
Abstract
A series of hydrophobically modified sodium polyacrylates (NaPACn) grafted with linear alkyl side chains with 10 to 18 atoms of carbon (n) has been synthesized. FT-IR and 1H-NMR characterization proved that the alkyl grafts were attached to the polymer backbone via the amide link and the grafted amount was around 3 % (mol). The aqueous solutions of NaPACn were investigated by a score of methods including viscosity, fluorescence (steady-state and time-resolved), refractive index, and dynamic rheology. The results revealed that each NaPACn has a characteristic overlapping concentration (c*), which lowers as the grafted chain is more hydrophobic. Above c*, the viscosity deviates from that of sodium polyacrylate (NaPA) and solutions of high viscosity are obtained. The phenomenon is more evident as the alkyl chain increases in length and was attributed to the cross-association of the grafts. The NaPAC16 and NaPAC18 are viscoelastic solids, whereas the C10–C14 derivatives are viscoelastic liquids. The comportment of NaPAC16 and NaPAC18 is sustained by the positive yield stress and the high relaxation time values. Steady-state fluorescence unveiled intracoil hydrophobic microdomains (HMD), whereas dynamic fluorescence and refractive index showed the existence of both intra- and intercoil aggregates.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Water-soluble associative polymers (APs) are a class of compounds with hydrophilic backbone decorated with a few hydrophobic groups. If the hydrophobic groups are pendant, they can be randomly or bunch grafted, without or with spacer, and the resulted compounds are denoted as side-chain associative polymers. When the parent polymers are modified only at the ends, one obtains telechelics. The hydrophilic polymer can be natural or synthetic as well as neutral or charged. The hydrophobic group can be derived either from hydrocarbons or fluorocarbons. The synthesis of APs can be done by modification of a commercial polymer or by copolymerization of a water-soluble monomer with a hydrophobic one [1, 2].
In water, above a threshold concentration, the hydrophobic moieties undergo association and APs show peculiar properties such as spectacular increase of viscosity [3], gelation [4], drag reduction [5], and elasticity [6]. They result from the reversible association of grafts and make APs distinguishable from regular polymers without associative groups. As a result, APs triggered applications in many fields for instance, personal care products, cosmetics [7], carriers for protein, DNA and drugs [8–13], waterborne paints [14, 15], wastewater purification [16], enhanced oil recovery [17–20], etc.
On the other hand, the acquired data reveal that the properties of hydrophobically modified polymers are influenced by the molecular structure, the amount of grafted hydrophobic side chains, the distribution of grafts, and the spacer linking the hydrophilic and the hydrophobic parts [21–27]. Besides the mentioned papers, there is a huge work on the synthesis, characterization, properties, and applications of different classes of hydrophobically modified polymers. In this study, we are only interested to find out new information on the behavior of randomly modified with linear C10–C18 alkyl chains, so the literature we will refer to is mostly on this kind of polymers.
We are interested in these polymers because the specific behavior of hydrophobically modified sodium polyacrylates (NaPACn) is given by the interplay between the electrostatic repulsions of the carboxylate groups and the attractions of the hydrophobic grafts. In aqueous solution, the hydrophobic groups try to avoid the contact with water and form intra- and intermolecular aggregates. Strong associations are obtained by increasing the alkyl chain length, because the modified polymers are less soluble in water [22, 27, 28].
Hydrophobically modified polyacrylates in aqueous solutions were intensively investigated in the 1990s mainly by the Iliopoulos group [21, 22, 28]. For NaPAC18, it has been observed that above c*, the viscosity increases several orders of magnitude, both in water and in saline solutions [28]. The phenomenon was attributed to the interchain cross-linking of the grafted hydrophobic groups. Due to the very high tendency to aggregate, the microstructure of the high molecular weight NaPACn was hard to elucidate by 13C-NMR [21, 29]. To address this, NaPACn with low molecular weight was used [22]. The results by fluorescence, electrical conductivity, and 13C-NMR revealed that both the length of the alkyl chain and the modification degree are key factors in AP association. The effect of hydrophobicity on the viscosity of modified polyacrylates was also studied [30]. It has been found out that the alkyl chain must be longer than 8 atoms of carbon to detect any viscosity change. Other evident outcomes were that at low NaCl concentration the aggregation of hydrophobic groups is inhibited by the electrical repulsion of the backbone charges, whereas at high salt levels, the repulsions are screened and intramolecular association predominates. In spite of numerous studies devoted to APs, the formation of intra- and intercoil aggregates and their interplay is still a matter of debate.
Another issue was the structures developed by hydrophobically modified poly (sodium acrylates) in aqueous solution. By attaching fluorinated alkyls to PAA and using 19F NMR, it has been revealed that the associative polymers are structurally heterogeneous in water with free (F) and aggregated (A) pendant chains [31]. It was also found out that the proportion of F and A depends on polymer concentration [32]. For example, at a concentration of 0.3 wt.%, the grafts are about 40 % free, and up to 50 % as intracoil partial aggregates (PA) The F chains are fully and constantly exposed to water. In the PA state, a small number of side chains belonging to the same associative macromolecule are put together and build up hydrophobic microdomains. At 5.0 wt.% associating polymer, the proportion of F is unchanged, but intercoil aggregates do appear. They have micelle-like structure, containing about 25 side chains which belong to different associative polymers.
However, despite the fact that these polymers have been intensively studied, their detailed behavior in aqueous solution is not completely understood. Therefore, in the present study, we synthesized a series of water-soluble hydrophobically modified polyacrylates. They were obtained by grafting a parent PAA with C10–C18 linear alkyl side chains and were characterized by FT-IR and 1H-NMR. The work aims to acquire new data about the sodium salts of these compounds in water by using viscometry, dynamic rheology, fluorescence, and refractometry. Viscosity was used to find out the c* of each NaPACn. Dynamic fluorescence and refractometry were employed to explore the concentrations at which intra- and intercoil aggregates do form. The refractometric method is originally proposed to characterize associative polyacrylates because the technique is simple, accurate, expedient, and requires only minute material. The dynamic rheology tests revealed how important is to know how APs behave under deformation, especially at concentrations higher than c* and what elasticity they have. These investigations revealed that only by corroborating results from several techniques, it is possible to tailor materials with suitable characteristics for target applications.
