly and versatile, has not been reported for carbon-based nanocomposite brush fabrication.
Polymer/CNT brushes are typically formed via layer-by-layer assembly [12, 13] and solution polymerization [14] rather than emulsion polymerization. The key challenges with IEP are to reduce the tendency of nanoparticles to agglomerate and to improve interfacial attachment. The activation of grafting surface with functional groups is a prerequisite for covalent anchoring of macromolecules [6]. Molecular assembly has been reported to tether polymer brushes on organic, polymeric, and inorganic substrates such as poly(ethyleneterephthalate) [16], poly(vinyl chloride) [17], Al2O3 [18], Fe3O4 [19], ZnO [20], and carbon black [21].
Multi-walled carbon nanotubes (MWNT) are often tangled [22]; hence, an efficient functionalization method is needed to modify the CNT surface to enable synthesis of a polymer brush. Surface modifications are required [23], often via oxidation of MWNT with strong acids followed by treatment with methacrylamide [14] or 2-bromo-2-methylpropionyl bromide [24]. Aggressive chemical functionalization with strong acids at high temperatures introduces structural defects and inferior CNT properties [25]. We aim to modify CNT surfaces under moderate conditions by attaching carboxyl groups (−COOH) via a dual step approach: noncovalent followed by covalent functionalization [26]. Subsequently, the CNT–COOH will be converted to CNT–NH2 or “NH2-terminated CNT” using organosilane chemistry [27, 28]. Tetraethyl orthosilicate (TEOS) will enable functionalizing CNT surfaces with alkoxysilane molecules. Scalable in situ emulsion polymerization will be used to synthesize polymer brush nanocomposite from “NH2 -terminated CNT” substrate. The materials will be characterized for their morphology, structure, composition, thermal, and sorption properties.
Polymer brush synthesis
The process we developed to synthesize polymer brush grafted from surface modified “NH2-terminated CNT” is described below.
Functionalizing CNT
To improve the dispersibility and reactivity of CNT, we designed a two-step functionalization process, using both noncovalent and covalent methods. MWNT prepared by CVD method were first dispersed with the aid of sodium dodecyl sulfate (SDS) and ultrasonication to debundle CNT, and improve their dispersibility and efficiency for covalent functionalization. Subsequently, the CNT were treated with oleic acid for covalent functionalization at 50 °C, a temperature significantly lower than conventional functionalization with strong mineral acids [29]. Oleic acid [CH3–(CH2)7–CH = CH–(CH2)7–COOH] as a long chain unsaturated fatty acid has a weak C = C bond and is suitable for attachment with CNT. C = C bond scission under reaction conditions with external energy in the form of agitation and heat enables attachment of functional groups on CNT sidewalls [26].
Synthesis of NH2-terminated CNT
Since TEOS helps in creating monolayers on CNT surface [30] and acts as a cross-linking agent [31] to aid polymerization, CNT was first treated with TEOS. Next, APTES was used to impart initiating polymerizable sites and convert the oleic acid functionalized CNT to “NH2-terminated CNT”. Measured 300 mg of oleic acid functionalized CNT, 20 mL ethanol, and 3 mL ammonium hydroxide were dispensed into a 50 mL flask. The flask was then immersed in a sonication bath for 45 min. After sonication, 2 mL of TEOS was added to the solution dropwise for 15 min. The mixture was stirred for 24 h at room temperature, was filtered, and dried under vacuum for 24 h. The TEOS modified functionalized CNT was mixed with 10 mL ethanol, 1 mL ammonium hydroxide, and 1.5 mL APTES. The mixture was ultrasonicated for 2 min and stirred for 24 h at room temperature. Next, the contents were stirred continuously at 60 °C for 2 h and ultrasonicated for 5 min. Finally, the mixture was washed with excess ethanol and dried under vacuum for 24 h to obtain surface modified CNT.
