
Abstract
The characteristics of the local piezoelectric response of isotropic films of copolymers of vinylidene fluoride (VDF) were compared with 6 and 29 mol% tetrafluoroethylene (TFE) obtained by crystallization from a solution in acetone. Time dependence of the electric displacement response was analyzed after switching of the spontaneous polarization. A copolymer with a higher content of tetrafluoroethylene is characterized by higher values of electrical displacement and piezoelectric response. For interpretation of this fact, we used molecular mobility in amorphous phase dates. It is shown that the activation energy of local and cooperative liquid-like (in the amorphous phase) mobility is markedly lower in the copolymer with a higher content of TFE. Under identical conditions of the crystallization, both films of the copolymers lead to the formation of larger crystals of the polar phase and magnitude of a “long” period at a high content of copolymer of TFE. It is postulated that these structural parameters are responsible for the stable value of residual local piezoelectric activity. It is found that the rapid decay of the signal in the local piezoresponse of polarized films is controlled by the activation energy of the local and cooperative dynamics chains of the amorphous phase.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ferroelectric polymers are promising materials for organic electronics. Such polymers are found relatively recently [1, 2] and because there are new posts regularly about the interesting properties of this kind of materials. Organic polymers of this class have a number of specific properties: low density, low modulus, high breakdown voltage, etc. It is shown that such materials exhibit high values of the piezo-, pyro-, and electrostrictive constants. Creating a series of sensors having features that are not implemented based on classical inorganic ferroelectrics was reported [1–3]. The most studied and advanced in practical implementations are polymers based on polyvinylidene fluoride (PVDF), which is a flexible chain crystalline polymer with a simple chemical structure of the monomer unit. The latter circumstance contributed to uniquely characterize the structure and chain conformation of four crystallographic modifications: α-, β-, γ-, and αp-phases [1, 2, 4]. Recently, it is shown that by chemical or physical modifications, ferroelectric polymers based on PVDF can lead to a relaxor state. It was found that in this case, such polymers can be implemented with a capacitive energy storage fast reset to an external circuit [5]. Furthermore, they discovered unusual properties such as a giant electrostriction [6, 7] and electrocaloric effect [8].
These polymers are considered promising as materials for ferroelectric memory [9, 10] because of the potentially high information density. Indeed, the kinetic unit chain with several monomer units can serve as a memory cell, since the direction of the dipole moment (very large quantities) of such units in the control electric field can take two stable equilibrium positions. Essentially, the same situation occurs if the kinetic units in a ferroelectric polymer form domains. In this regard, the task of detailed research in these polymers poses a number of issues, among them, the character of crystallization and the formation of the domain structure and the revealed peculiarities of molecular mobility in a complex two-phase system. The mentioned polymers are characterized by the presence of at least two phases: amorphous and crystalline, with the fraction of the latter being usually ≈0.5. These phases at room temperature are different not only structurally but also in terms of the nature of their dynamics, which are principally different. As polymers belong to the class of flexible chain, the amorphous phase is characterized by low glass transition temperature (about −40 °C). This means that at room temperature (at which all experiments were carried out), the amorphous phase is in a liquid-like state, i.e., it is characterized by high free volume, wherein the cooperative movement prevails. According to the literature data [1, 2, 4], relaxation times of such dynamics at room temperature have a value of ∼0.1 μs. The specificity of crystalline polymers (which include and consider ferroelectric polymers) is that they cannot be attributed to the classical two-phase systems. Chain character of molecules and statistical laws lead to that the same chain can be, as in amorphous and crystalline phase. In such a situation can be expected to influence the dynamics of chains of amorphous phase on the process of switching domains crystalline phase.
In this paper, the structural characteristics of vinylidene fluoride (VDF) copolymers are compared with different contents of tetrafluoroethylene (TFE) crystallized under similar conditions from a solution in acetone. It is shown that there is a correlation between the quantitative characteristics of the surface morphology of the films produced and parameters of ferroelectric domains. It is shown that in the copolymer with a high content of TFE, morphology with higher “long” period and with thick crystals is formed. It was found that such copolymers have larger domains and the value of the initial signal of the local piezoelectric response in polarized films controlled by activation energy cooperative and local mobility of the copolymer.