Experimental section
Materials
All the chemicals used were of reagent grade. Poly (acrylic acid) (PAA) was supplied by Wako Pure Chemical Industries Ltd., Osaka, Japan, as a 25 % (wt.) aqueous solution, and the solid PAA was recovered by freeze-drying. n-Alkyl amines [decylamine (DA), dodecylamine (DDA), tetradecylamine (TDA), hexadecylamine (HDA), and octadecylamine (ODA)], dicyclohexylcarbodiimide (DCC), N-methyl-2-pyrrolidinone (NMP), sodium hydroxide, and pyrene were provided by Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany. All the chemicals were used without further purification. Solutions for measurements were prepared with water produced by a Millipore Simplicity UV system.
The SEC analysis of PAA
PAA was transformed in methyl ester for analysis by SEC. Recent work showed the difficulty and numerous errors of direct measurement of molecular weight of PAA by SEC [33]. On the other hand, esterification enables easier and precise measurement of molecular weight by SEC [34]. We used an earlier developed protocol, based on the Mitsunobu route, for the transformation of PAA in poly (methyl acrylate) [35]. The SEC analysis was performed at 25 °C in DMF with 50 mmol/L LiCl. The flow rate was of 1 mL/min, using the 7.5 × 50 mm PLgel 10 μm guard column in series with the 7.5 × 300 mm PLgel 10 μm MIXED-B analytical column, both from Agilent. Detection was done by Wyatt Heleos II MALS at 30 °C in series with Waters 410 RI at 35 °C/930 nm. The number average molecular weight (M n) recalculated of the PAA was of 166,000 Da and the polydispersity index M w/M n of 3.4 (see Supplementary Material).
Synthesis of NaPACns
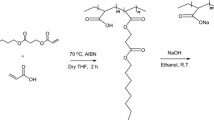
The hydrophobically modified polyacrylates were obtained by adapting the method originally described by Magny [36]. An example of synthesis is given below. A three-neck 250-mL round-bottom flask fitted with vertical condenser, magnetic stirring bar, and nitrogen inlet was placed into an oil bath, atop of a magnetic stirrer heating plate with temperature fixed at 60 °C. PAA 6 g (0.083 mol) and 100 mL of NMP were introduced into the flask. The stirring, heating, and nitrogen were put on. After the dissolution of PAA (about 24 h), 0.4728 g of DDA (0.0025 mol) and 0.635 g of DCC (0.0031 mol, 20 % excess) were added, and the mixture was left to react for 48 h. Then, the mixture was cooled to room temperature and filtered to remove the crystals of dicyclohexylurea. The product was neutralized and precipitated in 250 mL of NaOH (40 %). The precipitate was filtered and washed with 50 mL of hot NMP and methanol. The washing was repeated three times. The recovered precipitate was dissolved in water and dialyzed against Millipore water using dialysis membranes of cellulose acetate with molecular weight cutoff (MWCO) of 6000. The modified polymers were recovered by freeze-drying. Using this procedure, we obtained five samples of hydrophobically modified polyacrylates containing a theoretical amount of 3 mol% grafted n-alkyl side chains. Their chemical structure is shown in Scheme 1.

Molecular structure of NaPACn, x = grafted amount (see Table 1)
Methods
FT-IR spectra were collected on a Thermo Scientific Nicolet iN10 microscope. The resolution was set at 4 cm−1 and 32 scans were averaged for each spectrum.
1H-NMR spectra were carried out at 30 °C on a Varian Gemini 2000 spectrometer operated at 300 MHz. The sodium salts of polymers were dissolved in deuterium oxide (D2O).
13C-NMR spectra were recorded on a Gemini 2000 BB at 75 MHz, using 10 s recovery delay, 90 deg. pulse, 1 H decoupler mode “yyy,” 20 Hz rotation, and averaging 4096 transients, in D2O, using methanol as internal reference (49.5 ppm [37]).
The viscosities were measured with a SV-10 Vibro Viscometer (A&D Company Ltd.). The viscometer has two thin sensor plates driven with electromagnetic force at 30 Hz. They have a constant sine-wave vibration in reverse phase like a tuning-fork. Calibration was done with purified water. The temperature (23 °C) was controlled by Julabo F12-ED equipment.
Steady-state fluorescence spectra were recorded out on a Horiba Jobin Yvon FluoroMax 4P spectrofluorimeter. The slits were of 2 nm for excitation and 1 nm for emission. The decay measurements were performed on Edinburgh Instruments FLSP 920 spectrofluorimeter using the time-correlated single photon counting technique. The excitation source was a picosecond pulsed diode laser at 338.0 nm. The fluorescence emission was collected at 371 nm. The decay curves were solved in terms of a multiexponential model with the instrument analysis software. The lifetimes are obtained using a nonlinear least squares fitting program based on the Marquardt-Levenberg algorithm. The quality of the results was judged by the value of reduced chi-square, weighted residuals, and autocorrelation function of the weighted residuals. A fit was considered good when χ 2 is within the 0.8 to 1.2 range, and the plots of weighted residuals and autocorrelation function were randomly distributed around zero [38]. By these criteria, the decay curves were best fitted with a tri-exponential function.
A digital Abbe refractometer AR2008 (KRUSS, Germany) was used for refractive index measurements.
The dynamic rheology tests were carried out on a Kinexus Pro Rheometer (Malvern, UK). The samples were placed into two flat plates, the diameter of bottom plate was 55 mm, and that of the top one was 50 mm. The measurement type was a dynamic frequency sweep test in the 0.1–10 Hz range. All measurements were made within the linear viscoelastic regime. The temperature (23 °C) was controlled by Julabo CF41 cryo-compact circulator.
The Casson model (Eq. 1), describing the shear viscosity measurements for Newtonian or non-Newtonian materials, was implemented into the equation for dynamic shear measurement (Eq. 2) [39, 40].
where τ (Pa) represent the shear stress, γ (s−1) the shear rate, G″ (Pa) the loss modulus, Ω (rad/s) the angular frequency (Ω = 2 π f, where f (Hz) is the frequency), and k, k′ are Casson constants.
The theoretical relaxation time (λ) for dynamic shear measurement [39–41] was calculated with Eq. 3:
where η* (Pa·s), η′ (Pa·s), G′ (Pa), and λ (s) are the complex viscosity, dynamic viscosity, storage modulus, and relaxation time, respectively.
