
Abstract
We investigated the gelation process of 12-hydroxystearic acid (12-HSA)/dodecane gel (turbid gel), 12-HSA/xylene and 12-HSA/toluene gels (transparent gels) by using time-resolved synchrotron small-angle X-ray scattering technique. The fibers were developed via nucleation and growth mechanism, and the induction period was longer at higher temperatures. After the induction period, the fiber growth can be divided into two regimes. In the first stage, the scattering intensity increased with time without appearance of any crystalline peak, and in the second stage (001) peak appeared due to crystalline nucleation. In the former regime, the fibers grew with increasing the cross-sectional size, while in the latter regime, they grew with keeping it almost constant, i.e., with keeping the fiber thickness constant. The fiber thickness for the turbid gel (radius of the gyration R c of ∼100 Å) was thicker than that for the transparent gel (R c of ∼82 Å). The fractal dimensions of the fibrillar aggregates for the turbid gels (2.0–2.3) at various temperatures were larger than those for the transparent gels (1.4–1.6), suggesting that the structure of the former gels is more branching or more compact in comparison with the latter.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Studies of organogels using low-molecular mass organogelators (LMOGs) have received much attention because of a diverse range of applications such as food, cosmetic, and petrochemical industries. The material forms a three-dimensional network composed of fibrillar aggregates by self-assembly of LMOGs in an organic solvent, so that an organogel is formed [21, 23]. Formation and dissolution of the fibrillar network are usually thermally reversible, and therefore thermally reversible sol–gel transition occurs. So far numerous studies have been conducted on the structures of the fibrillar network. In many cases, the fibrillar size or the shape is significantly affected by kinds of solvents [19, 20]. Temperature dependence of the fibrillar structure may be different by the kind of solvents or organogelators. Huang et al. reported that the morphology of organogels comprised of 5α-cholestan-3β-yl N-(2-naphthyl)-carbamate (CNC) changes from a spherulite to fiber-like structure using optical microscopy, as temperature is increased [6]. Similar observation was reported for 12-hydroxystearic acid (12-HSA) gels in canola oil by Rodger et al. [13, 14]. On the other hand, Takeno et al. showed that the cross-sectional size and the size distribution of the fibers are not affected by change in temperature or composition for apparently transparent 12-HSA/toluene gels by using small-angle neutron scattering (SANS) [17], while the fibrillar network structures of apparently turbid 12-HSA/dodecane gels changes with the composition variation [18].
Studies on the gelation kinetics as well as structures of the organogels in the equilibrium state (or the steady state) are important in understanding of the gelation mechanism. So far some researchers have studied kinetics of organogel formation using circular dichroism [5, 6], fluorescence [5, 6], differential scanning calorimeter [12], synchrotron small-angle X-ray scattering (SAXS) [15, 17], and rheological measurements [5–7, 22]. Liu et al. supposed that the self-assembling process proceeds via nucleation-growth-crystallographic mismatch branching mechanism using optical and scanning electron microscopies for a Lauroyl-l-glutamic-α-γ-dibutylamide (LGBA) gel [7]. Furthermore, they reported that the structure with a fractal dimension of 2.15 was formed for the LGBA gel in iso-stearyl alcohol using viscoelastic measurements. Huang et al. indicated that the fractal dimensions of CNC gels became smaller at higher temperatures and for lower concentrations [5, 6]. Thus, although some studies have been devoted to the gelation kinetics of LMOGs, the influence of the solvent on the kinetics is not sufficiently clarified at the present stage. Besides, only few studies have been conducted on the dynamics of the structural formation at the very early stage of the gelation before nucleation or immediately after it, since the self-assembling process of LMOGs usually proceeds so fast. Synchrotron SAXS measurements are a very powerful technique to pursue such fast process, because highly intense synchrotron X-ray beam enables us to measure in a very short time (typically ≤ 1 s). In this study, we investigated the gelation kinetics using time-resolved synchrotron SAXS technique. Here, we will report on the gelation process of a 12-HSA/dodecane gel (turbid gel), and a 12-HSA/toluene and 12-HSA/xylene gels (transparent gel).
Experimental
Sample and sample preparation
12-HSA sample was purchased from Aldrich Chemical Co. (purity 99 %). After the sample at a certain composition was dissolved in dodecane, xylene, or toluene by raising temperature, it was put at the ambient temperature to prepare gel. Apparently, a turbid gel was formed in dodecane, while a transparent gel was formed in toluene or xylene.
Cloud points
We obtained the cloud points (dissolution–aggregation temperature) for 12-HSA/dodecane solutions by carrying out the light scattering (LS) experiments using a He-Ne laser with a wavelength of 632.8 nm. We measured the scattering profiles at various temperatures and compositions using a glass cell with a diameter of 12 mm. If the scattering intensity at a given temperature was sufficiently strong, we assessed that the solution was in the aggregated state. Otherwise, it was assessed to be in the homogeneous state.
Time-resolved SAXS
Synchrotron SAXS measurements were conducted at the beam line 15A (BL15A) of the Photon Factory at the High Energy Acceleration Research Organization in Tsukuba, Japan. The X-ray scattering intensity was detected with an image intensifier coupled to a CCD camera. The detected data were corrected for distortion of the CCD image and electrical background. The CCD images were circularly averaged to obtain the scattering intensity as a function of wave vector q, where q is defined as q = 4π sin(θ/2)/λ. Here θ and λ represent the scattering angle and the wavelength of X-rays used in this study, respectively. The scattering profiles are corrected for background scattering, strength of incident X-ray beam, and exposure time. The samples for the SAXS experiments were put into a borosilicate glass tube with a diameter of 1.5 mm and their volume was ∼0.025 cm3. Time-resolved SAXS measurements were carried out with the temperature-jump cell designed for investigation of a fast self-assembling process. The detail of the cell was described elsewhere [15]. After the sample was quenched from a sol state into a gel state, the SAXS data were captured as a function of time.
Results and discussion
Kinetics of the cross-sectional size of crystalline fibers
We present SAXS profiles of a 12-HSA/dodecane gel at various times after quench from 85 °C (sol state) into a target temperature (40 °C) in Fig. 1, (a) before crystalline nucleation and (b) after the nucleation. The melting point of the gel T m was 70.5 °C according to the previous result using a differential scanning calorimetry [18]. In the early stage, the scattering intensity increased with time without appearance of any peak as shown in Fig. 1a. At t = 18.6 s after T-jump, a scattering peak appeared at q ≅ 0.12 Å−1 as shown in Fig. 1b, which was attributed to (001) reflection as reported in the previous papers [16, 18, 20]. In this regime, although the scattering intensity increased with time, the profile showed almost the same shape in the log-log plot. After t ≅ 32 s the scattering profile showed no significant change due to gelation. Similar behavior was also seen at other temperatures. Figure 2 shows the scattering profiles of a 12-HSA/xylene gel at 36 °C at various times. Crystalline nucleation took place at t = 31.9 s and afterwards the profiles had almost the same shape in the double logarithmic plot, similar to 12-HSA/dodecane gels. Thus, irrespective of turbid gels or transparent gels, the gelation kinetics shows a similar behavior. Figure 3 presents the time t nc, when crystalline nucleation occurs, i.e., (001) peak appears, at various temperatures for 12-HSA/dodecane gels. t nc was remarkably longer at higher temperatures. This result is also consistent with that obtained for 7 wt.% 12-HSA/ toluene gels, as reported in the previous study [17].