Synthesis of PS/CNT polymer brush
Measured amounts of “NH2-terminated CNT” and 2 g SDS (Sigma Aldrich) were added to 40 mL deionized water in a conical flask to produce micelles. The mixture was sonicated for 15 min and stirred for 1 h at room temperature. Styrene (3.75 g) was added to the conical flask dropwise. Prior to use, the inhibitor (butylcatechol) was removed from styrene monomer (Sigma Aldrich). The mixture was heated to 70 °C and stirred continuously with a magnetic stirrer. At 70 °C, potassium persulfate (KPS) initiator (0.37 g) dissolved in 4 mL water was added to the flask. The mixture was then stirred for about 6 h at 70 °C until polymerization was complete. The product was washed with excess deionized water and ethanol to remove unreacted styrene, oligomers, and impurities. The product was filtered and dried under vacuum at 50 °C for 24 h to obtain dried and solid PS/CNT composite. PS/CNT composite samples having 1, 5, and 10 wt% “NH2-terminated CNT” were synthesized for testing with respect to the amount of styrene used. Based on the synthesis condition, the corresponding samples are denoted as PS/CNT (1 wt%), PS/CNT (5 wt%), and PS/CNT (10 wt%) polymer nanocomposites, respectively. A sample of pure PS was synthesized via emulsion polymerization using the monomer without CNT.
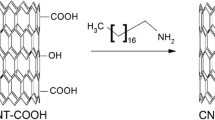
Figure 1 represents the developed reaction scheme starting with pristine CNT, showing the intermediate products during the stepwise process. The “NH2-terminated CNT” containing the active amine group has a basic nitrogen atom with a lone pair, wherein one or more hydrogen atoms can be replaced by a substituent such as an alkyl or aryl group [32]. Apart from their basicity, the dominant reactivity of amines derives from their nucleophilicity [33], which facilitates attachment with organic moieties.

Reaction scheme for synthesis of PS/CNT composite brush
Characterization
Fourier transform infrared (FTIR) spectrometry (Themo Scientific Nicolet 6700) was employed to identify the functional groups on the CNT and synthesized PS/CNT polymer brush to evaluate the efficiency of functionalization, surface modification, and polymerization steps. FTIR data were collected for pure PS and PS/CNT polymer brush nanocomposites for wavenumbers 700–4000 cm−1. X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi, Thermo scientific, UK), a surface-sensitive quantitative spectroscopic technique, was used to measure the elemental composition. XPS spectra were obtained by irradiating with a beam of x-rays while simultaneously measuring the kinetic energy and number of emitted electrons. XPS enabled elemental characterization of “NH2-terminated CNT” and PS/CNT polymer brush nanocomposite using monochromated Al Ka x-rays at 164 W (10.8 mA and 15.2 kV) with binding energy reference of C1s at 285.0 eV for adventitious hydrocarbons.
Transmission electron microscopy or TEM (JEM 1400, JEOL, Japan) for the surface modified CNT and PS/CNT polymer brushes was conducted with accelerating voltage till 120 kV. Dried and powdered composite samples were prepared with tenfold dilution in ethanol, and drop-casting method was employed to disperse the PS/CNT composites on a holey lacey 200 mesh carbon coated grid. Thermogravimetric analysis or TGA (DST-Q600, TA, USA) measurements were used to determine mass changes associated with thermal transitions and degradation for pure PS, pristine CNT, and PS/CNT polymer brush nanocomposites in a nitrogen environment at 15 °C min−1 with a temperature range of 25° –800 °C.
Differential scanning calorimetry or DSC (SEIKO SS2200) was used to determine the glass transition temperature (T g) of the synthesized PS and PS/CNT polymer brush nanocomposite samples (~5–7 mg) at 5 °C/min with a holding time of 5 min for every 40 °C increase in temperature. A purge gas stream of N2 provided an inert environment. The heat capacity (C p) values at the glass transition zone of thermal profiles were analyzed to determine the T g for the samples. Nitrogen physisorption measurements were conducted using an Autosorb-iQ (Quantachrome Instruments, USA) adsorption unit at a temperature of 77 K. The samples were first evacuated at 120 °C for 12 h prior to the analysis. Surface areas were calculated using the Brunauer—Emmett—Teller (BET) method in conjunction with the sorption isotherms over P/Po = 0.05–0.25. The total pore volume was calculated at P/Po = 0.99.