Samples and experimental techniques
Samples of a VDF–TFE copolymer with a molar composition of 94:6 (sample #1) and 71:29 (sample #2) were studied; the microstructure of the copolymer was previously characterized via a 19F NMR technique [11, 12]. The fraction of head-to-head (HHTT) chemical defects in the studied copolymer #1 is 4.2 mol%. For a copolymer of VDF–TFE of 71:29, the number of such defects was 2.5 mol%. The presence of TFE dyads in the chain are in an amount of 5 mol%. Isotropic films with a thickness of 20–40 μm were prepared via crystallization on a glass substrate at room temperature from a solution (8 wt% copolymer) in a low-boiling solvent (acetone). To reduce the surface roughness of the film, the glass substrate was polished. To provide thickness homogeneity, the substrate was adjusted in a horizontal plane.
To study the electric properties, 100-nm-thick aluminum electrodes were deposited on the films via thermal vacuum evaporation. High-voltage polarization and conductivity were measured with the use of a previously described system based on a modified Sawyer–Tower circuit [13].
The piezoresponse force microscopy (PFM) measurements were performed on a scanning probe nanolaboratory NTEGRA Prima (NT-MDT, Russia) equipped with internal lock-in amplifiers. Using conductive atomic force microscopy tips, it is possible to measure local electrical and topographical properties both simultaneously and independently. The investigations were performed with commercially available W2C- and Pt-coated Si tips. All PFM images were taken in the mode of amplitude contrast where the signals correspond to a real part (X = amplitude times cos(phase)) of the PFM signal obtained through lock-in amplifiers.
The radial field distribution is given by the following expression, which is obtained under the assumption that the tip is a charged sphere [14]:
Here, r is the distance from the tip along the surface, R is the probe tip radius, δ is the distance of the tip from the surface, C t is the capacitance of the system consisting of the tip and dielectric semi-infinite crystal, and ε c and ε a are the relative permittivity of the polymer sample along the polar and nonpolar directions, respectively.
For the quantitative data analysis, we applied an autocorrelation function method, which has been successfully used for the topographic data evaluation [15]. From the topographical image was built an autocorrelation function calculated by the following equation:
The average correlation length of the registered estimated distribution ratio is
where r is the distance from the central peak (nm), ξ is the correlation length (nm), and h is the exponent parameter (0 < h < 1) [16].
The same procedure was used for the evaluation of the correlation lengths of ferroelectric domains, which were detected by the linear piezoelectric response.
Results and discussions
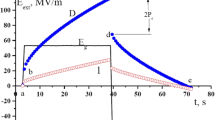
Figure 1 shows the time dependence of the electric displacement in the polymer films #1 and #2, when applying for a film rectangular bipolar pulse electric field. In accordance with Eq. (1) at a film thickness of about 20 μm and a poling potential in the field of 20 V by PFM method, the electric field is 17 MV/m. Thus, the fields used in the macroscopic measurements (Fig. 1) and in the PFM method are of the same order. In this connection, comparing data on the macroscopic hysteresis and local piezoelectric response after polarization can be carried out. As follows from the inset of Fig. 1, domain switching process has two stages: fast and slow.

Time dependences of electric displacement in the isotropic copolymer films (sample #1 (VDF–TFE 94:6) and #2 (VDF–TFE 71:29)) during the application of a rectangular electric pulse
First, after changing, the polarity of the polarizing field is characterized by a jump D, which has a switching time of the order of 10−3–10−7 s [17]. More interesting is the slow stage of change D, which takes tens of seconds. Analysis of this stage of the switching process was carried out taking into account that the ferroelectric material has a finite conductivity σ. In this case, the time dependence of D is described by the following equation [1, 2]:
where t is the time, τ s is the switching time of the spontaneous polarization, and m and n are the constants.