All the measurements were performed at room temperature (around 23 °C), except the 1H-NMR and 13C-NMR spectroscopy.
Preparation of NaPA and NaPACn solutions
The solutions were prepared at room temperature by dissolving the weighted amount of unmodified or modified polymer in water under magnetic stirring. For fluorescence measurements, the solutions were prepared by dissolving the polymers in pyrene saturated water having a pyrene concentration of 6.7 × 10−7 M. The samples were left to equilibrate overnight.
Results and discussion
Characterization of NaPACn
The NaPACn samples were qualitatively assessed by FT-IR and quantitatively evaluated by 1H-NMR spectroscopy. Because of overlapping the carbonyl band in Na salt over the amide I and II bands, NaPA and NaPACn could not be used for ascertaining the amide formation by FT-IR. Instead, we used the H-form of the polymers. Figure 1 illustrates the FT-IR spectra of PAAC18, PAAC10, and parent PAA. The bands around 1630 and 1550 cm−1 (shoulders on the carbonyl band of PAA) correspond to amide I and amide II vibrations in the spectra of PAAC10 and PAAC18. They are absent in the spectrum of PAA and prove that the polymer analogous reaction was successfully accomplished. The amide I and amide II characteristic vibrations were present in the spectra of all modified polymers.

FT-IR spectra of PAA, PAAC10, and PAAC18
1H-NMR spectroscopy gives quantitative information regarding the modification degree of PAA. Figure 2 comparatively shows the spectra of NaPA and NaPAC16. The spectra of all the others NaPACn are similar (data not shown). The peaks around 3.1 (corresponding to the methylene protons in α position from the amide group) and 0.8 ppm (attributed to the methyl protons at the end of the alkyl chain) confirm that PAA is successfully labeled with alkyl chains. The (CH2)n protons from the alkyl chain overlap with the backbone protons of PAA in the region 1–2.5 ppm and cannot be used to calculate the modification degree.

1H-NMR spectra of NaPA and NaPAC16
The modification degree was calculated taking into account the integral at 0.8 ppm corresponding to the CH3 protons from the alkyl chain of the amide, and the signal between 1 and 2.4 ppm of the protons from PAA backbone and the methylene side chain protons (see Fig. 2).
Table 1 summarizes the obtained degrees of grafting (x). The results show that NaPAC10 and NaPAC12 have closer grafting degrees to the theoretical value. Smaller grafting degrees, but higher than 2.0 % (molar), are obtained for NaPAC16 and NaPAC18, whereas NaPAC14 has the smallest amount of grafted side chains. The data are different from those obtained at PAA modification with aryl-alkylene-amines [42]. The differences can be attributed to the lower reactivity of fatty amines.
We investigated the association degree of the NaPACns by 13C-NMR. The results are listed in the Supplementary Material (SM). The spectra of the NaPACn at usual concentrations used in the present study were noisy and signals flat and wide for any interpretation, because of the wide molecular weight distribution (M w/M n = 3.4) and high number average molecular weight (166,000 Da). Using a higher polymer concentration, of about 8 % by weight, at room temperature or cooling at about 10 °C, we detected rather sharp signals of the associated grafts (methyl at 14.00 ppm and methylene at 22.68 ppm) (Fig. SM1 for NaPAC12). No signals of free grafts were detected. Gradually rising the temperature, the signals became wider and several species appear upfield showing a higher mobility of the chains, hence lesser association extent. At 65 °C, at least three different species can be detected distributed from extended domain of association (almost the same as at room temperature) to free species (Fig. SM2). At this relatively elevated temperature for aqueous systems, the associated species in large domain still prevail (about 55 % calculated from integrals in Fig. SM2) and only a very small proportion, about 18 %, of the grafts are free. We cycled the temperature and the entire pattern is reversible but with an equilibration of minimum 48 h. Moreover, the initial spectrum, at the first dissolution in deuterated water, evolves continuously at room temperature during the first 72 h. We attributed this behavior to the wide distribution together with the high average molecular weight of our polymers, in contrast with previous studies reporting on polymers having lower molecular weights and narrower distributions [21, 22, 29]. This behavior can be explained based on a memory effect, the chains keeping the entanglement from the solid state.
Viscosity
In the “Introduction,” we emphasized that the most important application of water-soluble associative polymers is to control the viscosity of water-borne fluids [14–20]. We measured the viscosity on our NaPACn specimens and on NaPA as well. The obtained results are presented in Fig. 3.

The change viscosity vs. polymer concentration for NaPA and NaPACn aqueous solutions (the lines are only guide for the eyes)
For NaPA, the viscosity increases in the dilute regime, but in the semi-dilute one, it grows very slowly denoting a typical behavior of “weak” polyelectrolyte [1]. The results for NaPACns are completely different. At low polymer concentration, all the curves deviate from that of NaPA. At first, the viscosity grows suddenly. Then, instead of flattening as for NaPA, it continues to rise with a slope that is steeper as the grafted alkyl chain is longer. This conduct is resembling to that of associative polymers [27, 28] and demonstrates that our modified polyacrylates belong to this class of compounds.
To discuss our viscosity data, one has to refer to previous works on associative polyacrylates published by Iliopoulos et al. [21, 31, 32, 36]. Addressing the problem of grafts distribution along the polymer chain, the authors proved that hydrophobically modified poly(sodium acrylates), prepared by grafting of PAA (having M n of 25,000 Da) in NMP, have random distribution of the hydrophobic groups along the polymer chain [21, 36]. Since our associative polymers were synthesized by the same method, it is very likely to assume that they have similar random distribution of the pendant alkyl chains.
A careful analysis by Newton method of the curves in Fig. 3 allowed us to determine the point on the NaPACns concentration scale at which the slope of viscosity changes. This point is the overlapping concentration (c*) and represents the concentration at which the intercoil aggregation occurs. The phenomenon primarily results from the intermolecular interaction of grafted alkyl side chains entailing intercoil aggregates. In addition, with the increase of the alkyl chain length, more intermolecular interactions do occur, leading to higher viscosities and more stable structures.