Time evolution of SAXS profiles for 12-HSA/dodecane gel at 40 °C in the regime before crystalline nucleation (a) and after the nucleation (b)

Time evolution of SAXS profiles for 12-HSA/xylene gel at 36 °C in the regime before crystalline nucleation (a) and after the nucleation (b)

Temperature dependence of the crystalline induction period for 12-HSA/dodecane gel
According to the classical homogeneous nucleation theory, the rate of nucleation J (=1/t nc) can be presented as a function of supersaturation ratio S and temperature T in the following form [1, 8, 24],
with
where C, V, R, and N A represent a constant, the molar volume of the gelator, the gas constant, and the Avogadro’s number, respectively. X and X * are the mole fraction of the gelator for the prepared sample and the equilibrium mole fraction at a given temperature, respectively, and γ is the interfacial tension. β equals to 16π/3 in the case of spherical nucleus. We obtained the dissolution–aggregation temperature at various compositions using the light scattering measurements in order to estimate X * at the measurements temperature of the SAXS experiments. The dissolution temperature can be often described by the van’t Hoff equation,
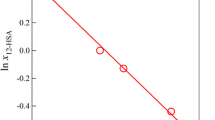
where ΔH d and ΔS d are the dissolution enthalpy and the dissolution entropy, respectively. Figure 4 presents the van’t Hoff plot of 12-HSA/dodecane solution obtained from the light scattering measurements. From the linear relation, we obtained ΔH d = 169 kJ mol−1, which is close to the value for 12-HSA/toluene gels [17]. We estimated X * at the temperature of the SAXS experiments from the van’t Hoff relation, and then obtained the supersaturation ratio S using the Eq. 2. Figure 5 shows the plot of ln J vs 1/(T 3 ln S 2) based upon Eq. 1. The plot shows a good linear relationship, from which we obtained the interfacial tension γ = 5.5 mJ m−2, assuming that γ and V are independent of temperature in the temperature range covered in this study. The value of the interfacial tension is close to that of lipid/fatty acid system [9–11].