Results and discussions
Functional group identification using FTIR
Figures 2 and 3 present FTIR data for pristine and functionalized CNT, and Fig. 4 presents data for synthesized nanocomposites. The FTIR spectra (Fig. 2a) for pristine CNT show that, as expected, no characteristic peak of any functional group is exhibited. For CNT functionalized with oleic acid, the FTIR spectra (Figs. 2b, 3b), showed a well-defined peak at 1709 cm−1, which corresponds to the presence of C = O moiety for the carboxylic acid group. The peaks in the 2800–3050 cm−1 region are characteristic of C–H stretches arising from SDS which were used to debundle the CNT at the preparatory step, and the shoulder band in the 3450–3500 cm−1 region is characteristic of carboxylic acid O–H stretches. The broad peak at 1150–1250 cm−1 is identified as the C–O stretching mode [34]. For the nonconjugated compounds, the C = C stretching vibration gives rise to a weak IR band in the range of 1640–1660 cm−1. The spectra of pure oleic acid obtained by Bronstein et al. [35] and Ding et al. [36] showed a shoulder peak at 1640–1660 cm−1 indicating the presence of C = C bond. This particular peak is not observed in our FTIR spectra (Fig. 3b) for oleic acid functionalized CNT, thus, indicating the scission of C = C bond of oleic acid in the substrate.

FTIR spectra of (a) pristine CNT, (b) CNT functionalized by oleic acid, (c) NH2-terminated CNT for 700–4000 cm−1 range

FTIR spectra of (a) pristine CNT, (b) CNT functionalized by oleic acid, (c) NH2-terminated CNT for 1000–1800 cm−1 range. Inset: Spectra for “NH2-terminated CNT” in transmittance zone 60–100 %

FTIR spectra of (a) pure PS, (b) PS/CNT (1 wt%), (c) PS/CNT (5 wt%), and (d) PS/CNT (10 wt%) polymer brush
FTIR spectra for “NH2-terminated CNT” (Figs. 2c, 3-inset) show a broad peak at 1000–1300 cm−1 wavenumber assigned to vibrations involving Si atoms in the organosilane group. In particular, the peak at 1040–1100 cm−1 is assigned to Si − O − C stretching vibration. The band at 1230 cm−1 is assigned to the disorder-induced modes of the silica network, and such vibration modes have been identified at 1200–1250 cm−1 in sol-gel silica or Si:O:C:H films arose from TEOS. The presence of this band indicates the formation of a monolayer during CNT modification with TEOS before reacting with APTES. The absorption band observed for “NH2-terminated CNT” (Fig. 3c) at 1530–1600 cm−1 wavenumber range suggests N–H bending contributed by ‘–NH2’ group [37]. It is noteworthy that FTIR spectra for “NH2-terminated CNT” (Figs. 3c and 3-inset) do not show any peak at 1709 cm−1 which corresponds to C = O bond specific to carboxylic acid, which was evident in CNT functionalized by oleic acid. This absence suggests that the carboxylic acid group (−COOH) attached to CNT due to oleic acid functionalization reacts with APTES; an observation which supports the corresponding reaction step (Fig. 1). However, the adsorption peak evident for “NH2-terminated CNT” (Fig. 3c) at around 1655 cm−1 can be designated to C = O bond for amides [38]. Moreover, the shoulder band in the 3450–3500 cm−1 region for CNT functionalized by oleic acid (Fig. 2b) representative as characteristic for carboxylic acid’s O-H stretches is absent in the FTIR spectra for “NH2-terminated CNT” (Fig. 2c). These observations are consistent with the reaction scheme (Fig. 1) that suggests termination of hydroxyl group for oleic acid functionalized CNT when reacted with APTES. Thus, the FTIR spectra for pristine and modified CNT (Figs. 2 and 3) confirm the reaction scheme presented by Fig. 1 for CNT surface modification.
The FTIR spectra of pure PS and PS/CNT polymer brush (Fig. 4) confirm the efficiency of the IEP process. Figure 4d shows a strong peak at 1100 cm−1 for PS/CNT composite when the highest CNT concentration (10 wt%) was used compared to those of other nanocomposites with lower concentration (Fig. 4b, c). This peak arises from the silane modified nanotubes, i.e., “NH2-terminated CNT” and represents Si − O − C stretching vibration. The amplitude of this peak is proportional to the CNT concentration of the composites, thereby confirming the attachment of PS to surface modified CNT. Figure 4b–d also shows the signature peak arising from “NH2-terminated CNT” via N-H bending at 1600 cm−1 for PS/CNT nanocomposites irrespective of CNT concentration; which was not observed for pure PS (Fig. 4a). The peak at 2850–3100 cm−1 wavenumber represents both aliphatic and aromatic C-H stretching within that zone, which is a signature peak for pure PS (Fig. 4a). This type of peak was not identified for “NH2-terminated CNT” (Fig. 2c); but were identified for PS/CNT nanocomposites (Fig. 4b–d). These confirm the attachment of PS on the surface of “NH2-terminated CNT”. At 10 wt% CNT concentration (Fig. 4d), the amplitude for this peak is smaller compared to those for composites with 1 wt% and 5 wt% CNT (Fig. 4b, c), due to higher CNT content compared to the base polymer (PS).