The obtained c* values of grafted polymers determined through the Newton method are presented in Table 2. One may observe that c* decreases as the alkyl chain length becomes longer (i.e., as the hydrophobicity increases) like was also observed for hydrophobically modified poly(acrylic acid) [43]. The decrease is almost linear with the number of carbon atoms of the alkyl chain, being similar with the trend previously observed on surfactant and surfactant–polymer systems [44–47]. If the c* is expressed as molar label content (c l*, see Table 2), one can observe that this concentration also decreases with the alkyl chain length, but the C14 is the exception. It appears that this particular NaPACn needs lower label content to aggregate. This is not the true because the intercoil aggregate formation is determined by how strong the hydrophobicity is (the length of alkyl chain) and to a lesser extent by its amount. The label content is important only on the intracoil aggregation as we will show below. We consider that C14 is an exception to the general behavior of the investigated NaPACn because it has a lower label degree obtained by synthesis (perhaps there were some experimental errors in the synthesis for this batch). In the following, we will account this and discussion will be done accordingly.
Fluorescence
Compelling information on the hydrophobically modified polyacrylates in solution can be obtained by pyrene fluorescence. Pyrene is by far the most popular dye used in fluorescence experiments because of sensitivity to the polarity of the host microenvironment and long lifetime [48–51]. In steady-state fluorescence, the ratio of the first (I1, λ = 371 nm) to the third (I3, λ = 381 nm) vibronic peak in the emission spectrum of pyrene (the so-called polarity index, I1/I3) gives information about the polarity of the medium where the probe is located (for example, I1/I3 is 1.8–1.9 in water and 0.6 in hydrocarbons) [49]. The lifetimes in time-resolved fluorescence bring supplementary information because pyrene has longer lifetimes in hydrophobic environments than in water [52, 53].
Figure 4a shows the change of polarity index with polymer concentration. For NaPA, the polarity index is almost equal to 1.7 within the whole range of investigated polymer concentration. This proves that the NaPA does not have hydrophobic microdomains to accommodate pyrene inside. The case for NaPACns depicted in the same figure is totally different. At low polymer content, the I1/I3 is almost constant irrespective of alkyl chain length. As the polymer concentration increases, I1/I3 decreases, meaning that pyrene is located in a less polar medium. The polymer concentration at which the decrease occurs depends on the label length and is specific for each NaPACn. In surfactant systems, the decrease in I1/I3 points out the onset of micelle formation [48, 49, 54, 55]. In our case, the decline of polarity index is related to the formation of hydrophobic domains by the aggregation of grafted side chains. Because the polymer concentrations are much lower than c* (which marks the formation of intercoil microdomains), the graft aggregation should occur inside the same polymer coil. This concentration at which the polymers started to form intracoil aggregates was not previously revealed by steady-state fluorescence. In order to elucidate the mechanism of association, NaPaCn of low molecular weight, in aqueous solutions were investigated by fluorescence quenching of pyrene, electrical conductivity, and 13C-NMR [22]. The results on anionic polyelectrolyte was compared with those obtained on the cationic surfactant dodecytrimethyl ammonium chloride (DTAC). The study revealed that the hydrophobicity (both alkyl chain length and modification degree) plays a very important role in the polyelectrolyte aggregation, and the association is less cooperative than that of ionic surfactants. The concentration at which the polymers started to form intracoil aggregates could not be accurately determined by steady-state measurements [22]. This is because the authors chose to take the aggregation concentration similar to that for DTAC which has a high CMC (2 × 10−2 M or around 0.5 %). However, there is no general agreement on the detection of the concentration at which an amphiphilic compound starts to aggregate in aqueous solution by steady-state fluorescence measurements (at the beginning, at the middle, or at the end of the polarity index vs surfactant concentration curve decline) [56]. Anyway, the aggregation concentration value depends on the method employed [45, 46, 57]. Generally, the values obtained by spectroscopic methods (dye solubilization, fluorescence) are lower than those given by “classical” methods (surface tension, electrical conductivity) due to the interaction of the dye/fluorescent probe with the monomeric surfactant [58].

The variation of polarity index with polymer concentration (a) and label content (b) for the investigated systems. Inlet: Detailed view of the I1/I3 decrease at low label concentrations (the lines are only guide for the eyes)
For the present systems, as expected, the longer the grafted alkyl chain, the lower polymer concentration for intracoil aggregation of labels, i.e., the concentration of intracoil aggregate formation follows the order: C10 > C12 > C14 > C16 > C18. One may see from Fig. 4 (especially from the Figure’s inlet) that the concentrations are very close for C10 and C12 and for C16 and C18 because the label contents are very similar for the respective polymers (see Table 1) and, as was already mentioned, the amount prevails hydrophobicity in the intracoil association. Even the decline is very similar in-between the two pairs of polyacrylates as the label content is alike. An unexpected fact is that the polarity index does not level off as for the micellar systems [48, 55–57, 59; Aricov L, Băran A, Simion EL, Gîfu IC, Anghel DF (30 August to 2 September 2015) fluorescence investigation of hydrophobically modified polyacrylates in aqueous solution and in the presence of surfactants; International Symposium on Amphiphilic Polymers, Network, Gels and Membranes, Budapest, Hungary]. Instead, it decreases to values lower than those of sodium dodecyl sulfate anionic surfactant, but close to those of fatty alcohols determined by us, 0.87 and 0.55 for 1-decanol and 1-dodecanol, respectively. This does not imply that the C10 and C12 intercoil aggregates are more hydrophobic than those of the others polyacrylates. It means that these aggregates are less tightly packed and the probe more easily reaches them and penetrates inside as the viscosity is lower for the two polymers (see Fig. 3). The behavior of I1/I3 for the NaPAC14 is different: it starts to decrease from a label concentration in-between those of C12 and C16 (according to the hydrophobicity), but the decline is smooth, and extends over almost a decade of concentration. This can be attributed to a less cooperative association of the polymer that has the lowest label content. It means that a smaller hydrophobic attraction should prevail almost the same electrostatic repulsion as in the other NaPACns [59] (see also figure of pH in the Supplementary Material).
It is known that pyrene has fluorescence lifetimes which depend on environment [52], so new insights into the aggregation of NaPACns were obtained by dynamic fluorescence measurements. Figure 5 illustrates the decay curve of NaPAC12, and the derived lifetimes of this polymer together with the lifetimes of the other NaPACns are given in Fig. 6. (Other decay curves and their fittings, together with the lifetimes, pre-exponential factors and reduced chi-square are presented in the Supplementary Material. For a better understanding of pyrene lifetimes in the investigated systems, their variation on polymer concentration is also presented in the Supplementary Material.).