Contribution to the conductivity of the abovementioned carriers can give different reasons. Firstly, it is impurity carriers after synthesis (catalyst residues, etc.) which are always present in the initial polymer. To increase such a concentration of carriers in the copolymer VDF–TFE, authors [18] used the iodine vapor. It turned out that the increase in conductivity thus affected even at times of fast switching stage. In our opinion, in these polymers, other causes of additional carriers should be considered. Since the measurements are made at high fields, they can also occur due to, for example, the electrochemical reactions in the PVDF chains [19]. Carrier injection plays a more important role from the electrode material. Since we are dealing with a dielectric (albeit imperfect), it marked the charges that will be placed in the traps of a different depth. As a consequence, the space charge region will occur near the electrodes, creating an intrinsic field E i .
Movement of injected carriers under the influence of the local field should be characterized by low mobility, and therefore, to detect this process will require longer times. In this regard, long step increase D in Fig. 1 should characterize the time dependence of the formation of the space charge field. As can see from Fig. 1, with the same external field for copolymer #2, the value D in the second stage is higher than for sample #1. From Fig. 2, it follows that in sample #1, the field dependence of conductivity calculated by Eq. (4) is less pronounced.

Field dependences of the high-voltage conductivity in isotropic VDF–TFE copolymer films with a composition of 94:6 (curve #1) and 71:29 (curve #2)
Figure 3 shows the diffraction patterns in large and small angles that the two samples differ significantly. In both cases, the crystallization proceeds in a mixture of α-, β-, and γ-phases, but sample #2 of the lattice in the direction a- and b-axes is less densely packed [20, 21]. This can be seen from the shift of the main peak β-phase in the copolymer to lower angles (Fig. 3a). The average values of crystal sizes of the polar β-phase samples #1 and #2 are 8.6 and 19.4 nm, respectively [20]. From Fig. 3b, it can be seen that in both samples, lamellar crystals form stacks, which implemented the one-dimensional diffraction (along the c-axis lattice), characterized by the “long” period L. It is clearly seen that sample #2 is higher than sample #1, since it is for a maximum at the lower angle. The values of L in samples #1 and #2 are 7.0 and 19.1 nm, respectively [20].

Large-angle (a) and small-angle (b) X-ray diffractions for isotropic films of the VDF–TFE copolymer films with a composition of 94:6 (curve #1) and 71:29 (curve #2). Inset in (b) shows experimental data from the Lorentz correction
Figure 4 shows the temperature dependence of the time relaxation in Arrhenius coordinates, constructed according to a study of low-temperature dielectric relaxation [20].

Correlation diagrams for parameters of dielectric relaxations (above (Area I) and below (Area II) the glass transition point) in VDF–TFE copolymer films with a composition of 94:6 (curves #1) and 71:29 (curves #2)
As can be seen, local mobility (at low temperatures) is described by the Arrhenius equation and the activation energy for it is higher in sample #1. Above the glass transition, the temperature dependence of the relaxation times (frequency reorientation f) chains of the amorphous phase in both samples obeys the Vogel–Fulcher law:
where T 0 is the constant for the substance. As shown in [20], the parameter b reflecting the activation energy of the cooperative dynamics of liquid-like amorphous phase also is higher in sample #1 (see data in Table 1).
In accordance with the conclusions in [20], it should be associated with increased dipole–dipole interactions due to the higher concentration of polar groups in sample #1. Figure 5 shows the comparative curves of frequency-dependent components of the complex permittivity (ε*) and conductivity (σ) at room temperature for samples #1 and #2. It is seen that in sample #2, these values at low frequencies are much higher.