The van’t Hoff plot for 12-HSA/dodecane gels

The plot of ln J vs 1/(T 3 ln S 2) for 12-HSA/dodecane gels
Here let us analyze the time-evolution of the cross-sectional size of the fibers. For sufficiently long rod particles with the length L, the form factor P(q) can be described as follows [3, 4]
for 2π/L < < q, where I c(q) corresponds to the scattering intensity from the cross-section of the rod particles. I c(q) can be represented by
for 2π/L < < q < < 2π/R c , where A and R c are the area of the cross-section and the radius of gyration of the cross-section, respectively. In the case of circular cross-section, R c is related to the radius of the circular section R, by \( {R}_c=R/\sqrt{2} \). Figure 6 presents the plot of ln(Iq) vs q 2 at 35 °C (a) and at 50 °C (b) for 7 wt.% 12-HSA/dodecane gel. The plot of ln(Iq) vs q 2 shows a good linear relationship at both temperatures. We estimated R c from the slope of the plot. Figure 7a shows time course of R c at various temperatures for 12-HSA/dodecane gels. The time course of R c can be divided into two regimes, as well as that of the scattering profiles. At the early stage, R c largely increased with time, and afterwards became constant in the next stage. The crossover time t cr, at which R c started to level off, was longer at higher temperatures. For comparison, we show the value of t nc at each temperature as a dotted line in Fig. 7. It is shown that the dotted line quite agrees with the crossover time t cr. The result means that after crystalline nucleation took place, the fiber grew with keeping the thickness constant. Moreover, we can see that the fibers with almost the same thickness were formed at different temperatures, which agrees with the behavior of 12-HSA/toluene gel as reported in the previous paper [17]. Here it is important to notice that the plot of R c vs t has a small peak at t ≅ t cr, i.e., after crystalline nucleation took place, the cross-sectional size slightly decreased with time. This behavior may reflect the fact that the molecular packing became denser because of crystallization. In earlier studies it has been shown that hydrogen bonding between hydroxyl groups along the fiber axis and “head-to-head” contacts between carboxylic acid groups along the c-axis are formed in 12-HSA gels [20]. In the early stage where (001) reflection is not observed, growth of the fibers originated from formation of hydrogen bonding and self-assembly of alkyl chains driven by van der Waals interactions may dominantly occur, and subsequently the “head-to-head” packing may do. The (001) reflection seen in the next stage is caused by the neat “head-to-head” arrangement of the 12-HSA molecules. Next, we show the time-evolution of R c for 12-HSA/xylene or 12-HSA/toluene gels in Fig. 7b. The time-course of R c is very similar to that of dodecane gels, although the cross-sectional size is smaller. The R c values at the late stage were ∼82 Å for 12-HSA/xylene or 12-HSA/toluene gels, while they were ∼100 Å for 12-HSA/dodecane gels. The difference of the cross-sectional size between the transparent gel and the turbid gel may be because the driving force of the aggregation for the latter is stronger than that of the former. As a matter of fact, the melting point of the turbid gel are much higher than that of transparent gel (T m = 70.5 °C for 7 wt.% 12-HSA/dodecane gel, T m = 44.7 °C and 48.0 °C for 10 wt.% 12-HSA/toluene gel and 12-HSA/xylene gel, respectively) [18]. We present a plot of R c vs the time reduced by t cr in Fig. 8. The plot superimpose at various temperatures, indicating that the difference in temperatures essentially does not affect the growth mechanism of the fibrillar cross-section in the temperature range covered in this study, although it does the rate of the aggregation. Figure 9 shows the scattering profiles reduced by (001) peak intensity at various times in the gelation process after crystalline nucleation at 45 °C for 12-HSA/dodecane gel. The reduced profiles at various times almost superimpose, suggesting that the whole fibers in the network exhibit crystalline nature. The similar result was reported for 12-HSA/toluene gels in the previous paper [17]. The SAXS profiles in Fig. 9 have broad shoulders at ∼0.04 and ∼0.07 Å−1 due to the particle scattering of rods (fibers). The peak positions correspond to the radius of ∼125 Å in the cross-section of rods, if we calculate the scattering curve with the form factor of randomly oriented rod particles [3, 4, 17, 18]. The value is reasonable in comparison with that estimated with Eq. 5 in the low-q range, if we consider that the cross-section has a size distribution, and R c obtained with Eq. 5 corresponds to z-average.