The fluorescence decay curves for 0.2 % wt. NaPAC12 solution

The modification of fluorescence lifetimes with the label content (the lines are only guide for the eyes)
As mentioned in the “Experimental section,” the decay curves were best fitted with tri-exponential functions. The three lifetimes are as follows: the short one, τ1, belongs to the monomer which forms excimer (MAGRE [60]); the medium one, τ2, is attributed to the partially overlapped excimer; and the longest one, τ3, is ascribed to the isolated monomer [58, 60]. At low polymer content, the lifetimes of probe monomers are small because there are very few appropriate domains where they locate. The probe could be crowded in such microdomains and both quenching and excimer formation can occur (τ2 has high values). As more NaPACn is introduced into the system, more and larger hydrophobically microdomains (HMD) are formed and the probe localizes inside. The increase of the HMD number and size leads to increasing the distance between two probe molecules, diminishing the quenching and excimer formation. Therefore, both τ 1 and τ 2 decrease. The crowding of pyrene molecules into the small HMDs (their self-quenching) makes also τ 3 to have low values. At high NaPACn concentrations, the intercoil microdomains do form and τ 3 rises till a polymer concentration specific for each NaPACn, then it drops. This specific concentration is close to that at which viscosity starts to increase, i.e., c*. An explanation for the small values of τ 3 beyond c* could be that, at these high concentrations, extended interchain aggregates form, the solution viscosity becomes very high, and so the probe hardly reaches the HMD. Thus, short lifetime of pyrene monomers, but long excimer lifetime at low polymer content, means that there are some soft intracoil domains. The increase of isolated monomer lifetime and concomitantly the decrease of both MAGRE and excimer lifetimes imply more and/or viscous intracoil aggregates. An increase in polymer content and a depletion of free monomer lifetime mean both a higher viscous environment and/or intercoil aggregates (i.e., polymer content exceeds c*). Beside the abovementioned comments, there is no general conclusion on the behavior of τ 1 and τ 2 and system hydrophobicity, as the MAGRE behavior determines that of the excimer. Anyway, it seems that at high polymer content (above c*), both lifetimes are larger for the less hydrophobic polymers (except C14). This can be attributed also to the system viscosity which, at high polymer level and hydrophobicity, impedes the excimer formation or may impose a short distance between probes leading to the self-quenching. On the other hand, the free monomer lifetime behavior depends on label content. For C10 and C12, as the concentration approaches c* and the system became more viscous, the lifetime starts to decrease. The higher value for C10 also reveals that the intercoil domains are less tightly packed than those of C12 polymer (the C10 viscosity is lower than that of C12; see Fig. 3). Though at concentration lower than c* τ 3 depends of label quantity, it is also determined by the hydrophobicity for C14–C18 polymers: it is larger as the hydrophobicity increases, with the largest value for NaPAC18. This was also observed by time-resolved fluorescence quenching on 10 % NaPACn (n = 12 and 18) with MW of 5000 and graft degree of 5 % [22]. The large values and smaller difference between pyrene monomer lifetime in those two polymers solutions can be due to the method (the quencher may interact with the polyacrylates, so the probe lives in different media). Also, those polymers had small MW and label quantity was higher than in our study. The highest τ 3 value for our NaPAC18 may be due to the more tightly and perhaps larger microdomains (intra- and intercoil) formed by this polymer, so the probe better accommodates inside. As mentioned above, when the systems become more viscous, the probe hardly approaches the HMDs leading to a decrease of pyrene monomer lifetime.
Refractometry
Refractometry is a method largely used in the study of surfactant solutions [61–65]. It is able to detect changes of micelle size and shape and to determine the regular and the second CMC of the surfactants. The application of refractometry to surfactants relays on its dependence on concentration. For instance, when very little surfactant is present in water, it locates at the air-solution interface and the refractive index is almost constant or changes very little. As more amphiphile is introduced into system, the surface saturates and the compound begins to aggregate leading to the increase of refractive index. As more and more surfactant is added into solution, micelles change the size and shape entailing the change of the refractive index. The technique was applied not only to surfactant systems but also to a poly(vinyl pyrrolidone-vinyl acetate) hydrophobically associating copolymer, and c* was originally determined [66].
Figure 7a illustrates the refractometric results obtained on our NaPACns. At low polymer content, where there is very little label aggregation, the refractive index is almost constant. As intracoil aggregates start to form, the refractive index begins to increase. It has to be stressed that this concentration coincides or is very close to that at which I1/I3 starts decreasing (see Fig. 7b; the curves for the other investigated systems are similar and are presented in the Supplementary Material). As more polymers are added, the repulsive electrostatic forces loose the coil, some intrachain domains may open, and the labels start to form intercoil aggregates. These new type of aggregates may have different interior viscosity and structure that leads to the increase of both the refractive index and viscosity (see Fig. 3). This concentration at which the refractive index has another change of slope is very close to c*. Beyond it, more and possibly larger intercoil aggregates are formed leading to higher viscosity and refractive index values. It should be mentioned that at the highest polymer concentrations used in the study, the NaPACn are gels.

The variation of refractive index with polymer concentration for the investigated systems (a) and a comparison with polarity index variation for NaPAC10 (b) (the lines are only guide for the eyes)
Dynamic rheology
We performed dynamic rheological measurements below c*, at twice c* and at a polymer concentration of 3 % (wt.). The idea was to acquire information about the viscoelasticity of our polymers. The storage modulus (G′ elastic component) and the loss modulus (G″ viscous component), rise with the alkyl chain length and concentration, for all performed tests. Below c*, the viscoelastic behavior of the polymers was liquid like (G′ < G″, data not shown). At twice c*, only NaPAC18 has solid-like response (G′ > G″, see Supplementary Material). For this reason, we chose for further rheological tests a polymer concentration of 3 % (wt.), which is much higher than c*. The results are presented in Fig. 8. For NaPA, NaPAC10, NaPAC12, and NaPAC14, the elastic component was smaller than the viscous component, meaning that the solutions have fluid character. The viscoelastic behavior of NaPAC16 or NaPAC18 is different over the entire angular frequency range. They show higher elastic than viscous component and both components are independent of applied frequency denoting a gel-like behavior. This is in contradiction with the less hydrophobic homologs with C10, C12, and C14 chains. The association of longer (C16, C18) pendant groups gives rise to stronger, more crowded intermolecular hydrophobic junctions, reflected in the solid-like response of the respective polymers. Moreover, among the studied associative polymers, only those with C16 and C18 groups present yield stress, which is known to be a measure of the strength and number of hydrophobic junctions formed in aqueous solution [67]. In a similar manner, associative poly(vinyl alcohols) with various alkyl levels were found to present yield stress for higher hydrophobic contents [40].