Frequency dependences of the dielectric permittivity (a), dielectric loss (b), and conductivity (c) for VDF–TFE copolymer films with a composition of 94:6 (curves #1) and 71:29 (curves #2)
Noted above, difference of real and imaginary component of ε* (Fig. 5a, b), it is advisable to associate with components ε′ and ε″ ionic conductivity (ε i ′, ε i ″), which give an addition in the values from initial the polymer ε p ′ and ε p ″. This can be expressed as Eqs. (6) and (7):
With this approach, the ionic components, component ε*, will have the form [22]:
where v 0 is the concentration of mobile ions with charge q, d is the thickness of the sample, D 0 is the diffusion coefficient, f is the frequency of electric field, E d is the activation energy of ionic diffusion, and W is the dissociation energy of impurity molecules.
In our opinion, a special role in the relations shown in (8) and (9) is played by the value E d , which is included under the exponent. Recall that the copolymers are considered two-phase systems, where there are both crystals and disordered phase. The latter is characterized by an increased free volume and ionic impurities because it will be localized in the amorphous phase. As shown above, at room temperature dynamics, it is implemented as cooperative and local movements with relaxation times ∼10−7 s (Fig. 4). In this regard, the activation energy of the ion diffusion parameters must be controlled by the molecular mobility of the chains of the amorphous phase, where ionic impurities are located. The carrier can be transformed into a new equilibrium position (neighboring area of free volume), which is formed by the cooperative movement segments of the amorphous phase. This means that the E d has to depend on the activation energy of molecular motion in the amorphous phase. From Table 1, it can be seen that the activation energy for both local and cooperative mobility in copolymer #2 is considerably lower than in copolymer #1. Following the above activation energy of the ion diffusion, copolymer #2 should be lower than copolymer #1. In accordance with Eqs. (8) and (9) for copolymer #2, it is to be accompanied by an increase in low-frequency values of ε′ and ε″, which is observed in Fig. 5a, b.
From Fig. 5c, it can be seen that the low-frequency values of ac conductivity σ in copolymer #2 are also higher than those in copolymer #1. In accordance with this latter, the copolymer should be characterized by a higher Maxwell relaxation time τ:
where ε 0 is the electric constant and ε а is the dielectric permittivity of the matrix containing a of inclusions with conductivity σ and form factor A. Data in Fig. 5c and relation (10) show that the time of forming of space charge in the copolymer of VDF–TFE of 71:29 (sample #2) has to be lower than in the copolymer 94:6 (sample #1). Note that in the field, the space charge Ei is formed by the movement of the “free” charge. Low mobility of the free charge is the cause of high values of the Maxwell relaxation time, and therefore, for the dielectric thickness d 0 at a volume charge density ρ(x), E i has the form
In our experiments, it is shown that ρ(x) (and thus E i ) is a function of not only the coordinates (x) but also the time t. As the resulting field, E is the vector sum of the field source E g and the field E i experimentally recorded curves D(t) (at constant E g) describe the kinetics of the formation of the space charge (E i). Comparison of the curves I and II in Fig. 1 shows that in this case, the above kinetics of sample #2 is more rapid than in sample #1. Assuming the same composition in both ionic impurities, copolymers may be realized due to the higher mobility of carriers in the copolymer of VDF–TFE of 71:29. As shown in Table 1, it is conditioned by lower the activation energies of molecular mobility in the amorphous phase (αa) process.
The same reasons are also responsible for a stronger field dependence of the high-voltage conductivity of sample #2 copolymer (Fig. 2). It should be emphasized that the characteristics of high-voltage polarization of the considered copolymers should also depend on their degree of crystallinity. From the data in Fig. 5b, from the slope of the low-frequency curve ε″, parameter s is calculated [20]. For these polymers, it is shown that s decreases with an increase of the degree of crystallinity [20]. From Table 1, it follows that parameter s in sample #1 is lower than that in sample #2, indicating a lower crystallinity in the latter sample. Increasing the volume fraction of the disordered phase with high mechanical compliance of copolymer #2 may also lead to more high-voltage conductivity (Fig. 2). It should be remembered, however, that copolymer #2 is characterized by lower coercive fields than copolymer #1 [11, 12]. This fact should also be considered when analyzing the field dependence of high-voltage conductivity.
