
Abstract
Segmented urethane elastomers (SUE) with polytetramethylene oxide soft segments, toluene diisocyanate (TDI)/methylene-bis-o-chloroaniline (MOCA) hard segments, TDI/ MOCA + TDI/butanediol (BD) and TDI/MOCA + isophorone diisocyanate/MOCA mixed segments were investigated. A microphase separation in SUE was adjusted by the variation in the composition of the hard segments or preparation conditions. It was shown that non-linear relationships of SUE’s thermo-mechanical, thermal and mechanical properties on the composition of hard segments, including an extremal one, were typical. Basic dependence types of SUE strength on the stretching rates (direct, reciprocal, extremal) were determined at various stages of the elastomer structure evolution. A significant increase in the SUE strength was found at a low degree of microphase separation. A strong effect of interaction in the soft phase on the physicochemical properties of block copolymers was shown. An improvement of the miscibility between hard and soft phases and a loose structure of the hard phase promote a stabilisation of the strength value in a wide range of the stretching rates. The effect of the strength increase equals to 30–50 %.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Segmented urethane-containing elastomers (SUEs), including polyurethanes, polyurethane ureas and polyurethane epoxides, are a unique class of polymers, the structure and properties of which can be regulated in wide limits [1–10].
The majority of segmented polyurethanes (SPUs) and segmented polyurethane ureas (SPUUs) are heterogeneous block copolymers. Polymer chains of such materials consist of uniform soft segments and uniform hard segments [1, 3, 11–13] or mixed segments (multiblock copolymers) [14–20].
The difference in the polarity of soft and hard segments leads to microphase separation, followed by the formation of hard segment domains [3, 21–23]. Hard domains play the role of reinforcing fillers in SUEs and cross-linking points in a physical network [24, 25].
The ultimate physicо-mechanical properties of the elastomers under strain represent important criteria for a material’s ability to withstand applied loading and strain. Hard domains are primarily responsible for the high strength of heterogeneous SUEs [3, 23, 26, 27], whereas the soft matrix plays a subordinate role [24–27].
The concept of reorganisation of highly strained structures and the SUE hardening mechanism [3, 23, 26–31] enabled the explanation of the mechanical behaviour of many elastomers characterised by a high degree of microphase separation.
Recently, an inverse strength–strain rate dependence, as opposed to a direct one observed in the case of single-phase cross-linked elastomers, was shown to be typical for heterogeneous SUEs. The tensile strength of heterogeneous SPUUs (and SPUs) can be increased to 1.5–2-fold at lower stretching rates [32]. Under the same conditions, the strength of single-phase polyurethanes decreases by one order of magnitude [26]. The available data suggest that soft and hard phases influence the rate dependence of strength in opposite manners for the investigated block copolymers.
The effect of the interchain interaction in the soft phase and the effect of microphase separation on the character and extent of variation in ultimate SUE physico-mechanical properties were comprehensively considered in a wide range of stretching rates.
This article consists of two parts. The first part presents results of an investigation into the ultimate physico-mechanical properties of block copolymer versus the stretching rate dependence while its structure evolves under various time–temperature conditions. Polyurethane urea containing the same blocks (SSHS-type) was used as a test subject. The chemical composition of the elastomer was not varied during the tests. The second part describes the behaviour of two polymer systems containing mixed hard segments. Heterogeneous systems featuring various hard segment solubilities in the soft polymer phase were investigated. A variation in the ratio between various hard segments enabled the control of interchain interactions in this phase and in the microphase separation of hard and soft segments.
This article is a logical continuation of our previous study on diblock copolymers with polyether soft segments as well as urethane and urethane urea hard blocks (segments) [32].
Experimental section
Materials
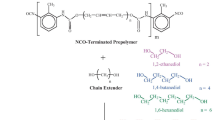
The following compounds were used as initial components for the synthesis of SUEs: 2,4-toluene diisocyanate (TDI) (BASF, Germany), isophorone diisocyanate (IDI) (Evonik Chemistry Ltd., Russia), oligotetramethylene oxide diol (PTMO; BASF, Germany) with Mn = 1000 g mol−1 as a chain extender, methylene-bis-o-chloroaniline (MOCA) (Mitsubishi International Corp., Japan) and butanediol-1,4 (BD) (Sigma-Aldrich). TDI, IDI and MOCA (99 % purity) were used without purification. Water was removed from PTMO and BD at 90 °C while stirring for 7 h under a vacuum of 0.2 kPa. Tributyl phosphate (TBP) used for swelling of elastomers samples was obtained from Sigma-Aldrich.
The synthesis was performed as a two-step process, with preliminary preparation of prepolymers: FP-1 on the base of TDI and PTMO and FP-2 on the base of IDI and PTMO. The prepolymers were produced by the reaction of diisocyanates and PTMO (NCO/OH = 2.06), with the reaction temperature kept at 60–70 °C for FP-1 and 75–85 °C for FP-2, while stirring. The content of NCO groups in prepolymers was determined using the ASTM D2572-80 standard method.
Two series of samples were produced, that is, the SPU series with urethane HS1 hard segments and with urethane urea HS2 segments and the SPUU series with urethane urea HS2 and HS3 hard segments. Polyether soft segments in both sets of experiments were the same.
To synthesise SPU, prepolymer FP-1 and various blends of MOCA with BD, at a ratio of NCO/(OH + NH2) = 1.05, were used. The prepolymer was pre-stirred for 30 min at 60 °C under vacuum (with residual pressure of 0.2 kPa within the reactor). Then, the melted MOCA, or its blend with BD, was injected into the reactor; in the case of one chain extender, it was only BD that was injected. The blend of prepolymer with the chain extender was stirred for 3 min at 60 °C under vacuum, and then it was cast onto 2 mm-thick metal moulds and kept at 90 °C for 3 days. Conversion completeness of NCO groups was controlled using Fourier transform infrared spectroscopy (FTIR) spectroscopy. A series of additional SPU* samples, obtained for comparison, was prepared using PTMO with Mn = 2000 g mol−1, instead an analogous oligomer with Mn = 1000 g mol−1.
The chain extender (MOCA), FP-1, FP-2 and their blends at NCO/NH2 = 1.0 were used to produce the SPUU series. Owing to the low reactivity of FP-2, the curing duration for the samples at 90 °C was 4 days. A model polyurea (PU) was synthesised in order to analyse the FTIR spectra of samples. This synthesis, on the basis of IDI and MOCA, was performed in strict accordance with the technique described in [33], at a molar ratio of NCO/NH2 = 1.0.
The SPUU-2 composition was additionally prepared on the basis of FP-1 prepolymer and of MOCA at a ratio of NCO/NH2 = 0.96. Almost complete conversion of the NCO groups in such a system was already observed after only 1 day at 25 ± 1 °C, owing to the excess of NH2 groups in the reaction mix. The given polyurethane urea was used to study the behaviour of the block copolymer while its structure evolved. The SPUU-2 samples were kept at 25 °C for different time periods. Then, their physico-mechanical properties and the glass transition temperature of the soft phases were examined. Some samples were kept at 25 °C for 12 days, and then one day at 90 °C. Chemical structures of the HS1 hard segments and of the HS2 and HS3 urethane urea segments are shown below.
HS1 (based on TDI and BD)