The FTIR spectra (Fig. 4) coupled with Table 1 confirm that styrene was successfully grafted from surface modified CNT as shown by the identified bonds, formed on the nanocomposites and contributed by the individual functional groups. These results validate the grafting and the IEP process for polymer brush synthesis from surface modified CNT.
XPS analysis
Figures 5 and 6 show the XPS analysis for “NH2-terminated CNT” and PS/CNT (10 wt%) polymer brush, respectively. The XPS total survey (Fig. 5a) demonstrates the elemental identification of C, O, Si, and N which confirms the structural elements of “NH2-terminated CNT” sample. In addition, the high resolution N1 s spectrum (Fig. 5b) at binding energy 399.51 eV illustrates the “–NH2” bonding of this material [39–40]. These observations are in agreement with the reaction scheme proposed in Fig. 1.

XPS analysis for “NH2-terminated CNT”: a XPS total survey analysis, b high resolution N1 s spectrum

XPS analysis for PS/CNT (10 wt%) polymer brush nanocomposite: a XPS total survey analysis, b high-resolution C1s spectrum
The PS/CNT (10 wt%) polymer brush, was analyzed via XPS to examine the efficiency of the nanocomposite synthesis process. Figure 6a shows the elemental species C, O, Si, and N which highlights the structural components of the sample. High resolution C1s spectrum (Fig. 6b) shows the binding energy at 285.81 eV, which identifies the bonding configuration of “–CH2-NH-“[41]. The high resolution C1s spectrum (Fig. 6b) also confirms the bond “C-N-C″ in the region 285.7–286.0 eV and C–O at 286.6–286.7 eV [42–43]. The binding energy at 284.5 eV confirms the presence of CNT in the polymer/CNT nanocomposite [44–45] as noted by the high resolution C1s peak (Fig. 6b). Moreover, since the peak for C = O is not noticeable at its characteristic binding energy of 287.9–288.1 eV [43] in the high resolution C1s spectra (Fig. 6b), we conclude that the C = O moiety is absent from the polymer brush nanocomposites. This suggests the scission of this particular bond during the synthesis. Hence, the XPS analysis is in agreement with our proposed reaction scheme for polymer brush synthesis via IEP process.
Morphological characteristics of PS/CNT nanocomposites
TEM image of “NH2-terminated CNT” is shown in Fig. 7a. The CNTs are debundled, once surface modification is implemented, and as expected the tubes appear hollow with no noticeable impurities. Figure 7a reveals that the mean diameter of CNTs is approximately 10–15 nm. Apart from the nature of the dispersion, the image also shows a monolayer around the CNT surface which is in agreement with the FTIR spectra for this sample. The monolayer is suitable for supporting polymerization of styrene throughout the CNT surface to form polymer brush acting as a cross-linking agent.

TEM images of: a “NH2-terminated CNT”; b PS/CNT (1 wt%); c PS/CNT (5 wt%); and d PS/CNT (10 wt%) polymer brush
Figure 7b–d present TEM images for PS/CNT nanocomposites grafted from “NH2-terminated CNT”. We note the consistent and superior dispersion of CNT with no agglomerates within the composite. These confirm the efficiency of CNT surface modification and the polymer synthesis process. The thickness of PS grafted CNT within the composite increased to approximately 50–75 nm as a result of polymer attachment. Images of the composites show that the polymer (PS) is attached throughout the CNT surface and is noticeable for a single nanotube in Fig. 7c. The TEM images of the synthesized nanocomposite in this work confirm the brush structure via attachment of polymer moieties on the CNT surface [46].
Thermal properties of synthesized nanocomposites
The high thermal conductivity of CNT enhances the thermal properties of the composite and prevents degradation of the surrounding polymer when superior dispersions of CNT are obtained [47]. Because of the enhanced thermal properties, polymer/CNT nanocomposites are useful in high temperature applications such as in heat sink for electronics, motors, and tires. We expect to achieve improved thermal properties for the synthesized nanocomposites due to superior dispersion and deagglomeration of CNT with their efficient attachment to polymer brush as shown by TEM images.