Figures 6 and 7 show the topography (a), out-of-plane PFM (b), and second harmonic PFM images (c) on sample #1 and #2 at room temperature, respectively. The value of surface roughness was about 32 nm for sample #1 and 54 nm for sample #2. Comparison of the sizes of the domains determined via PFM (Figs. 6d and 7d) and the crystallites in our film shows that the former is one to two orders of magnitude greater. Hence, the domains observed in our film via PFM must include both crystals and regions of the disordered phase.

Surface topography (a), local piezoelectric response (b), second harmonic PFM image (c), and the respective signal profiles (d) in copolymer film VDF–TFE (composition of 94:6) along the line scans indicated in (a)–(c) and distributions (e) of the local piezoelectric response and second harmonic signals

Surface topography (a), local piezoelectric response (b), second harmonic PFM image (c), and the respective signal profiles (d) in copolymer film VDF–TFE (composition of 71:29) along the line scans indicated in (a)–(c) and distributions (e) of the local piezoelectric response and second harmonic signals
Figures 6e and 7e compare the distribution of the local piezoresponse signal for the VDF–TFE films. Only one peak shifting to negative (for VDF–TFE composition 94:6) and positive (for VDF–TFE composition 71:29) d 33 values in Figs. 6e and 7e is also an indicative of the local self-polarization effect at the nanoscale.
Autocorrelation image (Fig. 8b) was obtained from the original topography image (Fig. 8a) by following transformation by Eq. (2). In order to estimate the mean size of the correlation length (ξ), we averaged the autocorrelation image over all in-plane directions and then approximated it by Eq. (3). The same calculation procedure was used for out-of-plane PFM images for both samples.

Topography (a) and corresponding autocorrelation image (b) of VDF–TFE copolymer film with a composition of 71:29 (sample #2)
The corresponding autocorrelation functions showing a difference in the correlation lengths in polymer samples #1 and #2 for topography images are shown in Fig. 9a. The average “topography” correlation length for sample #1 is 138.5 ± 0.3 nm and for sample #2 is 250.2 ± 1.4 nm. Under the same conditions, crystallization of both films less hindered dynamics chains of the copolymer #2 at its crystallization may be responsible for the formation of these surface features. It should be emphasized that the characteristics of the parameters’ surface topography correlate well with the size of the areas of polymer having long-range order. Indeed, in copolymer #2, the scale marked roughness substantially higher and higher dimensions as crystallites and supramolecular structures are, i.e., “long” periods [20].

The corresponding autocorrelation functions for topography (a) and ferroelectric domains (b) in VDF–TFE copolymers showing differences in the correlation lengths at different compositions of the copolymer films (sample #1 (94:6) and sample #2 (71:29))
In the next step, we calculated the correlation length of ferroelectric domains in the polymer samples. From Fig. 9b, it can be seen that the size of the ferroelectric domains in the surfaces of both films is different, and for sample #2, it is also higher than for sample #1.
The comparison of the curves in Fig. 9a, b shows that for sample #1, the correlation parameters for “topography” and the average domain size are almost the same. In the case of copolymer #2, domain size is much lower than the average “topography” parameter. The reason is not completely clear, but it can be assumed that the magnitude of dipole interactions significantly affect the dimensions of the domains, including at the surface. In sample #2, the magnitude of these interactions will be reduced through the introduction in chains of PVDF large concentration of nonpolar comonomer of TFE. The presence in this copolymer of groups of TFE dyads (see methodic part) leads to the formation of disordered PTFE nonpolar crystals [20]. All of this can be the cause of the “anomalous” decrease of the average domain size.
In order to study the effect of poling on the piezoresponse, samples #1 and #2 were first poled with DC = ±30 V and later scanned at 5 V peak-to-peak AC voltage. The results are presented in Figs. 10 and 11. Dark and bright rectangles correspond to the regions poled with +30 and −30 V, respectively (Figs. 10a–c and 11a–c). The application of the bias has resulted in reorientation of the polarization. Under similar conditions of polarization, the piezoresponse signal in sample #2 is higher than for sample #1 (Figs. 10d and 11d).