HS2 (based on TDI and МОСА)

HS3 (based on IDI and МОСА)

The structural formula of polymer chains of the SPU series block copolymers (while neglecting excessive NCO groups during the synthesis) can be presented as
where molar fractions of hard segments x 1 and x 2 are presented as the molar fractions of BD and MOCA diamine in the blend; x 2 is the molar fraction of HS1 urethane blocks as the total amount of hard segments. The molar fraction of the urethane urea segments was calculated as x 1 = 1 − x 2.
The structural formula of the SPUU block copolymers is as follows:
where n 1 and n 2 are the molar fractions of HS2 and HS3 hard segments and are given as the molar fractions of FP-1 and FP-2 prepolymers in the blend. The molar fraction of HS2 is calculated from the equation n 1 = 1 − n 2.
Compositions of the SUE samples of the SPU and SPUU series, as well as the concentrations of hard segments, are summarised in Table 1.
Methods
The glass transition temperature (T s g ) of the soft phase was determined by differential scanning calorimetry (DSC), using a DSC822e calorimeter (Mettler Toledo) at a scanning rate of 0.08 K s−1; the softening temperature (T h ) of the hard phase was determined by means of thermo-mechanical measurements, using the TMA/SDTA841e device (Mettler Toledo) at a scanning rate 0.05 K s−1 under a load of 0.015 MPa.
FTIR spectra in the area of carbonyl valence vibrations (between wave numbers ν = 1600 and 1760 cm−1) of the investigated samples were recorded using a IFS-66/S spectrometer (Bruker, Germany) with spectral resolution of 1 cm−1.
Mechanical tests were conducted at 25 °C on the Instron 3365 universal stretching machine with the Standard Video Extensometer in order to reliably measure the strain values of the samples. Tests were performed at a constant rate in 0.56–0.003 s−1 range. Tensile strength (σ k ), that is, the maximal stress value calculated for initial cross-sectional area of samples, the relative critical strain value [ε k (%)], 100 % modulus (E 100), that is, the stress value calculated for the initial cross-section of samples at the relative strain 100 % and the true tensile strength (f r = σ k ⋅ λ k ), where λ k = (ε k + 100)/100, were determined under various conditions.
The effective density of the spatial network conditioned by the hard segment domains and by the chemical cross-linkage (N dx ) was determined using the Cluff–Gladding–Pariser method [34], using equilibrium samples that had been swollen in toluene, which did not affect the hard domains [17, 24, 35, 42], and using samples swollen in TBP. High swelling in TBP completely destroys the domain structure of polymers [18, 25, 35, 42]. The total effective density N dx of the network was calculated from the results of equilibrium compression modulus values obtained under low-strain values (up to 5 %) for samples swollen in toluene to an equilibrium state. The effective density N x of the chemical network was calculated from the results for the samples swollen in TBP to an equilibrium state. Thus, the value of network density conditioned by hard domains was calculated as N d = N dx − N x .
Results and discussion
Diblock copolymer SPUU-2 with hard urethane urea segments
The results of mechanical testing of SPUU-2 polyurethane urea show unusual mechanical behaviour of the elastomer under different conditions. After 1 day of storage of the samples at 25 °C, their engineering strength (σ k ) decreases approximately 20-fold, as the stretching rate diminishes by two orders of magnitude. Four days later, the σ k versus the stretching rate υ dependence becomes extremal. After 12 days of storage at 25 °C, the strength properties of the material stabilise in a υ range from 0.56 s−1 (−lnυ = 0.58) to 0.003 s−1 (−lnυ = 5.81). An additional exposure of some samples to 90 °C for 1 day leads to a more pronounced σ k versus υ dependence. It is worth noting that the strength properties of the material do not improve after exposure to elevated temperature; they become even worse as the stretching rate accelerates. At this point, the rate dependence of E 100 weakens (Table 2).
It is interesting to note that strong hardening of elastomeric samples before rupture is observed after 4 days of exposure to 25 °C (Fig. 1). Rupture of elastomeric samples usually occurs under high strains. Therefore, discussions of elastomers hardening and the consideration of their strength dependence on various factors are better carried out using the f r (true strength) value than the σ k value [3, 32]. Henceforth, we use this approach, based on the strength theory of polymers.