TGA measurements for pure PS, pristine CNT, and PS/CNT polymer brush nanocomposites are shown in Fig. 8. We note that for pure PS (Fig. 8b), polymer degradation starts at about 370 °C and degrades substantially within 400–450 °C temperature. Whereas for PS/CNT polymer nanocomposites (Fig.8c–e), slightly greater weight loss is noted at 370–400 °C compared to pure PS. This is due to the presence of greater fraction of shorter polymer chain lengths in PS/CNT nanocomposites in comparison to pure PS. The weight average molecular weight for the polymer is 245,889 and 171,342 g/mol for pure PS and for PS with 1 wt% CNT, respectively. The shorter chains stem from restrictions to polymer chain growth in the presence of CNT [52]. However, at elevated temperatures (>450 °C) when substantial polymer degradation occurs, the PS/CNT composites are found to be less affected compared to pure PS due to enhanced heat dissipation by well-dispersed CNTs and their excellent thermal resistance. Thus, the TGA profiles demonstrate that the PS/CNT polymer brushes have improved thermal stability, which depends on CNT concentration (Fig. 8b). At 800 °C, the residual composite mass fractions were 0.17, 0.28, and 0.44 for CNT contents of 1, 5, and 10 wt%, respectively.

TGA curves of (a) pristine CNT, (b) PS, (c) PS/CNT (1 wt%), (d) PS/CNT (5 wt%), and (e) PS/CNT (10 wt%) polymer brush
The differential change in mass with respect to temperature (dw/dT, wt%/°C) for pure PS and PS/CNT polymer brushes with variable CNT concentrations are shown in Fig. 9 at 350–500 °C, which approximately corresponds to the degradation temperature range for PS. Figure 9b–d shows reduction in sample derivative weight as the CNT concentration increases. For pure PS (Fig. 9a), dw/dT reached about 3 wt%/°C at its maximum, whereas for PS/CNT polymer brush with 10 wt% CNT content (Fig. 9d), the value is below 0.5 wt%/°C. Similarly, the dw/dT values for 1 and 5 wt% composites (Fig. 9b, c) were lower than that for PS and are consistent with the level of CNT content. These confirm the superior thermal stability for the synthesized polymer brush nanocomposites with respect to the base polymer.

Derivative of weight degraded for (a) PS, (b) PS/CNT (1 wt%), (c) PS/CNT (5 wt%), and (d) PS/CNT (10 wt%) polymer brush in the temperature range 350°–500 °C
The addition of CNT to a polymer matrix typically increases the glass transition temperature (T g) when superior attachment of CNT and polymer is achieved [48–49]. This increment in T g is due to the immobility of polymer chains around the nanotube surface via interfacial interaction. Table 2 shows the glass transition temperatures determined using the DSC and subsequent analysis of the heat capacity, C p, at the glass transition zone. The T g of the PS/CNT polymer brush nanocomposites were found to increase substantially compared to pure PS as a function of CNT content: 4.8, 12.2, and 19.7 °C for 1, 5, and 10 wt% CNT in the nanocomposite, respectively. The relatively uniform dispersion and integration of CNT within the polymer matrix, confirmed by TEM data (Fig. 7), were primarily responsible for the enhancement in T g for the nanocomposites. These data further confirm the efficiency of our synthesis scheme.
Gas adsorption characteristics
Increased specific surface area and pore volume of materials correlate with enhanced gas adsorption properties of composites [50]. Well dispersed CNT with high specific surface area in a polymer composite is a promising candidate for gas adsorption. Figure 10 shows the nitrogen sorption/desorption isotherms measured at 77 K for pure PS & PS/CNT polymer brushes and Table 3 presents the BET surface area along with pore volume of the synthesized samples.

Nitrogen adsorption/desorption isotherm of (a) PS, (b) PS/CNT (1 wt%), (c) PS/CNT (5 wt%), and (d) PS/CNT (10 wt%) polymer brush
Table 3 shows substantial increase in BET surface area and pore volume with increase in CNT concentration for the synthesized polymer brush nanocomposites. Thus, with increase in the surface area and porosity, sorption ability of the composite is expected to be enhanced. The nitrogen adsorption/desorption isotherms shown in Fig. 10 support the result of enhancement in gas sorption capacity with increase in CNT content for the composites. These results demonstrate the effectiveness of superior dispersion of CNT coupled with the efficient IEP process.