Vertical component of the piezoelectric response of the film VDF–TFE (composition of 94:6) immediately after polarization at a voltage of ±30 V (a), piezoresponse image 20 min (b) and 40 min (c) after poling, piezoresponse signal profiles at various times after polarization (d), and relaxation dependences of the piezoelectric response signals for “positive” and “negative” poling regions (e)

Vertical component of the piezoelectric response of the film VDF–TFE (composition of 71:29) immediately after polarization at a voltage of ±30 V (a), piezoresponse image 20 min (b) and 40 min (c) after poling, piezoresponse signal profiles at various times after polarization (d), and relaxation dependences of the piezoelectric response signals for “positive” and “negative” poling regions (e)
If we assume that the piezocoefficient is proportional to the magnitude of the remnant polarization [1, 2, 17], the initial value of the piezoresponse in Figs. 10 and 11 should be proportional to D at the end of the cycle of polarization (Fig. 1). Indeed, in sample #2, it is higher in D (Fig. 1) and in the initial piezoresponse (Fig. 11). A more detailed analysis of the data of the local piezoelectric response should be reminded of the features of the mechanism of piezoelectricity in these ferroelectric polymers. Unlike inorganic crystals, the contribution from the classical changes of spontaneous polarization P under stress here is small [1, 2]. A high ratio of the amorphous phase with high compliance leads to macroscopic piezoelectric response that can make a great contribution to electrostriction and Poisson’s ratio [1, 2, 7].
where the first term takes into account the effect of electrostriction; the second term reflects the change in film thickness, due to the high compliance of the amorphous phase; and the third term takes into account only the “classical” polar crystal polarization change when applying for a film stress X.
To evaluate the contribution to the observed piezoelectric response from electrostriction signal measured for the first harmonic and the second harmonic frequency.
Measurements at the second harmonic can give important information about the properties of the material. For example, we obtain information about the contact potential difference [23]. With respect to the inorganic oxide ferroelectrics, the details of the signal at the second harmonic feel reduction of spontaneous polarization (fatigue) with increasing number of switching cycles [24]. As shown above in the investigated polymeric ferroelectrics, the “nature” of the signal at the second harmonic likely reflects the presence of the crystallites with another region and disordered phase with high mechanical compliance. Given these circumstances, the difference between the second harmonic signals for both copolymers will be analyzed.
Figures 6e and 7e show the last picture of the distribution of the signal on the selected scan area. As can be seen, samples #1 and #2 are significantly different. Previously, we have shown [25, 26] that both the copolymers at the selected film forming method crystallized in a metastable state. The copolymer composition VDF–TFE 94:6 (sample #1) shows metastability since the crystallization proceeds as a β- and γ-phase [25]. It is known that both have a noncentrosymmetric lattice phase [1, 2]. Therefore, the cellular structure of the quadratic pattern of piezoresponse and the doublet structure of the histogram (Fig. 6e) at the second harmonic is attributed to the electrostriction contribution of two types of amorphous phase which is on the borders with crystals β- and γ-phases. Indeed, polymorphic transition γ → β in the thermal recrystallization of the sample leads to a unimodal appearance of the histogram [25]. From Fig. 7e, it is clear that this type of histogram is characteristic for initial sample #2.
Since both samples crystallized under the same conditions, this means that the film of the copolymer of VDF–TFE of 71:29 (sample #2) was obtained in a state closer to equilibrium. Confirmation of this is obtained by IR spectroscopy, where absorption bands characteristic of the most energetically favorable α-phase were detected in sample #2 [20]. In these structural features arising in sample #2, we associate with it the characteristics of molecular mobility which influence the crystallization of the film at formation from solution. Lower activation energy cooperative (αa-) and localized (β-) mobility in sample #2 and will provide structure formation approaching equilibrium. As a consequence, a unimodal-distribution histogram at the second harmonic in sample #2 was obtained immediately after its preparation. For sample #1, this picture arises only after thermal recrystallization of the initial film [25]. In the latter paper, the authors have come to the conclusion that the observed domains in ferroelectric polymers include both crystalline regions and the region-disordered phase. This conclusion was based on the strong differences observed in crystallite sizes and domains.