Stress–strain plots for polyurethane urea SPUU-2 after the store under various conditions (υ = 0.056 s−1)
Analysis of the rate dependence of the true strength for SPUU-2 shows that all basic plots of the f r versus υ dependence (direct, inverse and extremal [32]) can be observed for the same block copolymer samples subjected to various storage conditions (Fig. 2a).

Effect of the store conditions on the rate dependence of polyurethane urea SPUU-2 for the true tensile strength fr (a) and the relative critical strain (b)
The appearance of characteristic bands of a carbonyl group in FTIR spectra of polyurethane ureas under investigation reveals important features of elastomer structural organisation at various stages of its formation.
The position of the carbonyl absorption band in the FTIR spectra of polyurethanes and polyurethane ureas can be shifted, to some extent, for compositions containing various diisocyanates, diamines and low-molecular-mass diols. Identification of C = O absorption bands was made using the results of spectral investigations of real and model compositions with urethane urea and urethane groups [16, 22, 27, 36–42].
The band at ν = 1640 cm−1 relates to the absorption of ordered hydrogen-bonded urea carbonyl groups [16, 22, 27, 37, 39]. Urea groups are bonded by H–bonds (self-associates of urea groups) and are localised in the hard segment domains. The intensity of this band can be used for a comparative evaluation of the microphase separation degree of hard and soft domains [16, 41]. The band at 1692–1693 cm−1 in the spectra of polyurethane urea can be related to the absorption of hydrogen-bonded, ordered C = O urethane and of free urea carbonyl [16, 36, 37, 41]. The band at 1730–1732 cm−1 is assigned to the absorption of free urethane carbonyl groups and the band at 1710–1711 cm−1 to hydrogen-bonded urethane carbonyl groups in the soft phase of the polymer [27, 36, 37]. The band at 1665 cm−1 is assigned to the absorption of hydrogen-bonded, disordered urea carbonyl [36].
At 25 °C, a low degree of microphase separation of hard and soft segments can be observed for SPUU-2 samples, especially the next day, which is distinctly evidenced by the corresponding bands of the FTIR spectrum (Fig. 3). Hard urethane urea segments are predominantly localised in the soft polymer matrix (carbonyl absorption bands at 1665, 1677, 1711 and 1730 cm−1). The absorption band associated with hydrogen-bonded, ordered urea carbonyl (1643 cm−1) is distinctly apparent 12 days later, after storage at 25 °C. However, the intensity of this band is lower compared with the C = O band at 1640 cm−1 after exposure of the polyurethane urea to temperature of 90 °C.

FTIR spectra for polyurethane urea SPUU-2 samples at various stages of the structure evolution
The low degree of microphase separation in SPUU-2 samples means that hard polar segments, that is, proton donors, are mainly located in the soft phase of the material. As a consequence, the glass transition temperature should increase. This increase should be greater for the lower storage times of polyurethane urea at 25 °C. In fact, this phenomenon is observed in the experiments (Fig. 4). Considerable reduction in the effective density of the physical network with hard domains, playing the role of cross-linking points, indicates a better miscibility of soft and hard phases. There are no chemical cross-linking points in the SPUU-2. All of the samples under investigation are soluble in TBP.