A plot of ln (Iq) vs q 2 for 12-HSA/dodecane gel at 35 °C (a) and at 50 °C (b)

Time dependence of R c for 12-HSA/dodecane gel (a) and for 12-HSA/xylene or 12-HSA/toluene gel (b)

A plot of R c vs t/t cr at various temperatures

The scattering profiles reduced by (001) peak intensity for 12-HSA/dodecane gel at 45 °C
Fractal analysis of fibrillar growth
It has so far been shown that the structures of the fibrillar network in low-molecular mass organogels often exhibit a fractal nature [5–7]. Weiss et al. [5] demonstrated that the gelation kinetics of organogels composed of a steroid type LMOG obeys the Dickinson’s fractal model, which was originally developed for colloidal aggregates [2]. According to the model, the time course of the volume fraction of the aggregates can be described as follows,
where A, ϕ eff, and d f are an experimental constant, the volume fraction of the aggregates, and a fractal dimension of the aggregates, respectively. In the case of our SAXS experiments, ϕ eff may be described by ϕ eff = (I q(t) − I q(0))/ (I q(∞) − I q(0)), where I q(t), I q(0) and I q(∞) represent the SAXS intensity at a given q, at time t, 0 and ∞, respectively. By replacing t with (t − t nc) in Eq. 6, we obtained d f of the aggregates from the slope of the graph of ln ϕ eff vs ln (t − t nc). Figure 10a and b present plots of ln ϕ eff vs ln (t − t nc) for 12-HSA/dodecane gel at T = 45 °C and 12-HSA/xylene gel at T = 36 °C, respectively, at a given q in the range from 0.01 to 0.07 Å−1. In both figures, a linear relation was obtained in the early stage of the gelation process, and the plot had almost the same slope at each q. This q-independent slope may reflect that the crystalline fibers grow with a dynamical self-similarity. Figure 11 presents the temperature dependence of the fractal dimension d f for 12-HSA/dodecane gel, and d f for 12-HSA/xylene gel and for12-HSA/toluene gel. d f for 12-HSA/dodecane gels showed almost the same values of 2.0 ∼ 2.3, independent of temperature and q. On the other hand, d f for 12-HSA/xylene gel and 12-HSA/toluene gel showed the value of 1.5 ∼ 1.6 and 1.4 ∼ 1.5, respectively, independent of q. Thus, fractal dimension of the turbid gel is quite larger than that of the transparent gel, which suggests that more branching or compact aggregates are formed in the turbid gel in comparison with the transparent gel.

The plot of data according to the Dinckinson’s model for 12-HSA/dodecane gel at 45 °C (a) and for 12-HSA/xylene gel at 36 °C (b)