The presence in the field dependence of the switching current of the spontaneous polarization components connected the amorphous phase [27] in sample #1 also confirms this conclusion.
In this paper, the average size of sample #2 crystal β-phase is ∼20 nm, which is a slightly different time domain size (∼150 nm). Given the degree of crystallinity below, this copolymer can be expected to increase the probability of entering in the abovementioned domain of the amorphous phase. Therefore, we can assume that the copolymer domain will include a large proportion of the amorphous phase. Since the latter, at ambient temperature, is in a liquid-like state after removal of the electric field, it will undergo a depolarization process. This process will be accompanied by a decrease in piezoelectric response signal (Figs. 10d and 11d). Indeed, in Figs. 10e and 11e, for both samples, decline is observed. To quantify the decay of the piezoresponse signal, a Kohlrausch–Williams–Watts (KWW) function was used:
where τ is the relaxation time and β is the parameter characterizing the distribution of relaxation times.
The analysis shows that the characteristic relaxation time for sample #2 is lower than that of sample #1 (Table 2). Unusual in our opinion is the fact that the copolymer of VDF with 6 mol% TFE (sample #1) is characterized by relaxation of almost one time, which is not typical for polymers.
The difference in the relaxation times again can be attributed to the difference in activation energy of liquid-like dynamics in both copolymers. If it is lower in copolymer #2, it is returned to an equilibrium state after the field is removed there should occur rapidly and this is indicated in the experiment.
A significant difference in the final values of the signal can be seen when comparing piezoelectric response relaxation curves in Fig. 9 for both samples. For sample #2, it has a sufficiently high value, while for sample #1, it is virtually zero. As noted above, sample #2 crystals are larger and have a long period. It is possible that these parameters are responsible for the structure and stability of the formed piezoelectric response. Partly, this conclusion is confirmed by the data of [23]. Here, for a sample of the copolymer of VDF–TFE of 94:6, it was shown that the size of crystals during heating increases and is accompanied by a simultaneous increase in stability of the piezoelectric response.
It should be noted that another pattern follows from Fig. 10 and Table 2. From the latter, it follows that the relaxation parameters of the film #1 after polarization potential of different signs do not match. Qualitatively, the same pattern is observed in the process of polarization (Fig. 1). Parameters of curve D(t) on slow stage of polarization are different for “+” and “–” field also. For example, at field +44 MV/m, the value σ = 119 pS/m is obtained, but at field −44 MV/m σ is already 88 pS/m. These differences should be associated with so that the efficiency of carrier injection and mobility holes and electrons may differ. This may lead to a variation in the space charge fields near the anode and cathode. This can lead to a marked feature polarization (Fig. 1) and depolarization (Figs. 10 and 11) for different signs of potential external sources.
Summary
The processes of polarization at both signs of an external potential in isotropic film copolymer VDF–TFE have different compositions with a metastable structure. The growth of the electric displacement after switching of the spontaneous polarization is associated with the formation of the space charge field. This field is formed by ionic impurities and injected carriers from the electrode materials. It is shown that the injected carriers from the anode and cathode are not equivalent in their mobility. Accordingly, the characteristics of the space charge differed, which includes such carriers. The strong metastable structures in the copolymer with a low content of TFE are accompanied by a “cellular” picture of the piezoelectric response signal at the second harmonic. It binds to the electrostriction contribution of two types of chains of the amorphous phase located at the borders with crystals β- and γ-phases.