DSC curves for polyurethane urea SPUU-2 samples
As mentioned above, the intensity of the absorbance band for hydrogen-bonded ordered urea carbonyl groups is characterised by a degree of microphase segregation between hard and soft segments. The results obtained with other methods (DSC, values of physical network density N d ) show the same tendency of microphase segregation in a polymer during the thermostatting process. Thus, one can see the full correlation between data obtained by various methods.
Enhancement of interchain interactions in the soft phase leads to drastic distinctions in the mechanical behaviour of block copolymers. This is distinctly apparent when comparing results of mechanical tests of samples at various stages of polyurethane urea structure evolution. At the initial stage of microphase separation (after 1 day of storage at 25 °C), the true strength value of the samples decreases steeply, whereas the stretching rate is lowered (Fig. 2a). The plot of the f r versus υ dependence is the same as the one for single-phase SUEs with labile physical networks [26, 32]. Relaxation of such network leads to a decrease in the strength of the material when the stretching rate decreases [32, 42]. The difference is that the relative critical strain of SPUU-2 samples increases as the stretching rate decreases (Fig. 2b).
At the next stage of microphase separation, the effective density of the polyurethane urea network, determined by the hard domains, increases (Table 2). The true strength of the samples increases significantly and the plot f r versus υ becomes extremal (Fig. 2a).
This phenomenon is explained in [32]. The extremal rate dependence of the strength for block copolymers can be explained by the superposition of two factors: (1) an increase in the orientation extent of hard segment domains under high strains, as the stretching rate decreases and (2) a decreased density of the labile physical network, owing to relaxation processes in the soft phase. Orientation of hard segments leads to hardening of the material, whereas relaxation of the labile physical network leads to a decrease in the strength with slow stretching. The hard phase mainly influences the mechanical behaviour of polyurethane urea at a high degree of microphase separation of hard and soft segments after exposure of samples to 90 °C. At high rates of mechanical loading, the orientation of hard segments in domains along the stretching direction is retarded, and this phenomenon leads to the inverse rate dependence of f r on υ for polyurethane urea.
The strength of heterogeneous elastomer decreases significantly as the stretching rate increases. The ratio K f(max) of the maximal value of the true strength (f r ) to its minimal value at υ = 0.56 s−1 (−lnυ = 0.58) equals 1.74 (Fig. 2a).
High strength properties of materials within the whole range of stretching rates can be attained at a low degree of microphase separation after 12 days of exposure to 25 °C. The true strength of polyurethane urea depends slightly on the stretching rate (K f(max) = 1.27). This is not paradoxical, and, in our opinion, a reasonable explanation can be given, taking into account the results of earlier investigations.
Thus, investigations into the mechanical behaviour of block copolymers show the strength properties of heterogeneous elastomers to be substantially determined by interchain interactions in the soft phase at the initial stage of structure formation; at the ultimate stage, these properties are determined by the domain structure. An optimal combination of domain structure and interchain interactions in the soft phase is attained at a low degree of microphase separation. Results of the conducted investigations and the ascertained regularities have constituted a basis for further studies of structure–property relationship of urethane-containing triblock copolymers of the SPU and SPUU series.
Triblock copolymer SPU with urethane urea and urethane segments
In the SPU series, the investigated samples differ in molar contents (x 2) of urethane segments (HS1) in hard blocks (urethane segments + urethane urea segments HS2), including samples with hard blocks (segments) of only one uniform type, that is HS1 (x 2 = 1) or HS2 (x 2 = 0). Samples were cured at 90 °C.
By using FTIR spectroscopy, the SPUs with uniform urethane segments were attributed to single-phase elastomers. There is no band at 1700–1703 cm−1 attributed to the absorption of hydrogen-bonded, ordered urethane carbonyl (C = O-associated groups of urethane segments localised in the hard phase of polymer [40]), in the spectrum of the sample at х 2 = 1.
The band at 1731 cm−1 for the absorption of free urethane carbonyl and the band at 1710 cm−1 for disordered urethane carbonyl are also observed (Fig. 5). A high degree of microphase separation, indicated by the intensive band at 1640 cm−1, is typical for polyurethane urea based on FP-1 and on MOCA. The intensity of this band decreases naturally after partial replacement of urethane urea segments with urethane segments. The band at 1700–1703 cm−1 does not appear. The glass transition temperature (T sg ) of the soft polymer matrix increases as the molar fraction of the urethane segments (x 2) in the hard blocks increases (Fig. 6 and Table 3). At this point, the effective density value of the physical network N d decreases and equals zero at x 2 = 1. Thus, urethane urea segments dissolve in the soft phase of the polymer. The softening temperature (T h ) of the hard phase decreases significantly at x 2 > 0.5, thus indicating imperfection of hard-domain structure (Table 3). Results of mechanical tests of SPU samples at the stretching rate of 0.28 s−1 are summarised in Table 3.

FTIR spectra for SPU samples with various molar fractions x 2 of urethane segments in the hard blocks

DSC curves for SPU samples with various combinations of hard segments
It is apparent that the critical strain of the material increases as the total effective network density (N dx ) decreases. At this point, the conditional modulus (Е 100) decreases significantly. The ε k and Е 100 values vary non-linearly, depending on the variable content of urethane blocks (x 2). The dependence of the true strength of SPU versus the composition of hard segments is extremal and has its maximum at equal quantities of urethane and urethane urea segments in the material (Fig. 7).