Temperature dependence of fractal dimensions obtained by the analysis of the Dinckinson’s model for various 12-HSA gels
Summary
We investigated the gelation kinetics for 12-HSA/dodecane gel (turbid gel) and 12-HSA/xylene gel or 12-HSA/toluene gel (transparent gel) using synchrotron small-angle X-ray scattering. The fibrillar cross-sectional size (radius of the gyration) of the former was ∼100 Å, while that of the latter was ∼82 Å. Before the crystalline nucleation occurred, the fibrillar cross-sectional size became larger with time. On the other hand, after the crystalline nucleation took place, the fibrillar cross-sectional size became constant, i.e., the fiber grew with keeping the thickness constant. The fractal analysis revealed that the fractal dimension of the fibrillar network for the turbid gel was 2.0 ∼ 2.3, while that for the transparent gel was 1.4 ∼ 1.6, suggesting that the former’s network structure was more branching or compact in comparison with that of the latter.
References
Cheng WT, Li ZB (2010) Nucleation kinetics of nesquehonite (MgCO3 center dot 3H(2)O) in the MgCl2-Na2CO3 system. J Cryst Growth 312:1563–1571
Dickinson E (1997) On gelation kinetics in a system of particles with both weak and strong interactions. J Chem Soc Faraday T 93:111–114
Feigin LA, Svergun DI, Taylor GW (1987) Structure analysis by small-angle X-ray and neutron scattering. Plenum, New York
Glatter O, Kratky O (1982) Small angle X-ray scattering. Academic Press, London
Huang X, Raghavan SR, Terech P, Weiss RG (2006) Distinct kinetic pathways generate organogel networks with contrasting fractality and thixotropic properties. J Am Chem Soc 128:15341–15352
Huang X, Terech P, Raghavan SR, Weiss RG (2005) Kinetics of 5 alpha-cholestan-3 beta,6-yl N-(2-naphthyl)carbamate/n-alkane organogel formation and its influence on the fibrillar networks. J Am Chem Soc 127:4336–4344
Liu XY Sawant PD (2002) Mechanism of the formation of self-organized microstructures in soft functional materials. Adv. Mater. 14:421
Mullin JW (2001) Crystallization, 4th edn. Butterworth-Heinemann, Oxford
Petelska AD (2011) Interfacial tension of bilayer lipid membranes. Centr Eur J Chem 10:16–26
Petelska AD, Figaszewski ZA (2011) Interfacial tension of the lipid membrane formed from phosphatidylcholine-decanoic acid and phosphatidylcholine-decylamine systems. J Membr Biol 241:103–108
Petelska AD, Naumowicz M, Figaszewski ZA (2007) Interfacial tension of the lipid membrane formed from lipid-fatty acid and lipid-amine systems. Bioelectrochemistry 70:28–32
Rogers MA, Marangoni AG (2008) Non-isothermal nucleation and crystallization of 12-hydroxystearic acid in vegetable oils. Cryst Growth Des 8:4596–4601
Rogers MA, Wright AJ, Marangoni AG (2008) Crystalline stability of self-assembled fibrillar networks of 12-hydroxystearic acid in edible oils. Food Res Int 41:1026–1034
Rogers MA, Wright AJ, Marangoni AG (2009) Nanostructuring fiber morphology and solvent inclusions in 12-hydroxystearic acid/canola oil organogels. Curr Opin Colloid Interf Sci 14:33–42
Takeno H, Kikuchi N, Kondo S, Dobashi T (2007) Rheological and structural studies on gelation of 12-hydroxystearic acid solution. Trans MRS-J 32:835–838
Takeno H, Maehara A, Kuchiishi M, Yoshiba K, Takeshita H, Kondo S, Dobashi T, Takenaka M, Hasegawa H (2012) Structural and thermal properties of unpurified and purified 12-hydroxystearic acid solutions. Sen'i Gakkaishi 68:248–252
Takeno H, Maehara A, Yamaguchi D, Koizumi S (2012) A structural study of an organogel investigated by small-angle neutron scattering and synchrotron small-angle X-ray scattering. J Phys Chem B 116:7739–7745
Takeno H, Mochizuki T, Yoshiba K, Kondo S, Dobashi T (2009) Self-assembling structures and sol–gel transition of optically active and racemic 12-hydroxystearic acids in organic solvents. Prog Colloid Polym Sci 136:47–53
Terech P, Pasquier D, Bordas V, Rossat C (2000) Rheological properties and structural correlations in molecular organogels. Langmuir 16:4485–4494
Terech P, Rodriguez V, Barnes JD, McKenna GB (1994) Organogels and aerogels of racemic and chiral 12-hydroxyoctadecanoic acid. Langmuir 10:3406–3418
Terech P, Weiss RG (1997) Low molecular mass gelators of organic liquids and the properties of their gels. Chem Rev 97:3133–3159
Wang RY, Wang P, Li JL, Yuan B, Liu Y, Li L, Liu XY (2013) From kinetic-structure analysis to engineering crystalline fiber networks in soft materials. PCCP 15:3313–3319
Weiss RG, Terech P (2006) Molecular gels materials with self-assembled fibrillar networks. Springer, Dordrecht
Zheng XF, Fu J, Lu XY (2012) Solubility and induction period study of asiaticoside and madecassoside in a methanol plus water mixture. J Chem Eng Data 57:3258–3263
Acknowledgements
The synchrotron SAXS measurements were performed under the approval of Photon Factory Program Advisory Committee (Proposals 2010G134 and 2012G121).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takeno, H., Mochizuki, T. A structural development of an organogel explored by synchrotron time-resolved small-angle X-ray scattering. Colloid Polym Sci 291, 2783–2789 (2013). https://doi.org/10.1007/s00396-013-3011-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-013-3011-0