Yuan et al. [50] reported the synthesis of porous organic polymers (POP) containing highly electron-deficient carborane components and achieved up to 780 mL/g nitrogen adsorption for a sample with a pore volume of 1.12 cm3/g and BET surface area of 1037 m2/g. Yang et al. [51] reported nitrogen adsorption by hypercrosslinked porous organic polymers to be 700 mL/g where the pore volume of the sample was 1.08 cm3/g and the total surface area was about 2065 m2/g. The nitrogen adsorption we achieved for our PS/CNT (10 wt%) polymer brush (950 mL/g) displays improvement over the polymer materials reported under similar conditions. We achieved significantly higher pore volume of 1.48 cm3/g compared to the reported samples based on polymers. Thus, for the polymer/CNT nanocomposites with satisfactory CNT dispersion and attachment with polymer nanostructures are obtained, gas adsorption can be enhanced significantly.
Conclusions
A novel approach was developed for synthesizing PS/CNT polymer brush composites, grafted from surface modified CNT, starting from pristine CNT in conjunction with in situ emulsion polymerization process. The multi-step CNT surface modification approach using oleic acid and subsequent organosilane chemistry was found to be highly efficient in achieving surface modified CNT which enabled superior CNT dispersion and its attachment to the polymer matrix during the polymerization process. The FTIR spectra confirmed the attachment of the reactive functional groups, and XPS analysis confirmed the elemental presence of moieties. The XPS survey also provided data on bond formation which was in agreement with the developed reaction scheme. TEM images showed debundled, uniform dispersion of CNT within the composites, and demonstrated formation of polymer brush grafted from CNT surface. The morphological characterization confirmed excellent integration of the CNT with the polymer matrix. The TGA profiles for the PS/CNT polymer composites validated the synthesis efficiency and displayed consistent and substantial improvement of thermal stability with increase in CNT concentration. Similar observations of glass transition temperature enhancement for the synthesized polymer brush nanocomposites over pure polymer were shown by the DSC results. Sorption/desorption experiments with the synthesized samples showed significantly enhanced BET surface area, pore volume, and gas adsorption/desorption capacity for the PS/CNT polymer brush with increased CNT concentration. The remarkable improvement in BET surface area and pore volume on incorporating CNT indicates the potential for PS/CNT brush in gas separation processes. The trend of property enhancement was found to be consistent even on incorporating a significant fraction of CNT (10 wt%), indicating that the CNT was well-dispersed and satisfactorily attached to the base polymer.
References
Ajayan PM, Linda SS, Braun PV (2003) Nanocomposite science and technology. WILEY-VCH, Weinheim, pp. 31–35
Azzaroni O (2012) Polymer brushes here, there, and everywhere: recent advances in their practical applications and emerging opportunities in multiple research fields. J Polym Sci A 50:3225–3258
Rastogi A, Paik MY, Tanaka M, Ober CK (2010) Direct patterning of intrinsically e-beam sensitive polymer brushes. ACS Nano 4:771–780
Barbey R, Klok HA (2010) Room temperature, aqueous post-polymerization modification of glycidyl methacrylate-containing polymer brushes prepared via surface-initiated atom transfer radical polymerization. Langmuir 26:18219–18230
Ayres N (2010) Polymer brushes: applications in biomaterials and nanotechnology. Polym Chem 6:769–777
Yameen B, Farrukh A (2013) Polymer brushes: promises and challenges. Chem Asian J 8:1736–1753
Jeon NL, Choi IS, Kim NY, Harada Y, Finnie KR, Girolami GS, Nuzzo RG, Laibinis PE, Whitesides GM (1999) The patterned polymer can be used as an etch resist to transfer the pattern into a Si substrate. Appl Phys Lett 75:4201–4203