The true tensile strength of SPU versus stretching rate and versus compositions of hard segments dependences (at υ = 0.28 s−1)

FTIR spectra of SPUU triblock copolymer containing various urethane urea segments and FTIR spectra of PU model polyurethane urea

DSC curves for SPUU series with various molar fractions n 2 of urethane urea segments HS3 in the total hard segments amount (HS2 + HS3)

The true tensile strength versus stretching rate dependence of SPUU samples containing variously composed hard segments

The true tensile strength of SPUU samples versus compositions of urethane urea segments dependence at variable stretching rate values
The f r versus (−lnυ) dependence shows an increase in SPU strength within a wide range of stretching rates (υ), from 0.56 s−1 (−lnυ = 0.58) to 0.003 s−1 (−lnυ = 5.81). The maximal strength increase is observed at high stretching rates. We succeeded in compensating the negative influence of the hard phase on the rate dependence of the elastomer strength by increasing the interchain interactions in the soft phase (resulting from the replacement of half of the urethane urea HS2 segments with urethane HS1 segments). The ratio with a maximal value of f r to its minimal reference value at υ = 0.56 s−1 (K f(max)) decreases from 1.68 to 1.28 (Fig. 7).
The obtained data correspond completely to developed ideas about the role of the soft phase in the hardening of the SUEs, and for its influence on the f r versus υ dependence of block copolymers.
Mechanical tests have shown the relative critical strain (ε k ) for SPUs with non-uniform hard segments to be dependent on the effective network density (N d ), as determined by the domain structure of the material. The density of the chemical network (N x ) is low (Table 3). Variations in the ε k values do not exceed 10 % at different stretching rates.
The results of the conducted investigations show unusual physico-mechanical properties of the SUE, including the fact that the strength of the material increases as the modulus value decreases considerably (Table 3 and Fig. 7). The total concentration of hard blocks in the investigated materials differs slightly after replacement of certain hard segments with others (Table 1). Thus, the properties of the elastomers were insignificantly influenced by the concentration of hard segments.
It is interesting to note that the increase twofold in the molecular mass of PTMO soft segments does not diminish the solubility of urethane segments in the soft polymer phase. FTIR spectra of SPU* with a molecular mass of PTMO segments equal to 2000 g mol−1 are close to the similar spectra of SPU samples in the carbonyl absorption area (Figs. 5). The band at 1700–1702 cm−1 is absent in the spectra of all samples. The urethane segments are dissolved in the soft phase of SPU*, which leads to an increase in the interchain interactions in this phase.

FTIR spectra for SPU* samples on the base of PTMO 2000 with various molar fractions x 2 of urethane segments in the hard blocks
The nature of the mechanical behaviour of triblock copolymer does not vary significantly with increasing molecular mass of the PTMO soft segments. However, the maximum strength is observed at a lower mole fraction of urethane segments HS1 composed of hard blocks (x 2 = 0.3). In this case, a considerable stabilisation of the heterogeneous elastomer strength values does not take place in a wide range of stretching rates (parameter K f = 1.41). Soft phase affects the strength of the material in a less extent, this can be seen by comparing the f r versus x 2 dependence for SPU and SPU* (Figs. 7).