The variation of storage and loss modulus for 3 % (wt.) NaPA and NaPACn solutions with angular frequency
It has been pointed out that our frequency sweep test revealed liquid- and solid-like behaviors. To obtain additional information, we studied the dependence of square root of G″ on the square root of Ω at a polymer concentration of 3 % (wt.). The results are shown in Fig. 9. The curves are fitted with Eq. 2 and the results are presented in Table 3.

The variation of square root of loss modulus for 3 % (wt.) NaPA and NaPACns solutions with the square root of angular frequency (the solid lines are data fitted with Eq. 2)
Figure 9 and Table 3 unveil that NaPA, NaPAC10, NaPAC12, and NaPAC14 have the G″0 nearly to zero. It means that they form little hydrophobic microdomains, by intermolecular association, and indicates a small number of network junctions. On the contrary, NaPAC16 and NaPAC18 have higher values of G″0, revealing stronger hydrophobic networks. The G0″ values also indicate that by increasing the hydrophobic alkyl chain length, more stable networks do result (see Table 3). Therefore, the modified polymers with C16 and C18 groups develop stronger networks than the others NaPACn (n = 10–14) and more energy is needed to break down their structures.
Another important parameter affected by the physical networks and molecular motions of the polymer is its relaxation time (λ). The parameter typically refers to the time needed to the polymer chains to return to equilibrium state after being stressed. The relaxation of non-Newtonian materials is not instantaneous as for Newtonian fluids [68]. For information about the relaxation time of our NaPACns, we extracted λ from Eq. 3 and its variation on angular frequencies is presented in Fig. 10.

The change of relaxation time of 3 % (wt.) NaPA and NaPACn solutions with angular frequency
One may observe that λ rises with the alkyl chain length and decreases with frequency. It means that, at 3 % (wt.), the elastic part of the systems increases with the length of alkyl chains. The NaPAC16 and NaPAC18 that present high values of yield stress and solid-like properties have the highest values of λ. This indicates an evident elastic effect. In contrast, the precursor and polymers with shorter alkyl grafts have lower λ. For them, the elasticity is not as prominent as for PAA grafted with C16 and C18. At low angular frequency, C10 has the highest relaxation time, followed by C12 and C14, the last having the smallest relaxation time. The difference between C10 and C12 is very small and that between them and C14 is larger, and one may admit that, at the applied frequencies, the behavior is dictated by the label content (see Table 1). On the contrary, at higher angular frequencies, the order is reversed, the behavior being imposed by the hydrophobic character. The NaPA gives a stable liquid system, having high relaxation times at low frequencies. At higher frequencies, it has the lowest relaxation times in agreement with its lack of hydrophoby, because it has no alkyl chains to form intercoil networks.
Conclusions
A series of thickening agents based on poly(acrylic acid) was synthesized. They were polyacrylates hydrophobically labeled with 10 to 18 carbon atoms. The label content determined by 1H-NMR was lower than 3 % (mol). The present work reveals that information on associative polymers in water can be obtained by a simple method like refractometry, as well as sophisticated but little material consuming ones (rheology, fluorometry). Dynamic fluorescence and refractive index data highlighted the occurrence of intracoil (at low polymer concentration) and intercoil aggregates (at higher NaPACn content, at c*). It was pointed out that the label content controls the intracoil aggregation, while the alkyl chain length determines the intercoil association. The overlap concentration (c*) decreases by increasing the hydrophobic character of the polymer. The study also revealed that grafting of small amounts of alkyl chains on PAA results in a new class of compounds with totally different properties. Thus, the polyacrylates labeled with C10–C14 have liquid-like behavior, while those grafted with C16 and C18 present solid-like character.
References
Winnik MA, Yekta A (1997) Associative polymers in aqueous solution. Curr Opin Colloid In 2:424–436
Barret JF, Calvet D, Collet A, Viguier M (2003) Fluorocarbon associative polymers. Curr Opin Colloid In 8:296–306
Volpert E, Selb J, Candau F (1998) Associating behavior of polyacrylamides hydrophobically modified with dihexylacrylamide. Polymer 39:1025–1033
Wu S, Shanks RA, Bryant G (2006) Properties of hydrophobically modified polyacrylamide with low molecular weight and interaction with surfactant in aqueous solution. J Appl Polym Sci 100:4348–4360
Camail M, Margaillan A, Martin I (2009) Copolymers of N-alkyl-and N-arylalkylacrylamides with acrylamide: influence of hydrophobic structure on associative properties. Part I: viscometric behavior in dilute solution and drag reduction performance. Polym Int 58:149–154
Yang Q, Song C, Chen Q, Zhang P, Wang P (2008) Synthesis and aqueous solution properties of hydrophobically modified anionic acrylamide copolymers. J Polym Sci Pol Phys 46:2465–2474
Somasundaram P, Chakraborty S, Qiang Q, Deo P, Wang J, Zhang R (2004) Surfactants, polymers and their nanoparticles for personal care applications. J Cosmet Sci 55:S1–S17
Leonard M, Rastello De Boisseson M, Hubert P, Dalençon F, Dellacherie E (2004) Hydrophobically modified alginate hydrogels as protein carriers with specific controlled release properties. J Control Release 98:395–405
Bromberg L (2008) Polymeric micelles in oral chemotherapy. J Control Release 128:99–112