Husseman M, Malmstreom EE, McNamara M, Mate M, Mecerreyes D, Benoit DG, Hedrick JL, Mansky P, Huang E, Russell TP, Hawker CJ (1999) Controlled synthesis of polymer brushes by “living” free radical polymerization techniques. Macromolecules 32:1424–1431
Matyjaszewski K (2012) Atom transfer radical polymerization (ATRP): current status and future perspectives. Macromolecules 45:4015–4039
Ding S, Floyd JA, Walters KB (2009) Comparison of surface confined ATRP and SET-LRP syntheses for a series of amino (meth)acrylate polymer brushes on silicon substrates. J Polym Sci A Polym Chem 47:6552–6560
Baum M, Brittain WJ (2002) Synthesis of polymer brushes on silicate substrates via reversible addition fragmentation chain transfer technique. Macromolecules 35:610–615
Liu Y, Zhao Y, Sun B, Chen C (2012) Understanding the toxicity of carbon nanotubes. Acc Chem Res 46:702–713
Pangilinan KD, Santos CM, Estillore NC, Rodrigues DF, Advincula RC (2013) Temperature-responsiveness and antimicrobial properties of CNT–PNIPAM hybrid brush films. Macromol Chem Phys 214:464–469
Ha JU, Kim M, Lee J, Choe S, Cheong IW, Shim SE (2006) A novel synthesis of polymer brush on multiwall carbon nanotubes bearing terminal monomeric unit. J Polym Sci A Polym Chem 44:6394–6401
Xu J, Xiao X, Zhang Y, Zhang W, Sun P (2013) RAFT-mediated emulsion polymerization of styrene using brush copolymer as surfactant macro-RAFT agent: effect of the brush copolymer sequence and chemical composition. J Polym Sci A Polym Chem 51:1147–1161
Bech L, Elzein T, Meylheuc T, Ponche A, Brogly M, Lepoittevin B, Roger P (2009) Atom transfer radical polymerization of styrene from different poly(ethylene terephthalate) surfaces: films, fibers and fabrics. Eur Polym J 45:246–255
Zou YQ, Kizhakkedathu JN, Brooks DE (2009) Surface modification of polyvinyl chloride sheets via growth of hydrophilic polymer. Macromolecules 42:3258–3268
Gorman CB, Petrie RJ, Genzer J (2008) Effect of substrate geometry on polymer molecular weight and polydispersity during surface-initiated polymerization. Macromolecules 41:4856–4865
Marutani E, Yamamoto S, Ninjbadgar T, Tsujii Y, Fukuda T, Takano M (2004) Surface-initiated atom transfer radical polymerization of methyl methacrylate on magnetite nanoparticles. Polymer 45:2231–2235
Fan XW, Lin LJ, Dalsin JL, Messersmith PB (2005) Biomimetic anchor for surface-initiated polymerization from metal substrates. J Am Chem Soc 127:15843–15847
Yang Q, Wang L, Xiang WD, Zhou JF, Tan QH (2007) Preparation of polymer-grafted carbon black nanoparticles by surface-initiated atom transfer radical polymerization. J Polym Sci A Polym Chem 45:3451–3459
Thostenson E, Li C, Chou T (2005) Nanocomposites in context. Compos Sci Technol 65:491–516
Vaisman L, Wagner HD, Marom G (2006) The role of surfactants in dispersion of carbon nanotubes. Adv Colloid Interf Sci 128–130:37–46
Kong H, Gao C, Yan D (2004) Functionalization of multiwalled carbon nanotubes by atom transfer radical polymerization and defunctionalization of the products. Macromolecules 37:4022–4030
Hilding J, Grulke EA, Zhang ZG, Lockwood FJ (2003) Dispersion of carbon nanotubes in liquids. J Dispers Sci Technol 24:1–41
Khan MU, Gomes VG, Altarawneh IS (2010) Synthesizing polystyrene/carbon nanotube composites by emulsion polymerization with non-covalent and covalent functionalization. Carbon 48:2925–2933
Sunkara V, Park DK, Hwang H, Chantiwas R, Soper SA, Cho YK (2011) Simple room temperature bonding of thermoplastics and poly(dimethylsiloxane). Lab Chip 11:962–965
Germain J, Hradil J, Jean MJF, Svec F (2006) High surface area nanoporous polymers for reversible hydrogen storage. Chem Mater 18:4430–4435
Sun YP, Fu K, Lin Y, Huang W (2002) Functionalized carbon nanotubes: properties and applications. Acc Chem Res 35:1096–1104
Min Z, Zhang X, He X, Chen L, Zhang Y (2010) A facile method to coat mesoporous silica layer on carbon nanotubes by anionic surfactant. Mater Lett 64:1383–1386
Bulla DAP, Morimoto NI (1998) Deposition of thick TEOS PECVD silicon oxide layers for integrated optical waveguide applications. Thin Solid Films 334:60–64
McMurry J, John E (1992) Organic chemistry, 3rd edn. Wadsworth, Belmont ISBN 0-534-16218-5