The true tensile strength of SPU* samples on the base of PTMO 2000 versus stretching rate, and versus compositions of the hard segments dependences (at υ = 0.28 s−1)
Triblock copolymers SPUU with diverse urethane urea segments
Polyurethane urea samples of the SPUU series contained different hard segments of HS2 and HS3 types. The selection of isophorone diisocyanate for the preparation of HS3 hard segments was intentional. It had previously been shown [14] that a “loose” (low-ordered) structure of the hard phase of SUE was formed when using isophorone diamine as a chain extender (ultimate mechanic properties of elastomer were not determined). The formation of a “loose” hard phase is conditioned by a cumbersome structure of this diamine that does not satisfy the regularity principle [14]. The structure of isophorone diisocyanate is similar. Taking into account this consideration, hard domains, unlike hard phases based on TDI and MOCA (with HS2 hard segments), can be expected to have analogous loose structures when using IDI and MOCA for the synthesis of polyurethane ureas.
Qualitative analysis of SPUU samples using FTIR spectroscopy enables important features in the elastomers structure with various molar fractions (n 2) of urethane urea segments (HS3) in hard segments to be revealed (Table 1). An intensive absorption band at 1640 cm−1 of hydrogen-bonded, ordered urea carbonyl is observed in the case of a sample with uniform HS2 hard segments based on TDI and MOCA. Replacement of 30 % of the HS2 segments with HS3 urethane urea segments in the polymer chains leads to a significant decrease in the intensity of this band (Fig. 8).
The band of hydrogen-bonded urea carbonyl shifts by 20 cm−1 (1660 cm−1) in the FTIR spectrum of polyurethane urea with HS3 hard segments (n 2 = 1). The validity of this band assignment is confirmed by the data from a PU model composition obtained through the reaction of IDI and MOCA (the band at 1662 cm−1) and by the data obtained from samples of polyurethane urea with hard segments based on diisocyanate and isophorone diamine [14]. The low-intensity band at 1705 cm−1 in the FTIR spectrum of the PU model can be assigned to the absorption of free carbonyl groups of urea groups (there are no urethane groups in the model).
There is also an absorption band of hydrogen-bonded urea carbonyl groups in the spectrum of a sample with equal quantities of different hard segments. No band at 1640 cm−1 was identified by analysis of second derivative of the spectral curve. Hence, the degree of microphase separation decreases in samples containing different hard segments in the polymer chains.
The glass transition temperature values of the soft phase of SPUUs with uniform hard segments (HS2 or HS3) vary slightly. In this case, an improvement in the compatibility of hard and soft segments should lead to extremal dependence of the glass transition temperature (T s g ) of polyurethane urea soft phase on the composition of urethane urea segments; this assumption was confirmed experimentally (Fig. 9 and Table 4).
A decrease in the degree of microphase separation leads to decreased values of the 100 % modulus. The minimal Е 100 value is observed at various stretching rates for compositions with equal quantities of different urethane urea hard segments. The relative critical strain increases slightly when the fraction of HS3 urethane urea segments decrease from n 2 = 1 to n 2 = 0 (material with HS2 urethane urea segments). In practice, the stretching rate does not influence the ε k value (Table 5).
The true strength f r of polyurethane urea with uniform hard segments can be higher than that of a composition with uniform urethane urea segments in a wide range of stretching rates (Fig. 10). The dependence of the extremal strength versus the composition of hard segments is more pronounced at higher stretching rates υ = 0.17, 0.28 and 0.56 s−1 (Figs. 11,12 and 13). Enhancement of interchain interactions in the soft phase leads to stronger hardening of triblock copolymers at fast stretching. This regularity is observed for all heterogeneous urethane-containing materials investigated in this work.
The presence of loose-structured hard phases in elastomers with urethane urea segments based on IDI and MOCA (n 2 = 1) is confirmed by the data obtained using several methods, including a low softening temperature of the hard phase (T h ) and high equilibrium swelling degree (Q) in low polar solvents (toluene). These data differ from those for polyetherurethane ureas with HS2 hard segments (Table 4, [32]). The N d parameter of the physical network of the SPUU cannot be determined by the swelling of samples in toluene when the hard phase of the material is loose structured.
Such a structure was found to lead to the weakening of the rate dependence of heterogeneous elastomer strength; however, the f r value of polyurethane urea based on PTMO, IDI and MOCA is considerably lower compared with the strength of analogous TDI-based polymers.
Enhancement of interchain interactions in polyurethane urea is also a reason for the reduction of the f r versus υ dependence of a material in the case of the minimal value of the K f(max) coefficient at n 2 = 0.5. The K f(max) versus n 2 dependence should be linear in the absence of this effect.
The conducted investigations show a strong influence of the soft phase on the strength value and on the strength–rate dependence for urethane-containing elastomers, and this may be useful in different application fields. It is not improbable that a similar effect is likely to be achieved for SUEs with mixed soft segments, provided that the solubility of hard segments in the soft phase would be significantly increased. Investigations in this field are of considerable interest and can be further continued.
Conclusions