Xiong SD, Li L, Jiang J, Tong LP, Wu S, Xu ZS, Chu PK (2010) Cationic fluorine-containing amphiphilic graft copolymers as DNA carriers. Biomaterials 31:2673–2685
Li L, Pan Q, Xiong SD, Xu ZS, Song GW (2011) Study on the interaction between a fluorine-containing amphiphilic cationic copolymer and nucleic acid by resonance light scattering technique. J Fluor Chem 132:35–40
Jeong B, Kim SW, Bae YH (2012) Thermosensitive sol-gel reversible hydrogels. Adv Drug Deliver Rev 64:154–162
Peddada LY, Garbuzenko OB, Devore DI, Minko T, Roth C (2014) Delivery of antisense oligonucleotides using poly(alkylene oxide)-poly(propylacrylic acid) graft copolymers in conjunction with cationic liposomes. J Control Release 194:103–112
Reuvers AJ (1999) Control of rheology of water-borne paints using associative thickeners. Prog Org Coat 35:171–181
Kastner U (2001) The impact of rheological modifiers on water-borne coatings. Colloid Surface A 183-185:805–821
Saitoh T, Ono N, Hiraide M (2012) Effective collection of hydrophobic organic pollutants in water with aluminum hydroxide and hydrophobically modified polyacrylic acid. Chemosphere 89:759–763
Taylor KC, Nasr-El-Din HA (1998) Water-soluble hydrophobically associating polymers for improved oil recovery: a literature review. J Petrol Sci Eng 19:265–280
Smeets M, Golombok M (2010) Application of shear induced structure materials for brine flooding in sandstone oil reservoirs. J Petrol Sci Eng 72:270–276
Wever DAZ, Picchioni F, Broekhuis AA (2011) Polymers for enhanced oil recovery: a paradigm for structure-property relationship in aqueous solution. Prog Polym Sci 36:1558–1628
Standnes DC, Skjevrak I (2014) Literature review of implemented polymer field projects. J Petrol Sci Eng 122:761–775
Magny B, Lafuma F, Iliopoulos I (1992) Determination of microstructure of hydrophobically modified water-soluble polymers by 13C-NMR. Polymer 33:3151–3154
Petit-Agnely F, Iliopoulos I, Zana R (2000) Hydrophobically modified sodium polyacrylates in aqueous solutions: association mechanism and characterization of the aggregates by fluorescence probing. Langmuir 16:9921–9927
Zhou H, Song GQ, Zhang YX, Chen J, Jiang M, Hogen-Esch TE, Dieing R, Ma L, Haeussling L (2001) Hydrophobically modified polyelectrolytes. 4 synthesis and solution properties of fluorocarbon-containing poly (acrylic acid). Macromol Chem Physic 202:3057–3064
Mylonas Y, Bokias G, Iliopoulos I, Staikos G (2006) Interpolymer association between hydrophobically modified poly(sodium acrylate) and poly(N-isopropylacrylamide) in water: the role of hydrophobic interactions and polymer structure. Eur Polym J 42:849–857
Zhang H, Xu K, Ai H, Chen D, Xv L, Chen M (2008) Synthesis, characterization and solution properties of hydrophobically modified polyelectrolyte poly(AA-co-TMSPMA). J Solut Chem 37:1137–1148
Hourdet D, Ducouret G, Varghese S, Badiger MV, Wadgaonkar PP (2013) Thermodynamic behavior of hydrophobically modified polyacrylamide containing random distribution of hydrophobes: experimental and theoretical investigations. Polymer 54:2676–2689
Alves L, Lindman B, Klotz B, Böttcher A, Haake HM, Antunes FE (2014) Controlling the swelling and rheological properties of hydrophobically modified polyacrylic acid nanoparticles: role of pH, anionic surfactant and electrolyte. Colloid Surface A 459:233–239
Wang KT, Iliopoulos I, Audebert R (1988) Viscometric behavior of hydrophobically modified poly(sodium acrylate). Polym Bull 20:577–582
Philippova OE, Hourdet D, Audebert R, Khokhlov AR (1997) pH-responsive gels of hydrophobically modified poly(acrylic acid). Macromolecules 30:8278–8285
Zhuang DQ, Da JCAH, Zhang YX, Dieing R, Ma L, Haeussling L (2001) Hydrophobically modified polyelectrolytes II: synthesis and characterization of poly(acrylic acid-co-alkyl acrylate. Polym Advan Technol 12:616–625
Furo I, Iliopoulos I, Stilbs P (2000) Structure and dynamics of water-soluble polymer aggregates as seen by 19F NMR spectroscopy. J Phys Chem B 104:485–494
Petit F, Iliopoulos I, Audebert R (1998) Aggregation of associating polymers studied by 19F n.m.r. Polymer 39:751–753
Lacík I, Stach M, Kasák P, Semak V, Uhelská L, Chovancová A, Reinhold G, Kilz P, Delaittre G, Charleux B, Chaduc I, D’Agosto F, Lansalot M, Gaborieau M, Castignolles P, Gilbert RG, Szablan Z, Barner-Kowollik C, Hesse P, Buback M (2015) SEC analysis of poly(acrylic acid) and poly(methacrylic acid). Macromol Chem Physic 216:23–37
Hoogenboom R, Schubert US, Van Camp W, Du Prez FE (2005) RAFT polymerization of 1-ethoxyethyl acrylate: a novel route toward near-monodisperse poly(acrylic acid) and derived block copolymer structures. Macromolecules 38:7653–7659
Nicolescu FA, Jerca VV, Dancus I, Petris A, Nicolescu TV, Rau I, Vlad VI, Vasilescu DS, Vuluga DM (2011) Synthesis and characterization of side-chain maleimide-styrene copolymers with new pendant azobenzene moieties. J Polym Res 18:1009–1016
Magny B (1992) Polyelectrolytes associatifs: methodes de synthese, comportement en milieu dilue et semi-dilue. These de Doctorat, l’Université Pierre et Marie Curie, Paris, France
Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Bercaw JE, Stoltz BM, Goldberg KI (2010) NMR chemical shifts of trace organics and impurities: common laboratory solvents and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 29:2176–2179
Lakowicz R Joseph (2006) Principles of fluorescence spectroscopy, 3rd edn. Springer, New York
Lyoo WS, Lee SJ, Kim JH, Noh SK, Ji BC, Kim BC (2004) Effect of molecular weight on the rheological properties of atactic poly(vinyl alcohol)/dimethylsulfoxide/water solution. J Appl Polym Sci 93:41–46
Hu Q, Huang G, Zheng J, Su H, Guo C (2012) Synthesis and rheological properties of hydrophobically modified poly(vinyl alcohol). J Polym Res 19:6–15