March J (1992) Advanced organic chemistry: reactions, mechanisms, and structure, 4th edn. Wiley, New York ISBN 0-471-60180-2
Peng H, Alemany LB, Margrave JL, Khabashesku VN (2003) Sidewall carboxylic acid functionalization of single-walled carbon nanotubes. J Am Chem Soc 125:15174–15182
Bronstein LM, Huang X, Retrum J, Schmucker A, Pink M, Stein BD (2007) Influence of iron oleate complex structure on iron oxide nanoparticle formation. Chem Mater 19:3624–3632
Ding X, Zhao J, Liu Y, Zhang H, Wang Z (2004) Silica nanoparticles encapsulated by polystyrene via surface grafting and in situ emulsion polymerization. Mater Lett 58:3126–3130
Lechevallier S, Hammer P, Caiut JA, Mazeres S, Mauricot R, Verelst M, Dexpert H, Sidney J, Ribeiro L (2012) Dexpert-Ghys APTES-modified RE2O3:Eu3+ luminescent beads: structure and properties. Langmuir 28:3962–3971
Akiyama Y, Fujit S, Senboku H, Rayner CM, Brough SA, Arai M (2008) An in situ high pressure FTIR study on molecular interactions of ketones, esters, and amides with dense phase carbon dioxide. J Supercrit Fluids 46:197–205
Briggs D, Seah MP (1990) Practical surface analysis, 2nd edn. Wiley, Chichester
Beamson G, Briggs D (1992) High resolution XPS of organic polymer, the scienta ESCA300 database. Wiley, Chichester
Emmanuel N, Lissouck O, René NNG, Hyppolite L, Christine P, Regis A, Louis-Max NM (2013) Green gluing of tropical wood part III: X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR) analysis of Frake and Ayous green wood’s glue line. AFM, France
Truica-Marasescu F, Wertheimer MR (2008) Nitrogen-rich plasma-polymer films for biomedical application. Plasma Process Polym 5:44–57
Briggs D, Brewis DM, Dahm RH, Fletcher IW (2003) Analysis of the surface chemistry of oxidized polyethylene: comparison of XPS and ToF-SIMS. Surf Interface Anal 35:156–167
Gorham JM, Woodcock JW, Scott KC (2015) Challenges, Strategies and Opportunities for Measuring Carbon Nanotubes within a Polymer Composite by X-ray Photoelectron Spectroscopy. NIST Special Publication, 1200–10
Petersen EJ, Lam T, Gorham JM, Scott KC, Long CJ, Stanley D, Sharma R, Liddle JA, Pellegrin B, Nguyen T (2014) Methods to assess the impact of UV irradiation on the surface chemistry and structure of multiwall carbon nanotube epoxy nanocomposites. Carbon 69:194–205
Advincula R, Brittain WJ, Caster KC, Ruhe J (2004) Polymer brushes: synthesis, characterization, applications. VCHWiley, Weinheim
Song PC, Liu CH, Fan SS (2006) Improving the thermal conductivity of nanocomposites by increasing the length efficiency of loading carbon nanotubes. Appl Phys Lett 88:153111–153113
Farbod M, Mobini N (2014) Physical properties, thermal stability, and glass transition temperature of multi-walled carbon nanotube/polypyrrole nanocomposites. Compos Interfaces 21:737–747
Sterzynski T, Tomaszewska J, Piszczek K, Skorczewska K (2010) The influence of carbon nanotubes on the PVC glass transition temperature. Compos Sci Technol 70:966–969
Yuan S, White D, Mason A, Liu D (2013) Porous organic polymers containing carborane for hydrogen storage. Int J Energy Res 37:732–740
Yang X, Yu M, Zhao Y, Zhang C, Wang X, Jiang J-X (2014) Remarkable gas adsorption by carbonized nitrogen-rich hypercrosslinked porous organic polymers. J Mater Chem A 2:15139–15145
Khan MU, Darestani MT, Gomes VG (2015) Structure and electrochemical properties of polystyrene/CNT nanocomposites. J Solid State Electrochem 19:3145–3156
Acknowledgments
Support from the University of Sydney is acknowledged for conducting part of the research. Assistance with the XPS analysis from UNSW is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Miftah U. Khan, Kakarla Raghava Reddy, and Theedanai Snguanwongchai contributed equally to this work.
Rights and permissions
About this article

Cite this article
Khan, M.U., Reddy, K.R., Snguanwongchai, T. et al. Polymer brush synthesis on surface modified carbon nanotubes via in situ emulsion polymerization. Colloid Polym Sci 294, 1599–1610 (2016). https://doi.org/10.1007/s00396-016-3922-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-016-3922-7