A non-traditional approach was used to study the influence of the soft phase on the ultimate physico-mechanical properties of heterogeneous urethane-containing elastomers. The basics of the approach were the conceptions of an interrelationship between the ultimate mechanical properties of variously structured polyurethanes and polyurethane ureas with the stretching rate thereof.
Properties of polyurethane ureas with alternating soft and hard segments were investigated within the process of evolution of its structure. Direct, extremal and inverse rate dependencies of the strength of urethane-containing elastomers were demonstrated without changing its composition. It was shown that the type dependence relates to the degree of microphase separation between the soft and hard segments within a material (tests were performed in a wide range of stretching rate values of 0.56–0.003 s−1).
It was found that diblock copolymers with high strengths can be obtained at a low degree of microphase separation between the hard and soft segments. Reinforcing the interchain interactions in a soft polymer matrix at the drop of concentration of the hard phase in urethane-containing elastomer promotes an increase in the strength value. A significant effect of the soft phase on the rate dependence of tensile strength and critical strains was shown for diblock copolymers, especially in the case of polyetherurethane urea with various microphase separation degrees.
It had previously been shown that a decrease in the stretching rate led to an elevation of the orientation degree of hard segments in the domains of polyurethane (polyurethane urea) under high strain. This phenomenon contributes to a larger strength of polymer at lower stretching rates. The relaxational nature of the labile physical network in the soft phase should lead to the opposite effect. Consequently, the effect of soft and hard phases on urethane-containing elastomer properties can be balanced for the strength value to be stable at different stretching rates.
Two series of new triblock copolymers with the same polytetramethylene oxide soft segments and various hard segments in the polymer chain were synthesised (1) with compositions containing poorly soluble urethane urea segments and urethane blocks soluble in the soft phase of the polymer and (2) heterogeneous elastomers comprising urethane urea segments of different types. Variations in hard segment compositions led to a variation in the content of polar hard blocks (proton acceptors in the soft phase of material) and enabled the control of the interchain interactions in this phase.
Both series of triblock copolymers were ascertained in order to show the dependence of the extremal strength on the composition of hard segments. In all cases, the highest hardening degree was achieved at an increased stretching rate, thus leading to a substantial reduction in the rate dependence of the strength. The dependence of the extremal 100 % modulus on the composition of hard blocks is caused by a decrease in the degree of microphase separation, and it is observed throughout the whole range of stretching rates. The results of these studies are in line with our developed ideas about the role of soft phases in the hardening effect and on the rate dependence of the ultimate physico-mechanical properties of heterogeneous urethane elastomers. The “loose” hard phase of segmented polyurethane urea has been ascertained to positively influence the stability of urea strength properties at various strain rates.
In this case, the increase in solubility of hard segments in the soft phase also allows the rate dependence of strength for urethane-containing elastomers to be reduced.
The ascertained regularities can find an application in a variety of technologies in order to produce urethane-containing block copolymers with improved physico-mechanical properties. Further studies can be aimed at ascertaining behavioural regularities of multiblock copolymers comprising mixed soft segments under various conditions of stretching.
References
Randall D, Lee S (2003) The polyurethanes book. Wiley, New York
Hepburn C (1992) Polyurethane elastomers. Elsevier Applied Science, London and New York
Petrović ZS, Ferguson J (1991) Polyurethane elastomers. Prog Polym Sci 16:695. doi:10.1016/0079-6700(91)90011-9
Lamba NMK, Woodhouse KA, Cooper SL (1998) Polyurethanes in biomedical applications. CRC Press, Boca Raton
Bagdi K, Molnar K, Wacha A, Bota A, Pukánszky B (2011) Hierarchical structure of phase-separated segmented polyurethane elastomers and its effect on properties. Polym Int 60:529. doi:10.1002/pi.3003
Fedoseev MS, Devyaterikov DM, Rybina GV, Meshechkina AE (2013) Curing of oligo(diene-isoprene-urethane-epoxy) oligomer in the presence of an active plasticizer, 1,2-epoxycyclopentane, and of its adducts with imidazoles. Russ J Appl Chem 86:1441. doi:10.1134/S1070427213090217
Unal S, Yilgor I, Yilgor E, Sheth JP, Wilkes GL, Long TE (2004) A new generation of highly branched polymers: hyperbranched, segmented poly(urethane urea) elastomers. Macromolecules 37:7081. doi:10.1021/ma049472e
Volkova ER, Tereshatov VV, Vnutskikh ZA (2010) Formation of polyurethane structural materials based on mixtures of oligoethers with different reactivities. Russ J Appl Chem 83:1372. doi:10.1134/S1070427210080082
Zhao CT, de Pinho MN (1999) Design of polypropylene oxide/ polybutadiene bi-soft segment urethane/urea polymer for pervaporation membranes. Polymer 40:6089. doi:10.1016/S0032-3861(98)00833-7
Hong Y, Guan J, Rujimoto KL, Hashizume R, Pelinescu AL, Wagner WR (2010) Tailoring the degradation kinetics of poly(ester carbonate urethane)urea thermoplastic elastomers for tissue engineering scaffolds. Biomaterials 31:4249. doi:10.1016/j.biomaterials.2010.02.005
Buist JM (1978) Developments in polyurethane-1. Applied Science Publishers, London
Wang CB, Cooper SL (1983) Morphology and properties of segmented polyether polyurethaneureas. Macromolecules 16:775. doi:10.1021/ma00239a014
Bagdi K, Molnár K, Sajó I, Pukánszky B (2011) Specific interactions, structure and properties in segmented polyurethane elastomers. Expr Polymer Lett 5:417. doi:10.3144/expresspolymlett.2011.41