Lyoo WS, Yeum JH, Kwon OW, Shin DS, Han SS, Kim BC, Jeon HY, Noh SK (2006) Rheological properties of high molecular weight (HMW) syndiotactic poly(vinyl alcohol) (PVA)/HMW atactic PVA blend solutions. J Appl Polym Sci 102:3934–3939
Anghel DF, Alderson V, Winnik FM, Mizusaki M, Morishima Y (1998) Fluorescent dyes as model “hydrophobic modifiers” of polyelectrolytes: a study of poly(acrylic acid)s labelled with pyrenyl and naphthyl groups. Polymer 39:3035–3044
Guo X, Abdala AA, May BL, Lincoln SF, Khan SA, Prud’homme RK (2006) Rheology control by modulating hydrophobic and inclusion associations in modified poly(acrylic acid) solutions. Polymer 47:2976–2983
Arai H, Murata M, Shinoda K (1971) The interaction between polymer and surfactant: the composition of the complex between polyvinylpyrrolidone and sodium alkyl sulfate as revealed by surface tension, dialysis, and solubilization. J Colloid Interf Sci 37:223–227
Mukerjee P, Mysels KJ (1971) Critical micelle concentrations of aqueous surfactant systems (NSRDS-NBS 36) US Government Printing Office, Washington DC
Van Os NM, Haak JR, Rupert LAM (1993) Physico-chemical properties of selected anionic, cationic, and nonionic surfactants. Elsevier, Amsterdam
Anghel DF, Saito S, Băran A, Iovescu A (1998) Interaction between PAA and nonionic surfactants with the same PEO but different hydrophobic moieties. Langmuir 14:5342–5346
Kalyanasundaram K, Thomas JK (1977) Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J Am Chem Soc 99:2039–2044
Dong DC, Winnik MA (1984) The Py scale of solvent polarities. Can J Chemistry 62:2560–2565
Karpovich DS, Blanchard J (1995) Relating the polarity-dependent fluorescence response of pyrene to vibronic coupling: achieving a fundamental understanding of the Py polarity scale. J Phys Chem-Us 99:3951–3958
Winnik FM, Regismond STA (1996) Fluorescence methods in the study of the interactions of surfactants with polymers. Colloid Surface A 118:1–39
Geiger MW, Turro NJ (1975) Pyrene fluorescence lifetime as a probe for oxygen penetration of micelles. Photochem Photobiol 22:273–276
Turro NJ, Kuo PL (1986) Pyrene excimer formation in micelles of nonionic detergents and of water-soluble polymers. Langmuir 2:438–442
Honda C, Itagaki M, Takeda R, Endo K (2002) Solubilization of pyrene in CnE7 micelles. Langmuir 18:1999–2003
Anghel DF, Saito S, Băran A, Iovescu A, Corniţescu M (2007) The aggregation of nonionic surfactants in the presence of poly(methacrylic acid). Colloid Polym Sci 285:771–779
Aguiar J, Carpena P, Molina-Bolivar JA, Carnero Ruiz C (2003) On the determination of the critical micelle concentration by the pyrene 1:3 ratio method. J Colloid Interf Sci 258:116–122
Anghel DF, Saito S, Iovescu A, Băran A (1994) Some critical points in the interaction between homogeneous non-ionic surfactants and poly (acrylic acid). Colloid Surface A 90:89–94
Pineiro L, Novo M, Al-Soufi W (2015) Fluorescence emission of pyrene in surfactant solutions. Adv Colloid Interfac 215:1–12
Băran A, Stîngă G, Anghel DF, Iovescu A, Tudose M (2014) Comparing the spectral properties of pyrene as free molecule, label and derivative in some colloidal systems. Sensor Actuat B-Chem 197:193–199
Seixtas de Melo J, Costa T, Francisco A, Maçanita AL, Gago S, Gonçalves IS (2007) Dynamics of short as compared with long poly(acrylic acid) chains hydrophobically modified with pyrene, as followed by fluorescence techniques. Phys Chem Chem Phys 9:1370–1385
Chiu YC, Chen LJ (1989) Refractive index of nonionic surfactant solutions containing polyoxyethylene mono-n-alkylether. Colloid Surface 41:239–244
Chahti A, Boukalouch M, Dumas JP, El Kinani A (2000) Phase transitions in colloidal systems. Part I: refractometric study of the binary system SDS + water or pentanol-1 + water and the ternary system water + SDS + pentanol-1 at 298.15 K. J Disper Sci Technol 21:525–535
González-Pérez A, Czapkiewicz J, Prieto G, Rodríguez JR (2003) Second critical micelle concentration of dodecyldimethylbenzylammonium chloride in aqueous solution at 25 °C. Colloid Polym Sci 281:1191–1195
Lee K, Kunjappu J, Jockusch S, Turro NJ, Widerschpan T, Zhou J, Smith BW, Zimmerman P, Conley W (2005) In: Sturtevant JL (ed) Amplification of the index of refraction of aqueous immersion fluids by ionic surfactants. Advances in Resist Technology and Processing XXII, Proceedings of SPIE Vol. 5753, SPIE, Bellingham, WA
Tan CH, Huang ZJ, Huang XG (2010) Rapid determination of surfactant critical micelle concentration in aqueous solutions using fiber-optic refractive index sensing. Anal Biochem 401:144–147
Taghizadeh MT, Foroutan M (2005) A study on P(VP-VA) hydrophobically associating behavior of water-soluble copolymers. Iran Polym J 14:47–54
Hong Z, Yongjun M, Hang W, Lin X (2010) Rheological properties of hydrophobically modified poly(acrylic acid) in mixed solutions. J Solut Chem 39:1243–1252
Huang J (1999) Extensional viscosity of dilute polymer solutions. Ph. D. Thesis, University of Toronto, Toronto, Canada
Acknowledgments
The research for this paper has been carried out within the research program Functional Complex Colloids of the “Ilie Murgulescu” Institute of Physical Chemistry, financed by the Romanian Academy. The authors gratefully acknowledge the support of the EU (ERDF) and Romanian Government allowing for acquisition of research infrastructure under POS-CCE O 2.2.1 project INFRANANOCHEM, no. 19/01.03.2009. This work is also supported by the PN-II-ID-PCE-2011-3-0916 Exploratory Research Project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(PDF 851 kb)
Rights and permissions
About this article

Cite this article
Aricov, L., Băran, A., Simion, E.L. et al. New insights into the self-assembling of some hydrophobically modified polyacrylates in aqueous solution. Colloid Polym Sci 294, 667–679 (2016). https://doi.org/10.1007/s00396-015-3825-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-015-3825-z