References
Kawai H (1969) Jpn J Appl Phys 8:975–976
Bergman JG Jr, McFee JH, Crane GR (1971) Appl Phys Lett 18:203
Kochervinskii VV (1994) Russ Chem Rev 63:367
Kochervinskii VV (1996) Russ Chem Rev 65:865
Chu B, Zhou X, Ren K, Neese B, Lin M, Wang Q, Bauer F, Zhang QM (2006) Science 313:334–336
Casalini R, Roland CM (2002) J Polym Sci : Part B: Polym Phys 40:1975–1984
Kochervinskii VV (2009) Crystallogr Rep 54:1146–1171
Li X, Qian X-S, Gu H, Chen X, Lu SG, Lin M, Bateman F, Zhang QM (2002) Appl Phys Lett 101:132903
Scott JC, Bozano LD (2007) Adv Mater 19:1452–1463
Asadi K, De Leeuw DM, De Boer B, Blom PWM (2008) Nat Mater 7:547–550
Kochervinskii VV, Glukhov VA, Sokolov VG, Romadin VF, Murasheva EM, Ovchinnikov YK, Trofimov NA, Lokshin BV (1998) Vysokomol Soedin A 30:1969
Kochervinskii VV, Murasheva EM (1991) Vysokomol Soedin A 33:2096–2105
Kochervinskii VV, Chubunova EV, Lebedinskii YY, Shmakova NA, Khnykov AY (2011) Polymer Science Ser. A 53:929–946
Rosenman G, Urenski P, Agronin A, Rosenwaks Y, Molotskii M (2003) Appl Phys Lett 82:103–105
Munoz RC, Vidal G, Mulsow M, Lisoni JG, Arenas C, Concha A (2000) Phys. Rev. B 62:4686–4697
Kiselev DA, Bdikin IK, Selezneva EK, Bormanis K, Sternberg A, Kholkin AL (2007) J Phys D: Appl Phys 40:7109–7112
Furukawa T (1989) Phase Transifions 18:143–211
Ikeda S, Fukada T, Wada Y (1988) J Appl Phys 64:2026–2030
Bihler E, Holdik K, Eisenmenger W (1987) IEEE Trans. Elect Insul EI-22(22):207–210
Kochervinskii VV, Malyshkina IA, Pavlov AS, Bessonova NP, Korlyukov AA, Volkov VV, Kozlova NV, Shmakova NA. J Polym Sci Polym Phys In press
Lovinger AJ, Davis DD, Cais RE, Kometani JM (1988) Macromolecules 21: 78
Uemura S (1974) J Polym Sci Polym Phys Ed 12:1177–1188
Collins L, Kilpatrick JI, Viassiouk IV, Tselev A, Weber SAL, Jesse S, Kalinun SV, Rodriguez BJ (2014) Appl Phys Letts 104:133103, 1–5
Murari NM, Hong S, Lee HN, Katiyar S (2011) Appl Phys Letts 99:052904, 1–3
Kochervinskii VV, Kiselev DA, Malinkovich MD, Pavlov AS, Kozlova NV, Shmakova NA (2014) Polym. Sci. A (Russia) 56:48–62
Kochervinskii VV, Kozlova NV, Bessonova NP, Shcherbina MA, Pavlov AS (2014) J Mater Sci Res 3:59–73
Kochervinskii VV, Pavlov AS, Kozlova NV, Shmakova NA (2014) Polymer Sci. A (Russia) 56:587–602
Acknowledgments
The work was carried out with financial support in part from the Ministry of Education and Science of the Russian Federation in the framework of Increase Competitiveness Program of MISiS, RFBR research project no. 14-03-00623 A. The studies are performed on the equipment at the “Materials Science and Metallurgy” Shared Facilities Center of the National University of Science and Technology “MISiS” (ID project RFMEFI59414X0007, contract no. 14.594.21.0007).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kochervinskii, V.V., Kiselev, D.A., Malinkovich, M.D. et al. Local piezoelectric response, structural and dynamic properties of ferroelectric copolymers of vinylidene fluoride–tetrafluoroethylene. Colloid Polym Sci 293, 533–543 (2015). https://doi.org/10.1007/s00396-014-3435-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-014-3435-1