Ahn TO, Jung SU, Jeong HM, Lee SW (1994) The properties of polyurethanes with mixed chain extenders and mixed soft segments. J Appl Polym Sci 51:43. doi:10.1002/app.1994.070510105
Makarova MA, Tereshatov VV, Strel’nikov VN (2010) Behavior in a humid medium of segmented polyurethane-ureas with dissimilar thermodynamically compatible and incompatible flexible blocks. Russ J Appl Chem 83:1360. doi:10.1134/S1070427210080069
Tereshatov VV, Tereshatova EN, Makarova MA, Tereshatov SV (2002) Influence of chemical structure and composition of mixed soft segments on the properties of elastomers with urethane–urea hard blocks. Polym Sci 44:275
Ishihara H (1984) Studies on segmented polyurethane-urea elastomers: structure and properties segmented polyurethane–urea having the binary segment components. J Macromol Sci Phys 22:763
Tereshatov VV, Strel’nikov VN, Makarova MA, Senichev VY, Volkova ER (2010) Structure and properties of segmented polyurethane-ureas with dissimilar soft blocks. J Appl Chem 83:1380. doi:10.1134/S1070427210080094
Mattia J, Painter P (2007) A comparison of hydrogen bonding and order in a polyurethane and poly(urethane-urea) and their blends with poly(ethylene glycol). Macromolecules 40:1546. doi:10.1021/ma0626362
Arun A, Gaymans R (2008) Tri-block copolymers with mono-disperse crystallizable diamide segments: synthesis, analysis and rheological properties. Polymer 49:2461. doi:10.1016/j.polymer.2008.03.049
Yamamoto T, Shibayama M, Nomura S (1989) Structure and properties of fatigued segmented poly(urethaneurea)s III. Quantitative-analyses of hydrogen-bond. Polymer J 21:895. doi:10.1295/polymj.21.895
Lee HS, Yoo SR, Seo SW (1999) Domain and segmental deformation behavior of thermoplastic elastomers using synchrotron SAXS and FTIR methods. J Polym Sci Polym Phys 37:3233
Christenson EM, Anderson JM, Hiltner A, Baer E (2005) Relationship between nanoscale deformation processes and elastic behavior of polyurethane elastomers. Polymer 46:11744. doi:10.1016/j.polymer.2005.08.083
Kontou E, Spathis G, Niaounakis M, Kefalas V (1990) Physical and chemical cross-linking effects in polyurethane elastomers. Colloid Polym Sci 268:636. doi:10.1007/BF01410405
Tereshatov VV, Tereshatova EN, Volkova ER (1995) Two types of physical networks in cross-linked segmented polyurethanes. Polymer Sci 37:1181
Smith TL (1974) Tensile strength of polyurethane and other elastomeric block copolymers. J Polym Sci Polym Phys 12:1825. doi:10.1002/pol.1974.180120907
Yeh F, Hsiao BS, Sauer BB, Michel S, Siesler HW (2003) In-situ studies of structure development during deformation of a segmented poly(urethane-urea) elastomer. Macromolecules 36:1940. doi:10.1021/ma0214456
Speckhard TA, Cooper SL (1986) Ultimate tensile properties of segmented polyurethane elastomers: factors leading to reduced properties for polyurethanes based on nonpolar soft segments. Rubber Chem Technol 59:405. doi:10.5254/1.3538208
Bonart R, Hoffmann K (1982) Orientation behavior of segmented polyurethanes as a function of foil preparation and the moment of determining the orientation. Colloid Polym Sci 260:268. doi:10.1007/BF01447964
Müller-Riederer G, Bonart R (1977) Orientierungsvorgänge bei der dehnung von polyurethan-elastomeren. Progr Colloid Polymer Sci 62:99. doi:10.1007/BFb0117099
Paiksung CS, Smith TW, Sung NH (1980) Properties of polyether poly(urethaneureas) based on 2,4-toluene diisocyanate. 2. Infrared and mechanical studies. Macromolecules 13:117. doi:10.1021/ma60073a023
Tereshatov VV, Makarova MA, Senichev VY, Slobodinyuk AI (2012) Interrelationship between ultimate mechanical properties of variously structured polyurethanes and poly(urethane urea)s and stretching rate thereof. Colloid Polym Sci 290:641. doi:10.1007/s00396-011-2585-7
Zhang S, Ren Z, He S, Zhu Y, Zhu C (2007) FTIR spectroscopic characterization of polyurethane-urea model hard segments (PUUMHS) based on three diamine chain extenders. Spectrochim Acta A 66:188. doi:10.1016/j.saa.2006.02.041
Cluff EF, Gladding EK, Pariser R (1960) A new method for measuring the degree of cross-linking in elastomers. Polymer Sci 45:341. doi:10.1002/pol.1960.1204514605
Tereshatov VV (1995) Variation of network parameters in segmented polyurethanes induced by tensile drawing. Polym Sci 37A:946
Luo N, Wang DN, Ying S (1996) Hydrogen bonding between urethane and urea: band assignment for the carbonyl region of FTIR spectrum. Polymer 37:3045
Luo N, Wang DN, Ying SK (1997) Hydrogen-bonding properties of segmented polyether poly(urethane urea) copolymer. Macromolecules 30:4405
Hofmann GR, Sevegney MS, Kannan RM (2004) A rheo-optical FTIR spectrometer for investigating molecular orientation and viscoelastic behavior in polymers. Int J Polym Anal Charact 9:245. doi:10.1080/10236660490920237
Wang SK, Sung CS (2002) Spectroscopic characterization of model urea, urethane compound, and diamine extender for polyurethane-urea. Macromolecules 35:877. doi:10.1021/ma011316
Seymour RW, Estes GM, Cooper SL (1970) Infrared studies of segmented polyurethane elastomers. I. Hydrogen bonding. Macromolecules 3:579. doi:10.1021/ma60017a021
Tereshatov VV, Makarova MA, Tereshatova EN (2004) Abnormal thermal and mechanical behavior of plasticized polyurethaneureas. Polym Sci 46:1332
Tereshatov VV, Senichev VY (1995) Stress–strain behavior of cross-linked polybutadiene urethanes. Polym Sci Ser A 37:702
Acknowledgments
This work was financially supported by the Russian Foundation for Basic Research, the Government of Perm Region (project 13-03-96000) and the Ural Branch of the Russian Academy of Sciences (project 12-T-3-1005).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tereshatov, V.V., Makarova, M.A., Senichev, V.Y. et al. The role of the soft phase in the hardening effect and the rate dependence of the ultimate physico-mechanical properties of urethane-containing segmented elastomers. Colloid Polym Sci 293, 153–164 (2015). https://doi.org/10.1007/s00396-014-3395-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-014-3395-5